
Abstract
2,3-butanediol is known to be a platform chemical with several potential industrial applications. Sustainable industrial scale production can be attained by using a sugarcane molasses based fermentation process using Bacillus subtilis. However, the accumulation of acetoin needs to be reduced to improve process efficiency. In this work, B. subtilis was genetically modified in order to increase the yield of 2,3-butanediol. Metabolic engineering strategies such as cofactor engineering and overexpression of the key enzyme butanediol dehydrogenase were attempted. Both the strategies individually led to a statistically significant increase in the 2,3-butanediol yields for sugarcane molasses based fermentation. Cofactor engineering led to a 26 % increase in 2,3-butanediol yield and overexpression of bdhA led to a 11 % increase. However, the combination of the two strategies did not lead to a synergistic increase in 2,3-butanediol yield.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
2,3-butanediol (2,3-BDO) is an industrially important molecule with several applications. It is utilized in the manufacture of printing inks, perfumes, fumigants, moistening and softening agents, resins, and lacquers. Apart from these uses, it can be utilized in the preparation of industrially important compounds such as methyl ethyl ketone and 1,3-butadiene [1]. It is produced in bulk quantities through chemical synthesis or fermentation routes [2]. Its sustainable production can be done by fermentation [3].
Bacillus subtilis has been known to produce 2,3-butanediol through the mixed acid metabolic pathway [4]. The benefits of using B. subtilis as producer strain are that it is a Generally Regarded As Safe (GRAS) microorganism and it has the ability to consume wide variety of sugars [5]. These qualities of the microbe enable 2,3-butanediol production from several feedstocks. Fermentative production of 2,3-butanediol by B. subtilis has been reported by using synthetic glucose, xylose, and sugarcane molasses [5–8]. The use of agricultural based feedstocks such as sugarcane molasses is a sustainable, economically viable and scalable way of producing 2,3-butanediol. In our previous study [6], we have shown the possibility of using sugarcane molasses for the production of 2,3-butanediol.
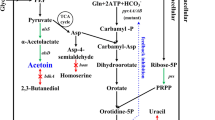
The 2,3-butanediol pathway involves the conversion of pyruvate to 2,3-butanediol. The sugars are converted to pyruvate by the glycolytic pathway. Then, the conversion of pyruvate to α-acetolactate is catalyzed by the enzyme α-acetolactate synthase (AlsS). Subsequently, acetoin is formed from α-acetolactate by the enzymatic action of α-acetolactate decarboxylase (AlsD) [5]. Acetoin is finally converted to 2,3-butanediol, and this reaction is catalyzed by the enzyme butanediol dehydrogenase (BdhA) [9] which involves the utilization of NADH (reduced form of nicotinamide adenine dinucleotide) [2]. The last step is also reported as the rate-limiting step in the pathway [5]. The formation of 2,3-butanediol is believed to be involved in regulating the NAD+/NADH ratio in the microbial cell [3].
One of the methods of strain improvement is cofactor engineering. It is employed for providing appropriate cofactor for increasing product formation. It may be done at the enzymatic reaction level, where the specificity of the enzyme to the cofactor is modulated or at the cellular level, where the intracellular concentrations of NADH or NADPH are modulated by cloning certain enzymes [10]. The intracellular regeneration of the reducing equivalent NADH has been reported previously for improving 2,3-butanediol production in various microbes. In Bacillus amyloliquefaciens, the improvement in 2,3-butanediol production is reported by using two different kinds of enzyme systems for cofactor regeneration, viz, overexpression of native glyceraldehyde 3-phosphate dehydrogenase [11] and heterologous expression of glycerol dehydrogenase [12]. In Escherichia coli, 2,3-butanediol production was enhanced by the heterologous expression of glucose dehydrogenase and formate dehydrogenase [13]. In B. subtilis, improvement in 2,3-butanediol production is seen by overexpression of a transhydrogenase by increasing the NADH pool [7].
The enzyme system formate dehydrogenase has several benefits over other regeneration systems and is widely used in industrial processes [14]. Formate dehydrogenase is responsible for the nearly irreversible conversion of formate to carbon dioxide with the concomitant formation of NADH. Formate is a cheap substrate and is not toxic to microbes. It is enzymatically converted to carbon dioxide which can be easily removed from the fermentation broth.
The over expression of butanediol dehydrogenase has been reported to be helpful for improving the conversion of acetoin to 2,3-butanediol in many organisms such as Lactococcus lactis [15], Saccharomyces cerevisiae [16, 17], B. amyloliquefaciens [11, 12], and B. subtilis [8]. In this work, metabolic engineering strategies such as cofactor regeneration and over-expression were tested for improvement of 2,3-butanediol yields in fermentation of sugarcane molasses using B. subtilis.
Materials and Methods
Bacterial Strains, Plasmids and Primers
B. subtilis 1A1 was obtained from Bacillus Genetic Stock Center (BGSC) and was used as the parent strain for genetic engineering. E. coli DH5α was used as an intermediate host for plasmid maintenance. The primers that are used in the study are mentioned in Table 1. The plasmids and strains are mentioned in Table 2.
Recombinant Plasmid and Strain Construction
The chromosomal DNA was isolated from Candida boidinii (MTCC228) using standard protocol (Promega, A1120). The formate dehydrogenase gene (fdh) was amplified from it by PCR using Primers P1 and P2. The gene fragment was ligated in BamHI-AatII digested vector pHCMC05 using Gibson assembly cloning kit (NEB, 5510S). The recombinant plasmid pHCMC-FDH was transformed in B. subtilis 1A1 by using protocol described by Vojcic et.al [18]. The transformants were selected on Luria Bernini (LB) agar plates possessing chloramphenicol at the concentration of 10 μg/ml. The mutant strain which episomally expressed formate dehydrogenase was referred to as the FDH mutant. Similarly, the control strain 1A1+05 was prepared by transforming empty vector pHCMC05 in B. subtilis 1A1 and resistance to chloramphenicol was used as criteria for transformant selection. The appropriate sequence of formate dehydrogenase gene and its flanking region within pHCMC05 was confirmed by DNA sequencing.
Two separate plasmids namely pMUTIN-GFP+ and pDG1662 were used for the construction of plasmid pDG1662-A. On isolation of genomic DNA from B. subtilis 1A1, the bdhA gene fragment was amplified by PCR from it using primers P3 and P4. The amplified gene fragment was ligated in KpnI-SpeI digested vector pMUTIN-GFP+ by using the Gibson Assembly cloning kit. The recombinant plasmid pMUTIN-bdh was extracted and used as template for further steps. Using the primer pairs P5 and P6, a cassette containing the erythromycin gene, bdh A gene, and lacI gene and their respective regulatory elements was amplified. This cassette was ligated in the EcoRI-SphI double digested vector pDG1662 using the Gibson Assembly cloning kit. The resulting recombinant plasmid pDG1662-A was transformed in B. subtilis 1A1 by the protocol described by Vjojec et.al [18]. The transformants were selected by growing them on LB plates containing erythromycin (4 μg/ml). Owing to the B. subtilis specific amyE up-down homology regions in pDG1662, the cassette was integrated at the amyE locus of the B. subtilis genome resulting in the BDH strain that had an additional copy of bdhA gene under the control of IPTG inducible Pspac promoter. The site of integration and the sequence of the integrated genes were confirmed by DNA sequencing. A mutant having both, the heterologous expression of fdh and overexpression of bdhA was also developed. It was constructed by transforming the BDH mutant with plasmid pHCMC05-FDH resulting in strain FDH+BDH. Similarly, a control strain was prepared by transforming the BDH mutant with empty plasmid pHCMC05 to form the strain BDH+05. The transformants conferred a joint resistance to both erythromycin (4 μg/ml) and chloramphenicol (10 μg/ml) and were selected by growing on LB agar plates having both the antibiotics.
Media and Culture Conditions
The seed medium composed of sugarcane molasses sugars 60 g/l (sucrose 43 g/l, glucose 6 g/l and fructose 11 g/l), corn steep liquor 15 g/l, yeast extract 5 g/l, ammonium sulfate 5 g/l and potassium dihydrogen phosphate 1 g/l. The fermentation medium composed of sugarcane molasses sugars 120 g/l (sucrose 77 g/l, glucose 19 g/l and fructose 24 g/l), soybean meal 9.5 g/l, corn steep liquor 15 g/l, and potassium chloride 2.9 g/l. A 5 % v/v inoculum was used for fermentation. The experiments were conducted in shake flasks at 15 % medium volume. Ten grams per liter sodium formate was spiked into the medium at 6 h after starting the fermentation. A 0.5-mM IPTG was added at 0 and 24 h of fermentation. Chloramphenicol was added in the plasmid carrying strains at a concentration of 10 μg/ml. The rpm was maintained at 150 using orbital shaker, and the temperature was maintained at 34 °C throughout the fermentation.
Analytical Methods
The sugars and acidic byproducts in the fermentation broth were analyzed by high-performance liquid chromatography (Agilent HPLC, 1200 series, Santa Clara, USA) equipped with RI detector at 40 °C. Organic acids and glycerol were measured using Aminex 87 HPX-H (300 × 7.8 mm) at 55 °C and 5 mM sulfuric acid as mobile phase. Fructose, glucose, and sucrose were determined using Aminex 87 HPX-N (300 × 7.8 mm) at 85 °C and 0.01 M di- sodium hydrogen phosphate as mobile phase. The flow rate of both mobile phases was 0.6 ml/min.
The analysis of acetoin, 2,3-butanediol, and ethanol was done by gas chromatography method. The fermentation broth was diluted with N, N Dimethyl formamide (DMF) with addition of isoamyl alcohol as internal standard and then quantified using a GC system (Agilent 7890 A, Santa Clara, USA) equipped with flame ionization detector and a 60 m ATTM-Wax capillary column (0.53-mm internal diameter, 1.0-um film thickness; Grace, USA). The operating conditions were as follows: helium used as carrier gas; the injector temperature and detector temperature were 220 and 240 °C, respectively; and the column oven temperature was maintained at 80 °C for 4 min, and then raised to 200 °C at the rate of 10 °C/min. The software Chemstation B03.02 was used for data acquisition and evaluation. The concentration of products was determined using response factor with respect to the internal standard.
Results
Expression of Formate Dehydrogenase in B. subtilis
NADH is involved in the conversion of acetoin to 2,3-butanediol [5]. The hypothesis tested here was that the increase in intracellular availability of NADH would lead to enhanced the conversion of acetoin to 2,3-butanediol. NADH has been regenerated in many microbial systems by heterologous expression of the enzyme formate dehydrogenase [13, 14]. Hence, it was cloned in Bacillus subtilis 1A1 to achieve a similar effect. As mentioned in the “Materials and Methods” section, the plasmid pHCMC05-FDH was constructed and transformed in B. subtilis 1A1 leading to the construction of the FDH strain. The site of ligation of fdh gene in the plasmid and the nucleotide sequence of the fdh gene were confirmed by DNA sequencing. A control strain, 1A1+05, was also constructed by transforming the parental, 1A1 strain with the empty pHCMC05 vector.
For cane molasses fermentation, sodium formate, being the substrate of formate dehydrogenase, was added to the fermentation medium for all the strains—FDH, 1A1+05, and the parent, 1A1. Product yields are shown in Table 3. Fermentation with the FDH strain showed a statistically significant increase in the 2,3-butanediol yield, both over 1A1 (26 %) and 1A1+05 (20 %). The decrease in the acetoin yield was significant over 1A1 (17 %) and marginally significant over 1A1+05 (11 %). However, the increase in 2,3-BDO yields was more than proportional to the decrease in acetoin yield as seen by the marginally significant increase in the 2,3-butanediol+acetoin yields—11 % in comparison to 1A1 and 10 % in comparison to 1A1+05. All the strains FDH, 1A1+05, and 1A1 had similar lactic acid yields.
Combined Expression of Formate Dehydrogenase and Butanediol Dehydrogenase in B. subtilis
The strain was developed further by over expressing the enzyme bdhA based on the benefits observed in increasing the conversion of acetoin to 2,3-butanediol [5]. For this, initially, a B. subtilis 1A1 strain over expressing the bdhA gene was constructed. A copy of the Pspac promoter followed by the bdhA gene was integrated into the genome of B. subtilis 1A1 at the amyE locus using an integration vector, pDG1662, resulting in the BDH strain. The site of integration in the genome and the nucleotide sequence of the transformed cassette were confirmed by DNA sequencing. The plasmid pHCMC05-fdh was transformed in the BDH strain resulting in the strain FDH+BDH. This strain was constructed to jointly express the fdh for NADH generation and overexpression of bdhA which were both controlled by the IPTG inducible Pspac promoter. A control strain, BDH+05, was also constructed by transforming the BDH strain with the empty pHCMC05 vector.
Cane molasses fermentation (Table 4) with the FDH+BDH strain showed a statistically significant increase in the 2,3-butanediol yield, both over 1A1 (29 %) and BDH+05 (20 %). The decrease in the acetoin yield was significant both over 1A1 (28 %) and BDH+05 (20 %). Increase in the 2,3-butanediol+acetoin yields was marginally significant both over 1A1 (9 %) and BDH+05 (8 %). There was marginally significant reduction in the yields of lactic acid by the FDH+BDH strain over 1A1 (12 %) and BDH+05 (10 %).
To evaluate the synergistic effect of bdhA overexpression in the FDH+BDH strain, the yields of this strain were compared with the yields of FDH strain (Table 4). Here, the FDH+BDH strain only showed marginal reduction in the yield on acetoin by 13 % and no significant incremental effect on yield of 2,3-butanediol and 2,3-butanediol+acetoin over the FDH strain. This implies that the contribution towards the increase in 2,3-butanediol yields of bdhA overexpression is minimal in comparison to the expression of fdh.
Comparison of Strains in the Presence and Absence of Sodium Formate
Since the contribution of bdhA over expression in the FDH+BDH strain was minimal, the cane molasses fermentation results of BDH and 1A1 strains were compared (Table 5). Initially, the fermentation of the strains was done in the presence of sodium formate—conditions similar to those required for the FDH+BDH strain. No significant changes were observed in the yields of 2,3-butanediol, acetoin, 2,3-butanediol+acetoin, and lactate. However, in the absence of formate, the BDH strain showed a statistically significant 11 % increase in the yield of 2,3-butanediol over parental strain, 1A1. Change in acetoin yield was not statistically significant. A 7 % increase in the total yield on 2,3-butanediol+acetoin was marginally significant in the BDH strain. FDH strain showed no significant change in the yields of 2,3-butanediol, acetoin, and 2,3-butanediol+acetoin over the parental strain, 1A1 (Table 5), in the absence of formate.
Comparison of the two scenarios (presence and absence of sodium formate as indicated in Table 5) for 1A1 strain showed that the presence of formate led to a substantial increase (14-fold) in lactate accumulation. 2,3-butanediol yield decreased by 34 % in the presence of formate. Acetoin yield increased by 33 %. In the presence of formate, a 20 % decrease was seen in the carbon flow through the 2,3-BDO pathway as indicated by the yields on 2,3-butanediol+acetoin. Similar results were seen for the BDH strain—7 fold increase in the lactate accumulation, 36 % decrease in 2,3-butanediol yield, 50 % increase in acetoin yield and 23 % decrease in 2,3-butanediol+acetoin yield. Identical results were seen for the FDH strain as well—6 fold increase in the lactate accumulation, 17 % decrease in 2,3-butanediol yield, no change in acetoin yield and 13 % decrease in 2,3-butanediol+acetoin yield.
Discussion
2,3-butanediol can be viably produced from sugarcane molasses by using B. subtilis. However, the major byproduct in the above process is acetoin [6]. Acetoin is a metabolic intermediate from which 2,3-butanediol is produced in B. subtilis. Its accumulation leads to a decrease in the yield of 2,3-butanediol. The enzymatic step in the conversion of acetoin to 2,3-butanediol involves the action of butanediol dehydrogenase [9] which is coupled with the transfer of hydrogen from the cofactor NADH to acetoin resulting in 2,3-butanediol [5]. The cause of acetoin accumulation can be attributed to the rate limitation at this step leaving some amount of acetoin being unconverted. One of the approach in which acetoin could be minimized is by facilitating its conversion to 2,3-butanediol. The current work was undertaken to study the effect of metabolic engineering approaches to reduce the rate limitation imposed at this crucial enzymatic step for increasing the conversion of acetoin to 2,3-butanediol. In order to improve the strain, two strategic changes were made in the genetic makeup of B. subtilis 1A1—cofactor engineering and overexpression of bdhA.
In this work, the cloning of formate dehydrogenase from C. boidinii in B. subtilis caused a marked improvement in the yield of 2,3-butanediol and decrease in the yield of acetoin from sugarcane molasses. Enhancement in the yield of 2,3-butanediol+acetoin indicates an enhanced carbon inflow in the 2,3-butanediol pathway. This effect could be attributed to the presence of increased levels of intracellular NADH that leads to the derepression of alsS which is in turn controlled by an intracellular repressor molecule, Rex [19]. The enzyme, AlsS, is the first enzyme of the 2,3-butanediol pathway that mediates the conversion of pyruvate to the first pathway intermediate, α-acetolactate [4]. Increase in the enzyme availability may increase the conversion of pyruvate to α-acetolactate leading to an increase in the total carbon flowing through the pathway. The yield on lactic acid did not vary significantly among the strains. However, the conversion of acetoin to 2,3-butanediol was not complete and may be due to insufficient copies of bdhA.
The strain was further enhanced by over expressing bdhA in the cofactor engineered strain of B. subtilis, as the over expression of butanediol dehydrogenase (bdhA) has been reported to improve 2,3-butanediol yields [8]. In the present work, fermentation of sugarcane molasses using the FDH+BDH strain showed an improvement in the yield of 2,3-butanediol and reduction in the yield of acetoin as compared to the control strains, BDH+05, and parental strains, 1A1 but not over FDH strain. Lactic acid was only marginally reduced in the FDH+BDH strain over BDH+05 and 1A1 strains and not reduced over the FDH strain. From this data, the contribution of bdhA overexpression seemed to be negligible as compared to cofactor engineering. The improvement in yields of the FDH+BDH strain over the control, BDH+05, and parent, 1A1, were largely due to the formate dehydrogenase activity and the derepression of the Rex controlled genes.
BDH strain was also constructed where the only genetic change was the overexpression of bdhA. The comparison of sugarcane molasses fermentation results of BDH and 1A1 strains in the presence of sodium formate led to no difference in any of the yields of 2,3-butanediol, acetoin, and lactate. This results in the non-performance of the over expression of bdhA. However, in the absence of sodium formate, the BDH strain shows an increase in the 2,3-butanediol yield over 1A1, no change in acetoin yield and increase in 2,3-butanediol+ acetoin yield. The improvement in the yield of 2,3-butanediol by the BDH strain in the absence of sodium formate could be because of the enhanced availability of intracellular enzyme copies of BdhA. This may lead to an increase in the flux of the rate-limiting step [8] where acetoin is converted to 2,3-butanediol, and this may explain the increase in 2,3-butanediol yield and no change in acetoin yield.
Another important change observed in the sugarcane molasses fermentation using the parental strain, 1A1 and BDH strain was that of the increase in lactic acid accumulation in the presence of sodium formate. Lactic acid formation in the presence of sodium formate is probably due to the derepression of ldhA. ldhA is a Rex repressed gene that is derepressed in the presence of extra NADH [20]. Increase in the availability of NADH could be due to the putative native formate dehydrogenase activity [21].
Utilization of NADH for lactic acid formation could have reduced its availability for acetoin conversion, and hence, the yield on acetoin was greater in conditions of the presence of sodium formate than its absence. The possible reasons for the non-functionality of bdhA overexpression strategy in the presence of sodium formate could be that some of the carbon is diverted towards lactate and therefore the native copy number of BdhA is sufficient to produce 2,3-butanediol. This reason could also explain the non-functionality of bdhA overexpression in FDH+BDH strain. In the presence of formate, the rate limiting step seems to be the availability of NADH and in the absence of formate, the rate limiting step seems to be the availability of the BdhA enzyme.
Conclusion
Bacillus subtilis was genetically modified to minimize the formation of acetoin in fermentation of sugarcane molasses. Cofactor engineering by the heterologous expression of formate dehydrogenase led to a 17 % decrease in acetoin. It also led to a 11 % increase in the carbon flow through the 2,3-BDO pathway, ultimately leading to a 26 % increase in 2,3-butanediol yield. Overexpression of bdhA also led to a 7 % increase in the carbon flow through the 2,3-BDO pathway, leading to a 11 % increase in 2,3-butanediol yield. However, the combination of the two strategies did not lead to a synergistic increase in 2,3-butanediol yield. The use of other cofactor engineering strategies with overexpression of bdhA may lead to further enhancements.
References
Garg, S. K., & Jain, A. (1995). Fermentative production of 2,3-butanediol: a review. Bioresource Technology, 51, 103–109.
Celińska, E., & Grajek, W. (2009). Biotechnological production of 2,3-butanediol—current state and prospects. Biotechnology Advances, 27, 715–725.
Ji, X. J., Huang, H., & Ouyang, P. K. (2011). Microbial 2,3-butanediol production: a state-of-the-art review. Biotechnology Advances, 29, 351–364.
Ramos, H. C., Hoffmann, T., Marino, M., Nedjari, H., Presecan-Siedel, E., Dreesen, O., Glaser, P., & Jahn, D. (2000). Fermentative metabolism of Bacillus subtilis: physiology and regulation of gene expression. Journal of Bacteriology, 182, 3072–3080.
Biswas, R., Yamaoka, M., Nakayama, H., Kondo, T., Yoshida, K., Bisaria, V. S., & Kondo, A. (2012). Enhanced production of 2,3-butanediol by engineered Bacillus subtilis. Applied Microbiology and Biotechnology, 94, 651–658.
Deshmukh, A., Mistry, S., Yewale, T., Mahajan, D., & Jain, R. (2015). Production of 2, 3-Butanediol from Sugarcane Molasses Using Bacillus subtilis. International Journal of Advanced Biotechnology and Research, 6, 66–79.
Fu, J., Wang, Z., Chen, T., Liu, W., Shi, T., Wang, G., Tang, Y. J., & Zhao, X. (2014). NADH plays the vital role for chiral pure D-(-)-2,3-butanediol production in Bacillus subtilis under limited oxygen conditions. Biotechnology and Bioengineering, 11, 2126–2131.
Yang T-W, Rao Z-M, Zhang X, Xu M-J, Xu ZH, Yang S-T (2013b). Effects of corn steep liquor on production of 2,3-butanediol and acetoin by Bacillus subtilis. Process Biochemistry, 48, 1610-1617.
Nicholson, W. L. (2008). The Bacillus subtilis ydjL (bdhA) gene encodes acetoin reductase/2,3-butanediol dehydrogenase. Applied and Environmental Microbiology, 74, 6832–6838.
Wang, Y., San, K. Y., & Bennett, G. N. (2013). Cofactor engineering for advancing chemical biotechnology. Current Opinion in Biotechnology, 24, 994–999.
Yang T, Rao Z, Zhang X, Xu M, Xu Z, Yang ST. (2013a). Improved production of 2,3-butanediol in Bacillus amyloliquefaciens by over-expression of glyceraldehyde-3-phosphate dehydrogenase and 2,3-butanediol dehydrogenase. PLoS One, 8, e76149.
Yang T, Rao Z, Zhang X, Xu M, Xu Z, Yang ST. (2015a). Enhanced 2,3-butanediol production from biodiesel-derived glycerol by engineering of cofactor regeneration and manipulating carbon flux in Bacillus amyloliquefaciens. Microbial Cell Factories, 14, 122-132.
Wang, Y., Li, L., Ma, C., Gao, C., Tao, F., & Xu, P. (2013). Engineering of cofactor regeneration enhances (2S,3S)-2,3-butanediol production from diacetyl. Scientific Reports, 3, 2643–2648.
Donk, W. A., & Zhao, H. (2003). Recent developments in pyridine nucleotide regeneration. Current Opinion in Biotechnology, 14, 421–426.
Gaspar, P., Neves, A. R., Gasson, M. J., Shearman, C. A., & Santos, H. (2011). High yields of 2,3-butanediol and mannitol in Lactococcus lactis through engineering of NAD+ cofactor recycling. Applied and Environmental Microbiology, 77, 6826–6835.
Ehsani, M., Fernández, M. R., Biosca, J. A., Julien, A., & Dequin, S. (2009). Engineering of 2,3-butanediol dehydrogenase to reduce acetoin formation by glycerol-overproducing, low-alcohol Saccharomyces cerevisiae. Applied and Environmental Microbiology, 75, 3196–3205.
Kim, S. J., Seo, S. O., Jin, Y. S., & Seo, J. H. (2013). Production of 2,3-butanediol by engineered Saccharomyces cerevisiae. Bioresource Technology, 146, 274–281.
Vojcic, L., Despotovic, D., Martinez, R., Maurer, K. H., & Schwaneberg, U. (2012). An efficient transformation method for Bacillus subtilis DB104. Applied Microbiology and Biotechnology, 94, 487–493.
Hartig, E., & Jahn, D. (2012). Regulation of the anaerobic metabolism of Bacillus subtilis. Advances in Microbial Physiology, 61, 195–216.
Larsson, J. T., Rogstam, A., & von Wachenfeldt, C. (2005). Coordinated patterns of cytochrome bd and lactate dehydrogenase expression in Bacillus subtilis. Microbiology, 151, 3323–3335.
Kunst, F., Ogasawara, N., Moszer, I., Albertini, A. M., Alloni, G., Azevedo, V., Bertero, M. G., Bessières, P., Bolotin, A., Borchert, S., Borriss, R., Boursier, L., Brans, A., Braun, M., Brignell, S. C., Bron, S., Brouillet, S., Bruschi, C. V., Caldwell, B., Capuano, V., Carter, N. M., Choi, S. K., Cordani, J. J., Connerton, I. F., Cummings, N. J., Daniel, R. A., Denziot, F., Devine, K. M., Düsterhöft, A., Ehrlich, S. D., Emmerson, P. T., Entian, K. D., Errington, J., Fabret, C., Ferrari, E., Foulger, D., Fritz, C., Fujita, M., Fujita, Y., Fuma, S., Galizzi, A., Galleron, N., Ghim, S. Y., Glaser, P., Goffeau, A., Golightly, E. J., Grandi, G., Guiseppi, G., Guy, B. J., Haga, K., Haiech, J., Harwood, C. R., Hènaut, A., Hilbert, H., Holsappel, S., Hosono, S., Hullo, M. F., Itaya, M., Jones, L., Joris, B., Karamata, D., Kasahara, Y., Klaerr-Blanchard, M., Klein, C., Kobayashi, Y., Koetter, P., Koningstein, G., Krogh, S., Kumano, M., Kurita, K., Lapidus, A., Lardinois, S., Lauber, J., Lazarevic, V., Lee, S. M., Levine, A., Liu, H., Masuda, S., Mauël, C., Médigue, C., Medina, N., Mellado, R. P., Mizuno, M., Moestl, D., Nakai, S., Noback, M., Noone, D., O’Reilly, M., Ogawa, K., Ogiwara, A., Oudega, B., Park, S. H., Parro, V., Pohl, T. M., Portelle, D., Porwollik, S., Prescott, A. M., Presecan, E., Pujic, P., Purnelle, B., Rapoport, G., Rey, M., Reynolds, S., Rieger, M., Rivolta, C., Rocha, E., Roche, B., Rose, M., Sadaie, Y., Sato, T., Scanlan, E., Schleich, S., Schroeter, R., Scoffone, F., Sekiguchi, J., Sekowska, A., Seror, S. J., Serror, P., Shin, B. S., Soldo, B., Sorokin, A., Tacconi, E., Takagi, T., Takahashi, H., Takemaru, K., Takeuchi, M., Tamakoshi, A., Tanaka, T., Terpstra, P., Togoni, A., Tosato, V., Uchiyama, S., Vandebol, M., Vannier, F., Vassarotti, A., Viari, A., Wambutt, R., Wedler, H., Weitzenegger, T., Winters, P., Wipat, A., Yamamoto, H., Yamane, K., Yasumoto, K., Yata, K., Yoshida, K., Yoshikawa, H. F., Zumstein, E., Yoshikawa, H., & Danchin, A. (1997). The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature, 390, 249–256.
Acknowledgments
The authors thank Dr. Anup Kadam (Analytical sciences department Praj Matrix-R &D center) for co-operation for sample analysis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Deshmukh, A.N., Nipanikar-Gokhale, P. & Jain, R. Engineering of Bacillus subtilis for the Production of 2,3-Butanediol from Sugarcane Molasses. Appl Biochem Biotechnol 179, 321–331 (2016). https://doi.org/10.1007/s12010-016-1996-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-016-1996-9