
Abstract
Exosomes (exos) are primarily responsible for the process of mesenchymal stem cells (MSCs) treatment for acute lung injury (ALI), but the mechanism remains unclear, particularly in altered microenvironment. Therefore, this study aimed to investigate the potential mechanism of exos derived from human umbilical cord mesenchymal stem cells (hucMSCs) primed with interferon-gamma (IFN-γ) on ALI and to propose a promising and cell-free strategy. This study extracted exos from hucMSCs supernatant primed and unprimed with IFN-γ marked with IFN-γ-exos and CON-exos, which were identified and traced. IFN-γ-exos administration to ALI models suppressed the NF-κB signaling pathway compared to CON-exos, which were quantified through western blot and immunohistochemical staining. Reverse transcription-quantitative polymerase chain reaction validated miR-199b-5p expression in the IFN-γ-exos and CON-exos treatment groups. Data analysis, a dual-luciferase reporter assay, and cell transfection were conducted to investigate the target binding between miR-199b-5p and Aftiphilin (AFTPH), with AFTPH expression analyzed via cell immunofluorescence and western blot. Co-immunoprecipitation was conducted for the interaction between AFTPH and NF-κB p65. The result revealed that miR-199b-5p was down-regulated in the IFN-γ-exos treatment group, which had a target binding site with AFTPH, and an interaction with NF-κB p65. Consequently, IFN-γ-exos inhibited the NF-κB signaling pathway in ALI in vitro and in vivo through the miR-199b-5p/AFTPH axis. Our results demonstrated new directions of novel and targeted treatment for ALI.
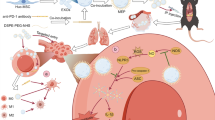
Graphical Abstract

The mechanism diagrams of the NF-κB signaling pathway in ALI regulated by IFN-γ-exos. Functional exosomes derived from hucMSCs primed with IFN-γ, which mediates miR-199b-5p targets AFTPH and negatively regulates the NF-κB signaling pathway to inhibit the production of NLRP3, GSDMD, caspase-1, and IL-1β, attenuating the inflammatory response on LPS-induced ALI.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute lung injury (ALI) is characterized by high morbidity, mortality, prolonged hospitalization, high costs, and a poor long-term prognosis post–discharge [1]. The fatality rate of ALI has markedly increased since the coronavirus disease-2019 pandemic [2], due to the complicated aetiopathogenesis of ALI [3], which is closely associated with inflammation, coagulation, alveolar epithelial damage, vascular endothelial dysfunction, etc. [4], and for which specific therapy is currently unavailable. Among the intricate pathological mechanisms, nuclear factor kappa-B (NF-κB)signaling pathway activation plays a crucial role in inflammation initiation and maintenance in ALI, which profoundly affects cellular transcription and translation processes, mediated by the NLRP3 inflammasome, which is composed of the NLRP3, and carried by the gasdermin D (GSDMD), which activates caspase-1 during cleavage [5, 6] and promotes the maturation of interleukin (IL-)-1β [7]. GSDMD is a cellular pyroptosis program executor and participant in highly inflammatory forms of cell death, and current results indicated that GSDMD is involved in NF-κB/NLRP3 signaling activation.
Currently, mesenchymal stem cells (MSCs) have been proven to perform curative impacts via paracrine pathways [8], which might interact with cells of interest and modify the path of the recipient by activating endogenous stem cells, inhibiting apoptosis, modulating inflammatory responses, reducing fibrosis, and facilitating chemotaxis [9]. Exosomes(exos) have been effectively used in treating a wide variety of disorders because of their unique capacity to transport and share intracellular chemical information [10]. Furthermore, exos derived from MSCs (MSC-exos) have properties comparable to those of host cells, which contain mRNA, microRNA, circRNA, lncRNA, proteins, lipids, and other active substances that participate in cell-cell communication [11, 12].
However, the microenvironment greatly affected the function of MSCs. MSCs are in a comatose state in the usual microenvironment, but their potent immune regulatory function is triggered in an altered microenvironment. A study [13] revealed that MSCs stimulated with inflammatory factors may enhance the activity of a specific MMP protein and cell migration, extravasation, and recruitment. Further, the immunosuppressive capabilities of MSCs may be well established and associated with indoleamine 2, 3-dioxygenase (IDO), soluble substances, such as hepatocyte growth factor, and transforming growth factor (TGF)-β [14, 15].
Interferon-gamma (IFN-γ) may be a priming trigger for the immunosuppressive effects of MSCs. Further, another study reported [16] that MSCs, primed with IFN-γ, remain active and effective in treating graft versus host disease. Previous research investigating [17] the changes in MSCs’ intracellular proteome and metabolomics after IFN-γ priming revealed that increased anti-pathogenic proteins synthesis prevented inflammation and fibrosis and enhanced survival. The International Society for Cell Therapy (ISCT) deemed the MSCs’ response to IFN-γ as the primary indicator of promoting the upregulation of immunosuppressive factors, such as IDO, programmed cell death 1 ligand 1, human leukocyte antigen, and antibacterial and antiviral factors [17, 18], while other pro-inflammatory cytokines and TLR agonists have influenced MSCs’ activities.
Our previous investigation, revealed that hucMSCs treatment can considerably reduce the inflammatory response in patients with ALI [19, 20]. Thus, we collected IFN-γ-exos and CON-exos, which were administrated in the ALI in vivo and in vitro to accurately explore the potential biological mechanisms of hucMSC-exos in an inflammatory microenvironment to find new targets for treating ALI.
Materials and Methods
Cell Experimental Design
Procell Life Science&Technology provided A549 and HEK293T cells, which were cultured in DMEM supplemented with 10% fetal bovine serum, 100 U/mL of penicillin, and 100 g/mL of streptomycin. The cells were maintained at 37 °C and 5% CO2. Lipopolysaccharide (LPS) was acquired from Sigma. A549 cells with inflammatory damage were constructed with 1 g/mL of LPS for 24 h.
We purchased hucMSCs from the Stem Cell Technology Application Research Center in Yunnan and maintained them in a special culture medium without exos for stem cells (System Biosciences, USA). IFN-γ was acquired from PeproTech Inc., America. Exos derived from hucMSCs primed or unprimed with 5 ng/mL of IFN-γ, which were noted as IFN-γ-exos and CON-exos, and centrifuged by ultrafiltration concentration and successive differential centrifugation methods [21], respectively, followed identification and applied to the LPS-induced ALI cell model, and groups were as described with CON, LPS, LPS + phosphate-buffered saline (PBS), LPS+CON-exos, and LPS+IFN-γ-exos. CON represented the normal control group. The LPS group consisted of A549 cells with inflammatory damage. The LPS+PBS group included A549 cells with inflammatory damage administered with PBS of equal volume for 24 h. The LPS+CON-exos group contained A549 cells with inflammatory damage administered with CON-exos at 50 μg/mL for 24 h. The LPS+IFN-γ-exos group involved A549 cells with inflammatory damage administered with IFN-γ-exos at 50 μg/mL for 24 h.
Animal Experimental Design
Animal experiments were performed under the Experimental Animal Care Guidelines. The C57BL/6 male mice (20 ± 2 g, Experimental Animal Center of Kunming Medical University, Yunnan, China) were raised in a special pathogen-free environment.
The ALI mouse model was constructed by infusing LPS of 2.5 mg/kg via the trachea for 6 h and treating with PBS, IFN-γ-exos, or CON-exos, as described for the CON, LPS, LPS+PBS, LPS+CON-exos, and LPS+IFN-γ-exos groups. The CON group represented the normal control group. The LPS group consisted of ALI mouse models. The LPS+PBS group included ALI mouse models treated with 25 μL of PBS buffer via the tail vein for 24 h. The LPS+CON-exos group consisted of ALI mouse models administered with 25 μL of CON-exos via tail vein for 24 h. The LPS+IFN-γ-exos group included ALI mouse models administered with 25 μL of IFN-γ-exos via the tail vein for 24 h. Sodium pentobarbital was used to induce deep anesthesia in the mice, and the lung tissue was extracted to execute the following analyses: the right inferior lung tissue was used to perform a western blot and the right inferior lung tissue was used to process IHC sections.
Identification and Tracking in Exos
This study investigated the morphology of exos using transmission electron microscopy (TEM) after isolating from the hucMSCs supernatant through ultrafiltration concentration and successive differential centrifugation. Simultaneously, we verified the exos by western blot of exosome surface marker antibodies CD63, CD81, TSG101. Finally, we fluorescently labeled the exos with PKH-26 to observe the tracing of exos in A549 cells.
Western Blot and Co-Immunoprecipitation Analysis
We used the following for western blot: CD63, CD81, TSG101, NF-κB p65, NLRP3, GSDMD, IL-1β, GAPDH, and β-actin rabbit monoclonal antibodies (Abcam, Cambridge, United Kingdom), AFTPH rabbit monoclonal antibodies (Novus Biological, USA), NF-κB p65 mouse monoclonal antibodies, immunoglobulin G (IgG) mouse antibodies and horseradish peroxidase-conjugated IgG rabbit antibodies (Proteintech Group Inc., Wuhan, China).
We used a lysed in a lysis mixture containing RIPA, phenylmethanesulfonyl fluoride, and protease and phosphatase inhibitor (Thermo Scientific, Massachusetts, USA) for 30 min at 4 °C to lyze the cells. The BCA protein assay kit (Beyotime Biotechnology, Shanghai, China) was used to determine protein content. We implemented 8%–10% Tris-HCL SDS-PAGE for protein extract electrophoresis. Afterward, we transferred the proteins onto a polyvinylidene fluoride (PVDF) membrane. After blocking and washing, the PVDF membrane underwent primary antibody incubation overnight at 4 °C, followed by secondary antibody incubation at room temperature for 1 h. Chemiluminescence detection was performed on the PVDF membrane.
NF-κB p65 mouse polyclonal and IgG mouse polyclonal were incubated with the total cell lysates overnight to develop IP groups for co-immunoprecipitation. The lysates were then spun with protein A/G beads (BioVision Inc., USA) for 4 h at 4 °C for co-immunoprecipitation. The beads were boiled for 10 min and then combined with SDS sample buffer (5X). NF-κB p65 and AFTPH expression were examined by western blot analysis.
Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
We purchased all reagents for RT-qPCR from Takara Biomedical Technology Co., Ltd., Beijing. The RNAiso for small RNA was used to extract total miRNAs in the LPS+CON-exos, LPS+IFN-γ-exos treatment, and cell transfection groups following the manufacturer’s instructions. The Mir-XTMmiRNA First-Strand Synthesis was used for reverse transcription synthesis, and TB Green Premix Ex TaqTM II (Tli RNaseH Plus) was utilized for qPCR. The following are the utilized primers in experiments: miR-199b-5p-forward 5′-GCCCCAGTGTTTAGACTATCTGTTC-3′; U6-forward 5′-CTCGCTTCGGCAGCACA-3′, reverse 5′-AACGCTTCACGAATTTGCGT-3′. The 2−△△Ct algorithm was used to determine the relative expression levels of miR-199b-5p and U6 as an internal reference.
Dual-Luciferase Reporter Assay
We used a dual-luciferase reporter assay to investigate the interaction between miR-199b-5p and AFTPH. Both the AFTPH-3′UTR-WT and AFTPH-3′UTR-MUT luciferase plasmids were designed, followed by transfection into HEK293T cells. The transfected cells were then incubated with both the miR-199b-5p-NC and mimic. The original medium was substituted with a fresh and full culture medium after a 6 h transcription period. We monitored the expression of firefly fluorescence in the cells using a microplate technique. The renilla luminescence production was then investigated when the cells were exposed to the Stop & and Glo® substrate.
Cell Transfect
We transfected A549 cells with 50 nM of miR-199b-5p inhibitor NC, miR-199b-5p mimic NC, miR-199b-5p inhibitor, and miR-199b-5p mimic (Guangzhou RiboBio Co., Ltd., Guangdong, China) for 8 h, respectively, and we changed the cultivation to fresh normal complete media for 24 h. The CON group was cultured under normal conditions. MiR-199b-5p mimics sequence was 5′- CCCAGUGUUUAGACUAUCUGUUG-3′(sense) and 5′-ACGUGACACGUUCGGAGAATT -3′ (antisense); MiR-199b-5p mimics NC sequence was 5′-UUCUCCGAACGUGUCACGUTT-3′(sense); MiR-199b-5p inhibitor sequence was 5′-GAACAGAUAGUCUAAACACUGGG-3′ and 5′-CAGUACUUUUGUGUAGUACAA-3′(antisense); MiR-199b-5p inhibitor NC sequence was 5′-ACAGAUAGUCUAAACACUGGGUU-3′(antisense). We used western blot to detect the expression of AFTPH and utilized RT-qPCR to identify the relative expression of miR-199b-5p in each group.
Immunofluorescence Staining
A549 cells were treated following the cell experimental design, followed by fixation in 4% paraformaldehyde (Biosharp Life Sciences, Anhui, China) for 20 min, broken with 0.5% Triton X-100, and closed with 10% goat serum (Biosharp Life Sciences, Anhui, China) for 2 h at ambient temperature. The cells were then incubated with AFTPH primary antibody (diluted to 0.25–2 g/mL) overnight at 4 °C, and the goat anti-rabbit IgG (H+L) fluorescent secondary antibody was incubated at room temperature for 1 h away from light. We placed the phalloidin reagent (Beyotime Biotechnology, Shanghai, China) on top and let it sit for 1 h at room temperature. A final 20 μL of anti-fluorescent quencher containing DAPI was utilized for nuclei labeling. Laser confocal fluorescence microscopy was used to investigate the intracellular fluorescence distribution and intensity, which was then assessed for relative fluorescence quantification using Image J software.
Immunohistochemistry (IHC)
The lower right lung tissue in every group was fixed using a 4% solution of paraformaldehyde for 24 h to perform IHC staining. After fixation, the tissue was processed through paraffin embedding, sectioned, and cut into slices with a 5-μm thickness. The slices underwent dewaxing, rehydration, and saponification. Subsequently, the slices were fixed with sodium citrate antigen, broken with 3% Triton X-100, and sealed at room temperature for 2 h with 10% goat serum. Afterward, NF-κB p65, IL-1β, and caspase-1 primary antibodies (diluted in 1:200) were left to incubate at ambient temperature for 1 h. HRP goat anti-rabbit IgG (H+L) was incubated at room temperature for 1 h. Sections were stained for DAB reagent following instructions. Finally, the sections were nucleofected, and images were analyzed through a white-light microscope.
Statistical Analysis
Statistical Package for the Social Sciences version 13.0 was used for statistical difference analysis. The data were all displayed with the mean ± standard deviation. The data analysis was repeated at least three times. The Shapiro-Wilk test, also known as the QQ plot, was used to normalize the data distribution. The two distinct sample t-test is applied to compare data from two groups. A one-way analysis of variance was used to assess the variations among three or more groups. Additionally, the P-value has to be <0.05, for a statistical difference to be declared.
Results
Isolation and Identification of CON-exos and IFN-γ-exos
We successfully isolated and identified CON-exos and IFN-γ-exos to investigate their function in ALI inflammatory injury. TEM was used to investigate CON-exos and IFN-γ-exos, and the structures were found to be double concave in shape (Fig. 1A). Subsequently, Fig. 1B shows that CON-exos and IFN-γ-exos that expressed CD63, CD81, and TSG101 level.Further, the A549 cells can successfully phagocytose them after CON-exos and IFN-γ-exos treatment (Fig. 1C). We have effectively extracted CON-exos and IFN-γ-exos as a consequence.

Isolation and identification of CON-exos and IFN-γ-exos. A Identification of exos and I -exos by TEM (scale bar=100 nm); B Measurement of protein levels of CD63, CD81, TSG101and in CON-exos and IFN-γ-exos by western blotting; C PKH-26 fluorescent staining detection (scale bar = 20 μm)
IFN-γ-exos Significantly Alleviates Inflammation in LPS-Induced A549 Cells
We measure inflammatory-related proteins to observe the association of IFN-γ-exos with inflammation in LPS-induced A549 cells. The results revealed increased NF-κB p65, NLRP3, GSDMD, and IL-1β expression, which decreased following IFN-γ-exos treatment in LPS-induced A549 cells (Fig. 2A–E). Hence, the IFN-γ-exos demonstrated significant capability in inhibiting the inflammatory reaction of A549 cells induced by LPS.

IFN-γ-exos significantly alleviates inflammation in LPS-induced A549 cells. A Protein band stripes of NF-κB p65, NLRP3, GSDMD and IL-1β expression in each group in LPS-induced A549 cells were detected by western blot; B–E Relative expression levels of NF-κB p65, NLRP3, GSDMD and IL-1β in each group. Differences between three or more groups were compared using a one-way analysis of variance (ANOVA). (ns: P > 0.05, *P < 0.05, **P < 0.01; ***P < 0.001)
IFN-γ-exos Alleviates Inflammation in ALI Mouse
We have constructed the mice model as the preliminary study to investigate the influence of IFN-γ-exos on inflammation in vivo. We discovered that IFN-γ-exos could suppress inflammation in ALI mice. IHC staining revealed that the IL-1β, caspase-1, and NF-κB p65 in lung tissues of mice were more obvious compared with the CON group after being induced with LPS. Further, IFN-γ-exos remarkably inhibited the expression of IL-1β, caspase-1, and NF-κB p65 in inflammatory lung tissues (Fig. 3A, B). Ultimately, we revealed substantially elevated NF-κB p65 and GSDMD levels in lung tissue following LPS induction. However, the effect of IFN-γ-exos was effectively reversed upon inhibition, as evidenced by western blot (Fig. 3C, D), indicating that functional exos that contain active substances may mediate NF-κB signaling pathway regulation.

IFN-γ-exos alleviates inflammation in ALI mouse. A Immunohistochemical staining of NF-κB p65, IL-1β, and caspase-1 expression in the lung tissues of mice in each group(scale bar=50μm); B Relative positive expression levels of NF-κB p65, IL-1β, and caspase-1. C Expression of NF-κB p65 and GSDMD in the lung tissues of mice was detected by western blot; D Relative expression levels of NF-κB p65 and GSDMD. Differences between three or more groups were compared using a one-way analysis of variance (ANOVA). (ns: P > 0.05, *P < 0.05, **P < 0.01; ***P < 0.001)
Influence of miR-199b-5p on Inflammatory Injury A549 Cells Treated with IFN-γ-exos
After a preliminary pre-experimental study, miR-199b-5p was revealed to be associated with inflammatory injury in acute lung injury, and the expression of miR-199b-5p was significantly down-regulated with IFN-γ-exos treatment compared to the CON-exos (Fig. 4A), maybe IFN-γ-exos contains a large number of functionally active compounds that may dramatically absorb miR-199b-5p expression in A549 cells under inflammatory injury will be further explored.

The target binding between miR-199b-5p and AFTPH. A Expression of miR-199b-5p in LPS+CON-exos and LPS+IFN-γ-exos measured by RT-qPCR; B Prediction analysis of miR-199b-5p and target gene of AFTPH via databases; C The results of double luciferase reporter gene analysis; D Relative expression of miR-199b-5p in transfected cells was measured by RT-qPCR; E Expression of AFTPH protein in cell transfection was detected by western blot; F Relative expression of AFTPH was assessed. Differences between each value were analyzed by the Student’s t-test and three groups were compared using a one-way analysis of variance (ANOVA). (ns: P > 0.05, *P < 0.05, **P < 0.01; ***P < 0.001)
The Target Binding Between miR-199b-5p and AFTPH
The possible binding site for miR-199b-5p and AFTPH-3′UTR was revealed using TargetScan (www.targetscan.org) and miRanda (www.micorrna.org) databases (Fig. 4B). We verified the interaction between miR-199b-5p and AFTPH using cell transfection and double luciferase reporter assay system. The miR-199b-5p mimic group demonstrated a significant reduction in fluorescence intensity compared to the miR-199b-5p-NC group in the AFTPH-3′UTR-WT group, indicating the role of miR-199b-5p on the AFTPH-3′UTR region. The difference in fluorescence intensity was not significantly different in the presence of miR-199b-5p mimic and miR-199b-5p-NC group after mutating the AFTPH-3′UTR. However, the fluorescence intensity was considerably less in the miR-199b-5p mimic group than in the AFTPH-3′UTR-WT group compare with the AFTPH-3′UTR-MUT group (Fig. 4C).
Furthermore, miR-199b-5p and AFTPH expression statistically remained between the CON group, the miR-199b-5p mimic NC group and the miR-199b-5p inhibitor NC group. The miR-199b-5p inhibitor and mimic groups demonstrated considerably decreased and increased miR-199b-5p expression, but with elevated and substantially reduced AFTPH protein expression, respectively (Fig. 4D–F). Therefor, miR-199b-5p might affect the transcription and translation stages of AFTPH.
AFTPH Expression in ALI In Vivo and In Vitro
Cell immunofluorescence and western blot analysis revealed that AFTPH protein expression was considerably diminished in the LPS group than in the CON group, while it was greater in the LPS+IFN-γ-exos group compared to the LPS+CON-exos group (Fig. 5A–D). The results indicated that AFTPH may be implicated in modulating the inflammatory reaction of IFN-γ-exos on LPS-induced cells.

Effects of IFN-γ-exos on the expression of the AFTPH. A Laser confocal microscopy was used to observe AFTPH-Cy5 fluorescence signals in A549 cells(scale bar = 50 μm); B Relative protein expression of AFTPH in cell immunofluorescence; C Protein stripe of AFTPH in each group in vivo was detected by western blot; D Relative expression of AFTPH in the lung tissues of mice. The interaction between AFTPH and NF-κB p65 protein was detected by co-immunoprecipitation. Differences between three or more groups were compared using a one-way analysis of variance (ANOVA). (ns: P > 0.05, *P < 0.05, **P < 0.01; ***P < 0.001)
The Interaction Between AFTPH and NF-κB p65
Co-immunoprecipitation results demonstrated the protein association between NF-κB p65 and AFTPH. IgG did not bind to NF-κB p65 and AFTPH protein in the IP group, while the NF-κB p65 precipitation experiment not only precipitated NF-κB p65 protein but also AFTPH protein, indicating that protein NF-κB p65 and protein AFTPH have protein interactions (Fig. 5E).
Discussion
Previous studies [19, 20] reported the undeniable therapeutic efficacy of hucMSCs on ALI. Immunomodulatory factors such as TGF-β and IDO [22, 23], have been demonstrated to minimize the expression of histocompatibility complex II (MHC II) and co-stimulatory molecules CD80 and CD86 when hucMSCs are incubated with certain IFN-γ concentration, indicating that immune plasticity exists in hucMSCs and the exertion is primarily dependent on exos for mediator distribution. Previous studies [24] have indicated that NF-κB signaling pathway inhabitation provides substantial protection against ALI, indicating a potential role of this pathway in the development of ALI. Consequently, we postulated that IFN-γ-exos would exhibit a more potent regulatory impact on the NF-κB signaling pathway in ALI, thereby initiating a comprehensive investigation by our entire research team.
The study revealed that LPS injury enhanced NF-κB p65, NLRP3, caspase-1, IL-1β, and GSDMD production. This indicated the NF-κB signaling pathway activation and NLRP3 cell pyroptosis, thereby subsequently amplifying the classical caspase-1 pathway. Consequently, GSDMD synthesis and secretion of IL-1β was elevated. In contrast, IFN-γ-exos dramatically decreased NF-κB and NLRP3 production. The findings imply, on the one hand, that exos mediate at least some of hucMSC capacity and implementation, and, on the other hand, that hucMSC microenvironmental tuning may be capable of targeting performance modification. NLRP3 inflammation is crucial in conventional Caspase-1-dependent pathway activation and amplification, including multiple molecules as triggers, such as K+ efflux, ROS stimulation, or lysosomal rupture. However, the stimulation processes of many inflammatory remained unknown. Several studies revealed that inhibiting NLRP3 inflammation is now a fundamental medicinal option for ALI. A previous study [25] revealed that NF-κB/TLR4 signaling pathway activation in ALI is also linked to t NLRP3 inflammation production. A study [26] revealed that TLR4 enhances the levels of caspase-1, NF-κB p65, NLRP3, and MyD88, while controlling NLRP3 inflammation activation and the inflammatory reaction of alveolar macrophages through the MyD88/ NF-κB pathway.
We performed the differential analysis of miR-199b-5p for LPS+IFN-γ-exos and LPS+CON-exos groups by RT-qPCR according to previous reports, and we revealed significantly down-regulated miR-199b-5p expression after IFN-γ-exos treatment. Therefore, miR-199b-5p, which might be used for inflammatory regulation of hucMSCs-exos primed with IFN-γ in LPS-induced ALI, was the focus of the study. Studies have indicated that miR-199b-5p was a tumor growth inhibitor in various cancer types. A study [27] revealed that miR-199b-5p overexpression suppresses prostate cancer cell proliferation and metastasis by regulating the DDR1-Erk pathway in vitro and in vivo. Further, a study [28] demonstrated that miR-199 contributes to DOX-induced cardiotoxicity via inhibiting TaF9-mediated autophagy and apoptosis. Furthermore, miR-199b-5p binds to its promoter area via a negative feedback regulatory ring mediated by Hes1 and is down-regulated by CpG island methylation upstream of its promoter region [29]. However, how miR-199b-5p relates to the inflammatory reaction in ALI remains unknown. We propose another hypothesis that miR-199b-5p, which is down-regulated by IFN-γ-exos resulting from ALI treatment, may be involved NF-κB signaling pathway regulation, in addition to NF-κB signaling pathway suppression by IFN-γ-exos in the above studies.
AFTPH, which was the miR-199b-5p downstream target gene, was found through the database study and verified by dual-luciferase reporter assay and cell transfection in this study. Mattera et al. [30] first discovered the presence of AFTPH in the trans-Golgi network and endosome-associated protein linkage system, bound to the heterotetrameric articulation through a network protein binding motif, and primarily involved in intracellular transport and cytokinesis based on combining with the adaptor protein ear domain of the adaptor complexes (AP) 1 and 2. A study [31] revealed the abundance of AFTPH in clathrin-coated vesicles in the brain and its involvement in the trafficking of synapses and synaptophysin. Additionally, a study [32, 33] revealed that NT-colon epithelial cells control NTR1 transportation into the cytoplasm via miR-133/AFTPH and are enhanced in the inflammatory intestine.
Law et al. conducted three studies on AFTPH, which focused on investigating the potential mechanisms of the NT/NTR1/miR-133a/AFTPH axes in colitis and dextran sodium sulfate-induced colonic tissue in mice [32, 33]. These studies found an increased neuro hypocretin (NT) and neuro hypocretin receptor (NTR1), as well as decreased miR-133a. Further, miR-133a can regulate the transcriptional levels of the downstream target gene AFTPH in both colitis mouse models [35,36,37] and patients with ulcerative colitis [34]. This regulation activates ERK [38, 39], AKT, MARK, and NF-κB signaling pathways in colonic epithelial cell models [40, 44]. Additionally, miR-133a and colorectal cancer development demonstrated a potential association between them [45, 46]. Consequently, the inflammatory response exerts a negative regulation on AFTPH. Our study revealed that IFN-γ-exos demonstrated superior anti-inflammatory effects in ALI. Therefore, we conducted co-immunoprecipitation assays to investigate the interaction between AFTPH and NF-κB p65 protein. Our results revealed a correlation between AFTPH and NF-κB p65 protein. The active components released during exocytosis. It is suggested topotentially modulate the regulation of AFTPH gene expression by targeting miR-199b-5p, causing a reduction in NF-κB signaling pathway activation. Hence, this could partially alleviate the inflammatory response in ALI. This indicates that differentially expressed miR-199b-5p in IFN-γ-exos might target the transcript stage of AFTPH, causing AFTPH protein expression upregulation. Consequently, it could negatively regulate NF-κB pathways and help suppress intracellular inflammatory responses to some extent. The discovery indicated that AFTPH protein expression is controlled negatively by NF-κB pathways, and is partially associated with the regulation of intracellular inflammatory responses.
The involvement of AFTPH in NF-κB nuclear transcription, its role in regulating the composition of specific vesicle populations at the proteins of the anti-Golgi lattice, and its association with endocytic uptake to develop vesicle populations for retrograde transport remains unknown. However, a study [39] revealed that AFTPH shares the same location as AP-1 in hippocampal neurons, along with synaptophysin. The result of Hirst et al. [47] revealed that AFTPH might play a role in categorizing the lysosomal protein histone D in cells other than neurons. Therefore, these problems must be further investigated. Furthermore, miR-199b-5p is down-regulated in the LPS+IFN-γ-exos group, indicating the presence of other active compounds such as circRNA and/or lncRNA in the exosome that act as sponge adsorbents in miR-199b-5p modulation. The physiological significance of exos is determined by the compounds that they transport. Exosomal RNA is frequently more diverse than other substances in exos and might reveal additional information regarding the cell of origin [48]. The presence of messenger RNA (mRNA), transfer RNA, ribosomal RNA, and miRNA is associated with gene and RNA regulation, RNA fragmentation, and RNA interference via exosomal transport, which are linked to the progression of diseases and various biological processes. Hence, we will proceed to do high-throughput sequencing of no-coding RNA from both kinds of exos.
The novelty of this study is that we investigated the potential molecular mechanism of IFN-γ-exos for ALI treatment, which mainly took the finding of a remarkable miR-199b-5p reduction after IFN-γ-exos treatment in the previous study as the cutting point, and revealed that miR-199b-5p is an upstream AFTPH regulator, which demonstrated has an NF-κB p65 protein interaction. Further, we speculate the involvement of the stability of the NF-κB complex, which subsequently regulates the nuclear transcription and transcriptional NF-κB levels. The current results provide no definitive explanation for the targeted regulatory relationship between AFTPH and NF-κB p65, but further studies will investigate deeper into the regulatory role of AFTPH on NF-κB p65.
Conclusion
In summary, our research confirmed the distinct expression of miR-199b-5p in IFN-γ-exos and CON-exos treatment, thereby demonstrating the role of miR-199b-5p in controlling AFTPH expression and its interaction with the NF-κB signaling pathway. Hence, IFN-γ-exos potentially facilitated miR-199b-5p’s targeting of AFTPH, hindering the NF-κB signaling pathway activation and reducing the inflammatory response in ALI. The results could potentially pave the way for a novel research direction in cell-free therapy focused on modifying the MSCs microenvironment. Additionally, AFTPH may appear as a crucial target for prospective treatment of ALI.
Data Availability
The datasets used in this work are accessible upon reasonable request from the corresponding author.
References
Huang, X., Zhang, R., & Fan, G., et al. (2020). Incidence and outcomes of acute respiratory distress syndrome in intensive care units of mainland China: a multicentre prospective longitudinal study. Critical Care, 24(1), 515. https://doi.org/10.1186/s13054-020-03112-0.
Martin, T. R. (2022). Lung injury and repair in coronavirus disease 2019-related acute lung injury. The American Journal of Pathology, 192(3), 406–409. https://doi.org/10.1016/j.ajpath.2022.01.001.
Beitler, J. R., Thompson, B. T., & Baron, R. M., et al. (2022). Advancing precision medicine for acute respiratory distress syndrome. The Lancet Respiratory Medicine, 10(1), 107–120. https://doi.org/10.1016/S2213-2600(21)00157-0.
Broz, P., & Dixit, V. M. (2016). Inflammasomes: mechanism of assembly, regulation and signalling. Nature Reviews Immunology, 16(7), 407–420. https://doi.org/10.1038/nri.2016.58.
Wang, Z., Zhang, S., & Xiao, Y., et al. (2020). NLRP3 inflammasome and inflammatory diseases. Oxidative Medicine and Cellular Longevity, 2020, 4063562. https://doi.org/10.1155/2020/4063562.
Lee, S., Suh, G. Y., & Ryter, S. W., et al. (2016). Regulation and function of the nucleotide binding domain leucine-rich repeat-containing receptor, pyrin domain-containing-3 inflammasome in lung disease. American Journal of Respiratory Cell and Molecular Biology, 54(2), 151–160. https://doi.org/10.1165/rcmb.2015-0231TR.
Rathinam, V. A. K., Zhao, Y., & Shao, F. (2019). Innate immunity to intracellular LPS. Nature Immunology, 20(5), 527–533. https://doi.org/10.1038/s41590-019-0368-3.
Han, Y., Yang, J., & Fang, J., et al. (2022). The secretion profile of mesenchymal stem cells and potential applications in treating human diseases. Signal Transduction and Targeted Therapy, 7(1), 92. https://doi.org/10.1038/s41392-022-00932-0.
Shi, Y., Wang, Y., & Li, Q., et al. (2018). Immunoregulatory mechanisms of mesenchymal stem and stromal cells in inflammatory diseases. Nature Reviews Nephrology, 14(8), 493–507. https://doi.org/10.1038/s41581-018-0023-5.
Wang, S., Lei, B., & Zhang, E., et al. (2022). Targeted therapy for inflammatory diseases with mesenchymal stem cells and their derived exosomes: from basic to clinics. International Journal of Nanomedicine, 17, 1757–1781. https://doi.org/10.2147/ijn.S355366.
Rani, S., & Ritter, T. J. A. M. (2016). The exosome - a naturally secreted nanoparticle and its application to wound healing. Advanced Materials, 28(27), 5542–5552. https://doi.org/10.1002/adma.201504009.
Brennan, M., Layrolle, P., & Mooney, D. J. (2020). Biomaterials functionalized with MSC secreted extracellular vesicles and soluble factors for tissue regeneration. Advanced Functional Materials, 30(37), 1909125. https://doi.org/10.1002/adfm.201909125.
Ries, C., Egea, V., & Karow, M., et al. (2007). MMP-2, MT1-MMP, and TIMP-2 are essential for the invasive capacity of human mesenchymal stem cells: differential regulation by inflammatory cytokines. Clinical Trials, 109(9), 4055–4063. https://doi.org/10.1182/blood-2006-10-051060.
Sheng, H., Wang, Y., & Jin, Y., et al. (2008). A critical role of IFNgamma in priming MSC-mediated suppression of T cell proliferation through up-regulation of B7-H1. Cell Research, 18(8), 846–857. https://doi.org/10.1038/cr.2008.80.
Kim, D. S., Jang, I. K., & Lee, M. W., et al. (2018). Enhanced Immunosuppressive Properties of Human Mesenchymal Stem Cells Primed by Interferon-gamma. EBioMedicine, 28, 261–273. https://doi.org/10.1016/j.ebiom.2018.01.002.
Polchert, D., Sobinsky, J., & Douglas, G., et al. (2008). IFN-gamma activation of mesenchymal stem cells for treatment and prevention of graft versus host disease. European Journal of Immunology, 38(6), 1745–1755. https://doi.org/10.1002/eji.200738129.
Wobma, H., Tamargo, M., & Goeta, S., et al. (2018). The influence of hypoxia and IFN-γ on the proteome and metabolome of therapeutic mesenchymal stem cells. Biomaterials., 167, 226–234. https://doi.org/10.1016/j.biomaterials.2018.03.027.
Krampera, M., Galipeau, J., & Shi, Y., et al. (2013). Immunological characterization of multipotent mesenchymal stromal cells-The International Society for Cellular Therapy (ISCT) working proposal. Cytotherapy, 15(9), 1054–1061. https://doi.org/10.1016/j.jcyt.2013.02.010.
Liang, B., Chen, J., & Li, T., et al. (2020). Clinical remission of a critically ill COVID-19 patient treated by human umbilical cord mesenchymal stem cells: A case report. Medicine, 99(31), e21429. https://doi.org/10.1097/MD.0000000000021429.
Lu, K., Geng, S. T., & Tang, S., et al. (2022). Clinical efficacy and mechanism of mesenchymal stromal cells in treatment of COVID-19. Stem Cell Research & Therapy, 13(1), 61. https://doi.org/10.1186/s13287-022-02743-0.
Zhang, Y., Bi, J., & Huang, J., et al. (2020). Exosome: a review of its classification, isolation techniques, storage, diagnostic and targeted therapy applications. International Journal of Nanomedicine, 15, 6917–6934. https://doi.org/10.2147/ijn.S264498.
Croitoru-Lamoury, J., Lamoury, F. M., & Caristo, M., et al. (2011). Interferon-γ regulates the proliferation and differentiation of mesenchymal stem cells via activation of indoleamine 2,3 dioxygenase (IDO). PLoS ONE, 6(2), e14698. https://doi.org/10.1371/journal.pone.0014698.
Lin, W., Oh, S. K., & Choo, A. B., et al. (2012). Activated T cells modulate immunosuppression by embryonic-and bone marrow-derived mesenchymal stromal cells through a feedback mechanism. Cytotherapy, 14(3), 274–284. https://doi.org/10.3109/14653249.2011.635853.
He, S., Wu, L., & Sun, H., et al. (2022). Antioxidant biodegradable covalent cyclodextrin frameworks as particulate carriers for inhalation therapy against acute lung injury. ACS Applied Materials & Interfaces, 14(34), 38421–38435. https://doi.org/10.1021/acsami.2c05220.
Niu, X., Zang, L., & Li, W., et al. (2020). Anti-inflammatory effect of Yam Glycoprotein on lipopolysaccharide-induced acute lung injury via the NLRP3 and NF-κB/TLR4 signaling pathway. International Immunopharmacology, 81, 106024. https://doi.org/10.1016/j.intimp.2019.106024.
Luo, M., Hu, L., & Li, D., et al. (2017). MD-2 regulates LPS-induced NLRP3 inflammasome activation and IL-1beta secretion by a MyD88/NF-κB-dependent pathway in alveolar macrophages cell line. Molecular Immunology, 90, 1–10. https://doi.org/10.1016/j.molimm.2017.06.035.
Zhao, Z., Zhao, S., & Luo, L., et al. (2021). miR-199b-5p-DDR1-ERK signalling axis suppresses prostate cancer metastasis via inhibiting epithelial-mesenchymal transition. British Journal of Cancer, 124(5), 982–994. https://doi.org/10.1038/s41416-020-01187-8.
Yu, Y., Guo, D., & Zhao, L. (2022). MiR-199 aggravates doxorubicin-induced cardiotoxicity by targeting TAF9b. Evidence Based Complementary Alternative Medicine, 2022, 4364779. https://doi.org/10.1155/2022/4364779.
Garzia, L., Andolfo, I., & Cusanelli, E., et al. (2009). MicroRNA-199b-5p impairs cancer stem cells through negative regulation of HES1 in medulloblastoma. PLoS ONE, 4(3), e4998. https://doi.org/10.1371/journal.pone.0004998.
Mattera, R., Ritter, B., & Sidhu, S. S., et al. (2004). Definition of the consensus motif recognized by gamma-adaptin ear domains. Journal of Biological Chemistry, 279(9), 8018–8028. https://doi.org/10.1074/jbc.M311873200.
Burman, J. L., Wasiak, S., & Ritter, B., et al. (2005). Aftiphilin is a component of the clathrin machinery in neurons. FEBS Letters, 579(10), 2177–2184. https://doi.org/10.1016/j.febslet.2005.03.008.
Law, I. K., Jensen, D., & Bunnett, N. W., et al. (2016). Neurotensin-induced miR-133alpha expression regulates neurotensin receptor 1 recycling through its downstream target aftiphilin. Scientific Reports, 6, 22195. https://doi.org/10.1038/srep22195.
Law, I. K., Bakirtzi, K., & Polytarchou, C., et al. (2015). Neurotensin-regulated miR-133alpha is involved in proinflammatory signalling in human colonic epithelial cells and in experimental colitis. Gut, 64(7), 1095–1104. https://doi.org/10.1136/gutjnl-2014-307329.
Brun, P., & Mastrotto, C., et al. (2005). Neuropeptide neurotensin stimulates intestinal wound healing following chronic intestinal inflammation. American Journal of Physiology Gastrointestinal and Liver Physiology, 288(4), G621–G629. https://doi.org/10.1152/ajpgi.00140.2004.
Castagliuolo, I., Wang, C. C., & Valenick, L., et al. (1999). Neurotensin is a proinflammatory neuropeptide in colonicinflammation. Journal of Clinical Investigation, 103(6), 843–849. https://doi.org/10.1172/jci4217.
Eysselein V. E., Nast CCJZFGV. Neuropeptides and inflammatory bowel disease. 2009, 26(6):253. https://doi.org/10.1097/MOG.0b013e328331b69e.
Zhao, D., & Pothoulakis, C. J. P. (2006). Effects of NT on gastrointestinal motility and secretion, and role in intestinal inflammation. Peptides, 27(10), 2434–2444. https://doi.org/10.1016/j.peptides.2005.12.016.
Cui, W., Zhang, S., & Shan, C., et al. (2013). microRNA-133a regulates the cell cycle and proliferation of breast cancer cells by targeting epidermal growth factor receptor through the EGFR/Akt signaling pathway. The FEBS Journal, 280(16), 3962–3974. https://doi.org/10.1111/febs.12398.
Josse, C., Bouznad, N., & Geurts, P., et al. (2014). Identification of a microRNA landscape targeting the PI3K/Akt signaling pathway in inflammation-induced colorectal carcinogenesis. American Journal of Physiology Gastrointestinal and Liver Physiology, 306(3), G229–G243. https://doi.org/10.1152/ajpgi.00484.2012.
Zhao, D., Keates, A. C., & Kuhnt-Moore, S., et al. (2001). Signal transduction pathways mediating neurotensin-stimulated interleukin-8 expression in human colonocytes. Journal of Biological Chemistry, 276(48), 44464–44471. https://doi.org/10.1074/jbc.M104942200.
Bakirtzi, K., Hatziapostolou, M., & Karagiannides, I., et al. (2011). Neurotensin signaling activates microRNAs-21 and -155 and Akt, promotes tumor growth in mice, and is increased in human colon tumors. Gastroenterology, 141(5), 1749–1761.e1741. https://doi.org/10.1053/j.gastro.2011.07.038.
Zhao, D., Bakirtzi, K., & Zhan, Y., et al. (2011). Insulin-like growth factor-1 receptor transactivation modulates the inflammatory and proliferative responses of neurotensin in human colonic epithelial cells. Journal of Biological Chemistry, 286(8), 6092–6099. https://doi.org/10.1074/jbc.M110.192534.
Zhao, D., Kuhnt-Moore, S., & Zeng, H., et al. (2003). Neurotensin stimulates IL-8 expression in human colonic epithelial cells through Rho GTPase-mediated NF-kappa B pathways. American Journal of Physiology Cell Physiology, 284(6), C1397–C1404. https://doi.org/10.1152/ajpcell.00328.2002.
Zhao, D., Zhan, Y., & Zeng, H., et al. (2010). Neurotensin stimulates expression of early growth response gene-1 and EGF receptor through MAP kinase activation in human colonic epithelial cells. International Journal of Cancer, 120(8), 1652–1656. https://doi.org/10.1002/ijc.22407.
Ma, Y., Zhang, P., & Yang, J., et al. (2012). Candidate microRNA biomarkers in human colorectal cancer: systematic review profiling studies and experimental validation. International Journal of Cancer, 130(9), 2077–2087. https://doi.org/10.1002/ijc.26232.
Dong Y., Zhao J., Wu C. W., et al. MiR-133a activates the p53/p21 pathway and functions as a tumor suppressor in colorectal cancer by repressing RFFL. 2013, 11(9). https://doi.org/10.1158/1541-7786.MCR-13-0061.
Hirst, J., Borner, G., & Antrobus, R., et al. (2012). Distinct and overlapping roles for AP-1 and GGAs revealed by the “knocksideways” system. Current Biology, 22(18), 1711–1716. https://doi.org/10.1016/j.cub.2012.07.012.
Li, M., Zeringer, E., & Barta, T., et al. (2014). Analysis of the RNA content of the exosomes derived from blood serum and urine and its potential as biomarkers. Philosophical Transactions of the Royal Society of London Series B Biological Sciences, 369(1652), 20130502. https://doi.org/10.1098/rstb.2013.0502.
Funding
This work was supported by the National Natural Science Foundation of China (81960817 and 82260384) and the Kunming Science and Technology Bureau (2020-1 - H-011).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Guarantor, financial support, and manuscript review by C.Q. and H.W. Material preparation and data collection were performed by C.W., Y.Yang., and C.J. Data analysis: Y.Yang and Y.Yin. The first draft of the manuscript was written by C.W. and C.X. and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Ethics Approval
Animal experiments were performed in accordance with the Experimental Animal Care Guidelines and complied with the ethical criteria of the research ethics committee of the faculty of medicine at the Experimental Animal Center of Kunming Medical University, Kunming (Permission code: kmmu20230914).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wang, C., Yang, Y., Jiang, C. et al. Exosomes Derived from hucMSCs Primed with IFN-γ Suppress the NF-κB Signal Pathway in LPS-Induced ALI by Modulating the miR-199b-5p/AFTPH Axis. Cell Biochem Biophys 82, 647–658 (2024). https://doi.org/10.1007/s12013-023-01208-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12013-023-01208-2