
Abstract
Secreted frizzled-related proteins (sFRPs) constitute a family of proteins, which impede the Wnt signaling pathway. Upregulation of the Wnt cascade is one of the multiple facets of carcinogenesis. Herein, we report the expression, solubilization, purification, characterization, and anti-cell proliferative activity of a novel recombinant GST-tagged sFRP1 of human origin. sFRP1 was cloned into pGEX-4T2 bacterial expression vector, and the recombinant protein was overexpressed in Escherichia coli BL21 (DE3). It was solubilized from inclusion bodies with N-lauroylsarcosine and Triton X-100, before being purified to homogeneity using glutathione agarose affinity chromatography column. The purified protein was characterized using Western blotting, MALDI TOF-TOF, and circular dichroism spectroscopy analysis. Homology modeling and docking studies revealed that tagging GST with sFRP1 does not change the binding conformation of the cysteine-rich domain and hence, possibly does not alter its function. The novel anti-proliferative activity of GST-sFRP1 was demonstrated in a dose-dependent manner on two cancer cell lines, viz., HeLa (cervical cancer) and MCF-7 (breast cancer). Also, combination therapy of the protein with chemotherapeutic drugs resulted in enhanced anti-cancer activity. This opens up a new avenue in the application of recombinant sFRP1 for cancer therapeutics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Protein therapeutics has emerged as a novel therapeutic modality for the treatment of complex and diverse diseases, such as cancer. Intricately woven signaling networks have been targeted using humanized monoclonal antibodies designed against tyrosine kinase receptor HER2 [1] in metastatic breast cancers, vascular endothelial growth factor receptor in colorectal cancers [2], and epidermal growth factor receptor [3]. Another such promising target is the Wnt signal transduction pathway whose aberrant activation has been strongly associated with tumor formation [4]. The Wnt pathway, initiated by Wnt ligand binding to corresponding frizzled (Fzd) receptors, plays an active role in differentiation events during normal embryonic developmental processes. In normal adult cells, it is blocked by a family of glycoproteins called the secreted frizzled-related proteins or sFRPs [5–8].
Explorations into the world of sFRP superfamily and their implications in cancer have commenced only recently. These sFRPs have been found to share sequence similarity with the extracellular cysteine-rich domain (CRD) of frizzled receptors. Hence, they act as putative binding sites for Wnt proteins [9]. As sFRPs lack transmembrane domain for Wnt signal transduction within the cell, they serve to inhibit the Wnt pathway by blocking the binding of Wnt ligands to respective Fzd receptors [8]. However, in various cancers, such as, colorectal cancers [6], gastric cancers [10], invasive breast tumors [11], bladder cancer [7], and tumors of mesenchymal origin [5], these sFRPs have been found to be downregulated due to promoter hypermethylation. In absence of these sFRPs, beta catenin, which is a downstream molecule of the Wnt pathway, escapes proteasome degradation and accumulates in the cytoplasm. Thereafter, it translocates to the nucleus, where it is involved in transcriptional upregulation of genes activating cell proliferation (such as, c-myc, cyclin D1 [4]) or genes inhibiting apoptosis (such as, survivin [12]).
It has been established that sFRP1, sFRP2, sFRP4, and sFRP5 are the ones most commonly implicated in tumorigenesis [13], of which sFRP1, being the first discovered one, forms the stepping stone for investigations. Accumulating evidence suggests that sFRP1 is downregulated in colorectal, breast, ovarian, malignant mesothelioma, and lung cancer [5, 14–17]. Specifically in lung carcinoma, loss of heterozygosity has also been reported in the sFRP1 gene locus, which may contribute to the absence of gene expression [17]. Interestingly, chain terminating mutations have also been found to exist in colorectal tumors [14], in addition to epigenetic inactivation. sFRP1 transfection led to reduced colony formation and decreased transcriptional activity of β-catenin in non-small cell lung cancer [17]. However, a discrepancy has been reported in prostate tumor, whereby, elevated expression of sFRP1 has been observed, as compared to adult prostate [18]. This evidence correlated with the finding that when human prostatic epithelial cells were treated with sFRP1, they exhibited increased proliferation and decreased apoptosis. A similar discrepancy was also found in gastric cancer, where high levels of sFRP1 have been shown to promote tumor formation [19]. Recently, transfection studies have revealed growth inhibitory effects of sFRP1 on few breast cancer cell lines [20] and hepatoma cell lines [21] with wild type β-catenin or β-catenin containing point mutations, but not in those containing truncated β-catenin.
In this study, we have made an endeavor to delineate the impact of bacterially expressed GST-sFRP1 on cancer cell types. For this purpose, we optimized expression, purification, and refolding conditions before adding it onto the cells. To the best of our knowledge, this is the first expedition aimed at illustrating the anti-cell proliferative effect of bacterially expressed GST-tagged sFRP1 on cancer cells, which may have significant prospective in cancer therapeutics.
Material and Methods
Cell Culture
The cancer cell lines HeLa (cervical) and MCF-7 (breast) were obtained from the National Centre for Cell Science (NCCS), India. The cells were cultured under conventional conditions in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10 % fetal bovine serum (FBS), 100 U/ml penicillin, 10 mg/ml streptomycin, and maintained in humidified atmosphere in a 5 % carbon dioxide incubator at 37 °C.
Cloning of sFRP1
The 945 bp human sFRP1 gene was obtained from DNASU Plasmid Repository, USA in pDNR-Dual vector. The gene was amplified by polymerase chain reaction (PCR) using gene-specific forward primer 5′-GTAGGATCCATGGGCATCGGGCGCA-3′ with a BamHI overhang and reverse primer 5′-GTTCTCGAGTCACTTAAACACGGACTGAAAG-3′ with a XhoI overhang. Taq polymerase (Bioline) was used for the PCR amplification. Conditions for the PCR reaction were as follows: denaturation at 95 °C for 50 s, annealing at 60 °C for 40 s, and extension at 72 °C for 1.5 min. The amplified and eluted product as well as the bacterial expression vector pGEX-4T2 (Amersham) were then digested with the mentioned restriction enzymes and ligated using Quick Ligase Kit (NEB). The ligation product was transformed into E. coli DH5α. Sequencing was done by Xcelris Labs Ltd. for clone confirmation. Digestion was carried out with the above restriction enzymes BamHI and XhoI for the same purpose.
Expression of GST-sFRP1 in Escherichia coli BL21 (DE3)
The recombinant vector was transferred into E. coli BL21 DE3 (Institute of Microbial Technology, India). The same was then cultured in small scale in Luria-Bertani (LB) media, containing 100 μg/ml ampicillin. It was kept overnight in a 37 °C shaker incubator, at 180 rpm. This was used as inoculum at a dilution of 1:100 for 100 ml of the secondary culture, which was allowed to reach mid log phase (an O.D. of 0.6) before induction with 0.2 mM IPTG. The induced culture was kept at temperatures ranging from 16 to 37 °C in a shaker incubator to acquire maximum expression of sFRP1 protein. For each of the temperatures tested for protein expression, induction times were varied from 4 to 12 h (data not shown). Eventually, the cells were harvested by centrifuging under chilled conditions at 6500 rpm for 5 min. Cells were lysed in 10 mM phosphate buffered saline (PBS) and 1 mM PMSF by sonication. Samples were run on 12 % sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) to observe the expression of protein for the varying conditions of temperature and time duration mentioned previously.
Solubilization, Purification, and Refolding of GST-sFRP1 Protein
Maximum protein expression was achieved with induction at 28 °C for 8 h. These conditions were hence followed throughout. However, when the cells were lysed by sonicator and centrifuged, the expressed mammalian protein was obtained in the insoluble fraction, perhaps as inclusion bodies. In order to solubilize the protein from pellet fraction, several modifications were made to the lysis buffer composition and sonication conditions. Finally, maximum solubilization was attained under the following conditions. Pellet was resuspended in 10 ml lysis buffer containing 10 mM PBS, 1 mM EDTA, 0.32 % N-lauroylsarcosine, and 1 mM PMSF. Cells were then sonicated using a probe sonicator, while keeping the sample on ice. Lysed cells were centrifuged at 20,100g for 25 min at 4 °C; 1 % Triton X-100 was added to the supernatant, which was then stirred for 1 h in cold. Thereafter, it was diluted three times with 10 mM PBS and filtered using 0.45 μm syringe filter. Then, it was loaded batch-wise onto a glutathione agarose (Sigma) affinity chromatography column and incubated for 30 min each at 4 °C. The column was then washed with one column volume (approximately 12 ml) of 10 mM PBS for fourteen times before elution of desired protein. Elution buffer was composed of 15 mM L-reduced glutathione in 50 mM Tris–HCl (pH 9.5). All steps were performed under cold conditions. Samples were analyzed on 12 % SDS-PAGE. The protein was refolded by subjecting it to step-wise dialysis against Tris–HCl, pH 7.4 for 6 h. The buffer concentration was decreased from an initial 100 mM to a final 10 mM, with three rounds of buffer replacement. Concentration was done with PEG. After two changes of buffer during dialysis, the dialysis bag containing the protein was covered with PEG 8000, which absorbs water, and kept at 4 °C for half an hour. It was replaced by fresh PEG after every 5 min. At the end of this concentration procedure, the protein was again dialyzed against 10 mM Tris–HCl buffer, pH 7.4.
Homology Modeling and Docking Studies
The structures of cysteine-rich domain (CRD) of sFRP1, sFRP1, and GST-tagged sFRP1 were predicted using Protein Homology/AnalogY Recognition Engine V 2.0 (Phyre2) [22]. The structure of WNT7A, which has been found to bind with sFRP1, was predicted using I-Tasser server [23–25]. The structure of CRD-sFRP1 hence predicted was compared to that predicted by SWISSMODEL (http://www.proteinmodelportal.org/query/uniprot/Q6FHJ7) and the crystal structure of CRD of sFRP3 (PDB ID: 1ijxA) by overlapping the respective structures using PyMOL and generating a root mean square deviation value. This was done in order to validate the structure of CRD-sFRP1. This was then overlapped with the predicted structures of full length sFRP1 and GST-sFRP1 (again by using PyMOL), in order to compare the binding domain of CRD-sFRP1, full length sFRP1, and GST-sFRP1. Next, each of these three structures was docked with WNT7A using ClusPro web interface. The docked structures hence obtained were modified using PDBEditor and analyzed with PDBsum Generate. The generated data for docked structures, viz., CRD-sFRP1:WNT7A, sFRP1:WNT7A, and GST-sFRP1:WNT7A were compared.
Western Blot
Western blotting was done to validate the presence of GST-sFRP1 using anti-GST antibody. Purified GST-sFRP1 was electrophoresed on 12 % SDS-PAGE and transferred onto a PVDF membrane. After confirming the transfer with Ponceau S staining, the membrane was washed with phosphate buffered saline/Tween (PBST) and subsequent blocking was done with 3 % BSA in PBST (1 % Tween 20 in 10 mM PBS). Thereafter, the membrane was incubated overnight with monoclonal anti-GST primary antibody raised in rat (Sigma). Then, it was washed with PBST four times, before being incubated for 2 h with anti-rat IgG (whole molecule) peroxidase antibody produced in goat (Sigma). After thorough washing with PBST, the blot was developed using chemiluminescent peroxidase substrate (Sigma). GST protein alone was also purified by affinity chromatography and blotted as control following the same protocol. The uninduced cell lysate (negative control) and induced cell fractions were also blotted.
Secondary Structure Characterization with Circular Dichroism
The secondary structure of GST-sFRP1 was assessed with the help of circular dichroism spectra obtained with JASCO-815 spectrometer (Jasco, Japan). This was carried out in order to analyze whether the purified, dialyzed, and concentrated protein retained its secondary structure. After step-wise dialysis, the final concentration of Tris–HCl buffer was 10 mM (pH = 7.4). The concentration of GST-sFRP1 analyzed in spectrometer was 0.3 μM. Spectra were recorded for protein in cuvette of 0.2 cm path length, under constant nitrogen gas purging at a flow rate of 5 L/min and at a temperature of 25 °C maintained by circulating water-bath. Scanning was done from wavelength 240 to 190 nm at a speed of 50 nm/min, with four accumulations. Background spectrum of corresponding buffer was subtracted from the sample spectrum.
MALDI TOF-TOF Analysis
MALDI MS-MS analysis was performed in order to characterize and confirm sFRP1 protein by tryptic digestion. In situ gel digestion of protein band was carried out using Trypsin Profile IGD kit (Sigma) following manufacturer’s protocol. The processed sample was mixed in a ratio of 5:3 with matrix α-cyano-4-hydroxycinnamic acid and spotted onto analyzer plate. MS-MS analysis was done with 4700 Proteomics Analyzer with TOF/TOF Optics (Applied Biosystems), which uses a diode-pumped solid state class I laser. MS-MS data were acquired in automatic mode using acquisition method MS-MS 1 kV positive.
Cell Viability Assay
The effect of GST-sFRP1 was ascertained on two different cancer cell lines, viz., HeLa (cervical cancer) and MCF-7 (breast cancer). The protein was dialyzed, concentrated, and quantified by Bradford protein estimation assay before adding onto cells. Cells were seeded in 96-well plate at a density of 7000 cells per well in DMEM containing 10 % FBS. After allowing them to attach for about 8 h, media was removed and the protein was added in serum-free media at varying concentrations ranging from 1.6 to 20 nM. Under these conditions, cells were incubated at 37 °C in 5 % carbon dioxide incubator for 48 h. Thereafter, MTT assay was performed, where MTT (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyltetrazolium bromide) is converted to purple formazan crystals by dehydrogenase enzymes in the mitochondria of healthy, respiring cells. DMSO was added to dissolve the formazan and absorbance was measured at 550 nm. Background measurement was done at 650 nm. The experiment was also conducted with purified GST protein as control, up to a concentration of 40 nM, to eliminate any probability of its effect on cell viability. Percentage cell viability was measured by the following equation:
Combination Therapy
Combination therapy of GST-sFRP1 with conventional chemotherapeutic drugs, namely, cisplatin, under the commercial name Platin 50 (Cadila Pharmaceuticals Limited), and doxorubicin hydrochloride, under the commercial name Adriamycin (Pfizer), was also performed. Cisplatin was used in the range of 0.5–5 and 3–10 μg/ml, whereas doxorubicin was used in the range of 0.1–0.5 and 0.09–0.2 μg/ml, for HeLa and MCF-7 cells, respectively. The range, in which the effect was observed, is in accordance with the range reported in literature [26, 27]. Both HeLa and MCF-7 cells were tested for the above combinations with MTT assay following the same protocol. Control and treated cells were observed under a microscope (Nikon ECLIPSE TS100). Both HeLa and MCF-7 cells were treated with 12 nM of protein, alone and in combination with cisplatin (2 and 5 μg/ml, respectively) or doxorubicin (0.4 and 0.2 μg/ml, respectively).
Cell Cycle Analysis
Cell cycle analysis was performed using Fluorescence Activated Cells Sorter (FACSCalibur, BD Biosciences, NJ, USA) to measure the DNA content of cells upon treatment with GST-sFRP1, alone and in conjunction with chemotherapeutic agents, namely, cisplatin and doxorubicin. Cells were seeded at a density of around 105 cells per well, in a six-well tissue culture plate. After allowing the cells to grow for 8 h, media was replaced by serum-free media containing GST-sFRP1, cisplatin, doxorubicin, and combination of GST-sFRP1 with cisplatin/doxorubicin. For treatment of HeLa cells, concentrations of protein, cisplatin, and doxorubicin used for this experiment were 12 nM, 1 μg/ml, and 0.2 μg/ml, respectively. For MCF-7 cells, treatment was done with 20 nM GST-sFRP1, 5 μg/ml cisplatin, and 0.1 μg/ml doxorubicin. The concentrations were determined on the basis of results yielded in MTT assays. The dose of protein, exerting the maximum effect on cell viability, was chosen; for the drugs, a concentration at which around 80 % of the cells were viable was selected. Treatment was done for 48 h, after which, cells were collected by trypsinization. Floating cells in media were collected by centrifugation at 650×g for 6 min at 4 °C, whereas trypsinized cells were centrifuged at 450×g for 6 min at 4 °C. Cells were then fixed in cold, by adding 70 % ethanol drop-wise and stored at −20 °C. Subsequently, cells were centrifuged, washed with cold PBS, and incubated with 0.2 mg/ml RNase A (Amersham) at 37 °C for 1 h. Thereafter, propidium iodide (Sigma) was added at a final concentration of 10 μg/ml and incubated for 20 min in dark, at 4 °C. Cells were analyzed in FACSCalibur (BD Biosciences); 10,000 cells were acquired and analyzed for each sample, using the software CellQuest Pro.
Statistical Tests
Statistical tests were carried out for the results obtained for all MTT assays using Graphpad Prism 6 software. One-way or two-way ANOVA analysis was performed, as per requirement.
Results and Discussion
Cloning, Expression, and Purification of GST-sFRP1
The coding sequence of human sFRP1 comprising of 945 bp was subcloned into pGEX-4T2 bacterial expression vector as demonstrated in Fig. 1a. PCR amplification was carried out using pDNR-Dual vector harboring sFRP1 gene, as template DNA; the amplified band at the desired position is shown in Fig. 1b. The clone was confirmed by sequencing (the sequence is given in Fig. S1) as well as by digestion with the restriction enzymes BamHI and XhoI (Fig. 1c), as mentioned in the “Material and Methods” section.

a Schematic of the method followed for subcloning sFRP1 into PGEX-4T2. Agarose (1 %) gel analysis. b Lane 1 shows 1 kb DNA ladder and lane 2 shows sFRP1 gene amplified with gene-specific primers using pDNR-sFRP1 as template. c Lane 1 is the DNA ladder, lane 2 is undigested pGEX-sFRP1, and lane 3 is pGEX-sFRP1 digested with BamHI and XhoI
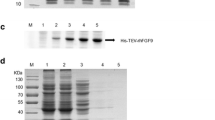
The recombinant plasmid containing N-terminal GST-tagged sFRP1 was eventually transformed into E. coli BL21 (DE3). Induction conditions were optimized, whereby maximum protein expression was found at 28 °C for 8 h (Fig. 2a). However, nearly the entire recombinant protein was present as inclusion bodies. Solubilization of this protein from the pellet fraction was attempted by various means, such as, cloning and expression in Rosetta-gami2(DE3) [28]; cloning in other bacterial expression vectors; induction at reduced temperatures and varying durations of time; and usage of detergents like Triton X-100, Tween 20, and sodium deoxycholate in different concentrations. Earlier investigations by different groups reveal the use of various detergents, which were modified and implemented in our study as mentioned above [29, 30]. However, urea and guanidine HCl, although found to be greatly effective in solubilizing proteins from inclusion bodies, were not tried, as their usage leads to a highly disordered protein structure that usually requires extensive refolding processes [31]. Also, the GST protein fusion system has been shown to enhance the stability and solubility of recombinant protein. Even in cases where it is not soluble, addition of detergents is sufficient for this purpose [32]. Here, substantial solubilization was accomplished with N-lauroylsarcosine and Triton X-100, using the protocol mentioned earlier in the “Material and Methods” section. A single band of GST-sFRP1 corresponding to molecular weight 61 kDa was obtained after purification, at the desired position in a 12 % polyacrylamide gel (Fig. 2b). The yield of purified protein, estimated by Bradford assay, was found to be approximately 0.5 mg from 100 ml of starter culture.

12% SDS-PAGE depicting expression and purification of GST-sFRP1. a Lane 1 shows the protein molecular weight marker (2–212 kDa), lane 2 shows the uninduced cell lysate of E. coli containing pGEX-sFRP1, lane 3 shows the expressed GST-sFRP1 in pellet fraction. b Lane 1 shows protein molecular weight marker and Lane 2 shows the purified single band at the legitimate position
Refolding of purified protein to obtain it in a functionally active form is an essential requirement for therapeutic applications. Since solubilization of proteins from inclusion bodies demands the presence of strong detergents, the protein itself gets completely unfolded in the process. Therefore, refolding is indispensible to attaining native conformation. For this purpose, different modes of dialysis have been previously used [31].
Homology Modeling and Docking Studies
Since functional activity of GST-sFRP1 has not been demonstrated yet, docking studies were performed to computationally confirm the functionality of GST-sFRP1, before conducting experiments in order to prove the same. As the only crystal structure available for the sFRP superfamily is that of the binding domain (CRD or cysteine-rich domain) of sFRP3 (PDB ID: 1ijxA), binding studies of sFRP1 with WNT could only be performed after predicting the structures of CRD-sFRP1, sFRP1, GST-sFRP1, and WNT7A. WNT7A was selected for the docking studies with sFRP1 as they have previously been shown to bind to each other. The 3-D structures for CRD-sFRP1 (Fig. 3a), sFRP1 (Fig. 3b), and GST-sFRP1 (Fig. 3c) were modeled using Phyre2, whereas WNT7A was modeled with I-TASSER server (Fig. 3d).

Three dimensional structures predicted with homology modeling servers. a CRD-sFRP1. b sFRP1. c GST-sFRP1. d WNT7A
CRD-sFRP1 hence predicted was confirmed by aligning with the structure of CRD-sFRP3 available in PDB in PyMOL (Fig. S2A) and that of CRD-sFRP1 predicted in the SWISSMODEL website (Fig. S2B). The structural integrity of the binding domain in the predicted whole molecule structure of sFRP1 as well as in GST-sFRP1 was confirmed by the same method (Fig. S2C and D, respectively). Also, the crystal structure of GST obtained from PDB (ID: 1UA5) was aligned with the predicted GST-sFRP1 to confirm the reliability of the predicted model (Fig. S2E). In each of the above cases, a root mean square deviation value was generated by PyMOL, which are detailed in supplementary figures. Thereafter, each of the predicted structures—CRD-sFRP1, sFRP1, and GST-sFRP1, was docked with WNT7A by means of the protein-protein docking server ClusPro 2.0 (Fig. 4a, b, c). This revealed that GST possibly did not participate in binding or hinder the binding of sFRP1 to WNT7A, as it remained separated.

Docking studies using ClusPro server. a. CRD-sFRP1 with WNT7A. b sFRP1 with WNT7A. c. GST-sFRP1 with WNT7A
The docked structures were then subjected to analysis of interacting residues using PDBsum Generate online server. The residues in the CRD responsible for interaction with WNT7A have been found to interact in a like manner in case of GST-sFRP1, which revealed that GST-sFRP1 may exhibit similar functional activity as sFRP1 alone. The summary of the results are outlined here in Table 1, while the details are given in Fig. S3, S4, and S5.
Western Blot
Western blotting with anti-GST monoclonal antibody generated legitimate band of GST-sFRP1 of approximately 61 kDa (Fig. 5). Purified GST alone was also blotted, which developed a band corresponding to the molecular weight of GST (Fig. 5). The uninduced cell lysate and induced cell fraction were blotted, as shown in supplementary information (Fig. S6).

Western blotting with anti-GST monoclonal antibody; Lane 1—purified GST, Lane 2—purified GST-sFRP1
Secondary Structure Characterization with Circular Dichroism
The secondary structure of GST-sFRP1, suspended in 10 mM Tris–HCl (pH 7.4), was characterized by far-UV circular dichroism. The spectrum was obtained with millidegree on the Y-axis, which was converted to mean residue ellipticity (mdeg.cm2dmol−1) by the formula
where θ is the ellipticity in millidegree, C is the concentration of the sample protein, n is the number of residues, and l is the path length [33]. Figure 6 shows the obtained spectrum. The secondary structure analysis was done by employing Yang’s reference, which revealed the α-helix content to be 20.7 %, β-sheet content to be 33.5 %, and random coil to be 29.5 %.

Far-UV circular dichroism spectra of GST-sFRP1 in 10 mM Tris–HCl at pH 7.4
Analysis of MALDI TOF-TOF Data
Soft ionization techniques in mass spectrometry have been employed in conjunction with database searching algorithms [34, 35] for protein identification and characterization since the past two decades. Here, MALDI coupled with time-of-flight analysis was used for characterizing the recombinant sFRP1; the mass spectrometry data is shown in Fig. 7. MS/MS study of each peptide fragment obtained by tryptic digestion of GST-sFRP1 generated a match with the MASCOT database, with a score of 104. This sequence similarity confirmed the identity of the protein.

MALDI TOF-TOF analysis of GST-sFRP1 upon tryptic digestion generated a peptide match of the following sequence KMVLPNLLEHETMAEVKQ
Cell Viability Assay
sFRP1 has been found to be downregulated due to promoter hypermethylation in various cancer types [5–7]. Hence, it can be hypothesized that its overexpression may lead to anti-proliferative activity by inhibiting the Wnt pathway. Binding of sFRP1 to Wnt ligands is known to inhibit the interaction of Wnt with corresponding frizzled receptors, which is necessary for Wnt signal transduction [10]. In non-small cell lung cancer, sFRP1 was demonstrated to inhibit the transcriptional activity of β-catenin [17], which is a downstream molecule in the Wnt canonical pathway. Although, the non-canonical pathway of Wnt has not been properly implicated in carcinogenesis, there is one report, for colorectal cancer, hypothesizing the same [14]. Also, treatment of sFRP1 with demethylating agents, like 5-aza-2′-deoxycytidine, restored the expression of sFRP1 [15, 36] and suppressed tumor growth [21]. However, to the best of our knowledge, the effect of bacterially expressed GST-tagged sFRP1 on mammalian cancer cells has not been assessed till date.
In our study, functional activity of recombinant sFRP1 was determined by adding it onto two different cancer cell lines HeLa and MCF-7. In both cases, the protein was found to display significant dose-dependent cell growth inhibitory effect. After 48 h of treatment, less than 60 % of cell viability was achieved with approximately 10 nM of GST-sFRP1 in case of HeLa (Fig. 8a) and 20 nM in case of MCF-7 (Fig. 8b), respectively. When purified GST alone was added onto cells, no decrease in cell viability was observed, up to a concentration of 40 nM (Fig. S7). The fact that treatment with recombinant sFRP1 alone displayed cell growth inhibition showed that it has the potential to contribute significantly to the domain of combination therapy for the treatment of cancer. Hence, we proceeded with the following experiments to test our hypothesis.

Effect of GST-sFRP1 in terms of reduction in percentage of viable cells as demonstrated by MTT assay. Statistical significance has been determined by one-way ANOVA, where *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. a HeLa cells. b MCF-7 cells
Combination Therapy
Combination therapy was attempted to check if administration of the protein enhances sensitization of cells towards conventional chemotherapeutic drugs. Till date, there has been no report of the combinatorial effect of sFRP1 with any anti-cancer drug. However, another member of the sFRP family, namely sFRP4, has been shown to significantly increase the sensitivity of transfected chemoresistant ovarian cancer cell lines to treatment with cisplatin [37].
Various concentrations of protein and drug were tested in order to optimize the ratio required for maximum impact on cell viability. Here, we have reported the optimized ratio obtained via MTT assays in case of HeLa and MCF-7, when treated with GST-sFRP1 along with cisplatin (Fig. 9a, b, respectively) and doxorubicin (Fig. 10a, b, respectively). Significant difference in cell viability was observed between treatment with only drug and that of drug with protein in each of the following cases.

Treatment of cell lines with increasing dosage of cisplatin alone and in combination with specified concentrations of GST-sFRP1. a in case of HeLa cells. b in case of MCF-7 cells. Statistical significance has been determined by two-way ANOVA, where *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001

Treatment of cell lines with increasing dosage of doxorubicin alone and in combination with specified concentration of GST-sFRP1. a in case of HeLa cells. b in case of MCF-7 cells. Statistical significance has been determined by two-way ANOVA, where *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001
Microscopic images of control and treated HeLa and MCF-7 cells have been illustrated in Fig. S8 and S9, respectively. Treatment with GST-sFRP1 did not seem to damage cell morphology, although the number of cells appeared to be decreased considerably. However, chemotherapeutic drugs caused significant damage to cells, which was augmented in case of combination therapy.
Cell Cycle Analysis
FACS-based analysis was performed to delineate the impact of GST-sFRP1 (alone and in combination with chemotherapeutic drugs) on cell cycle of HeLa (Fig. 11a, b) and MCF-7 (Fig. 11c, d) cells. Treatment of HeLa cells with protein substantially reduced the number of cells in G1 phase and increased the cell population in G2/M phase, as compared to untreated cells. A small population of cells (3.99 %) was found to appear in sub-G1 phase even in control cells, which was possibly due to incubation in serum-free media for 48 h. The percentage of this cell population was increased to 5.89 % in the HeLa cells treated with GST-sFRP1. Treatment with chemotherapeutic drugs significantly increased G2/M population, which was further augmented by combination therapy of the respective drugs with protein. In case of MCF-7 cells, GST-sFRP1 treatment resulted in a significant apoptotic cell population (7.49 %), which was further increased during combination therapy with either of the two drugs. Addition of either drug as well as combination therapy with protein caused a significant decrease in G1 population and increase in G2/M population in both cell lines. All the results indicated that the recombinant protein induced cell growth inhibition, partly through cell cycle arrest and partly through apoptosis in HeLa cells, whereas apoptosis seemed to be the more prominent mechanism in MCF-7 cells. Combination therapy was effective in augmenting the impact of the protein. These findings correlated with the results observed in MTT assays and morphology studies. It should be mentioned here that the concentrations of chemotherapeutic drugs used for this experiment were much lower than their respective IC50 dosage, in an attempt to reduce their adverse side-effects.

Flow cytometry-based cell cycle analysis. a HeLa cells treated with sFRP1 (12 nM), cisplatin (1 μg/ml), and their combination. b HeLa cells treated with sFRP1 (12 nM), doxorubicin (0.2 μg/ml), and their combination. c MCF-7 cells treated with sFRP1 (20 nM), cisplatin (5 μg/ml), and their combination. d MCF-7 cells treated with sFRP1 (20 nM), doxorubicin (0.1 μg/ml), and their combination
The development of resistance and severe side-effects of chemotherapy and radiotherapy have led to the evolution of gene therapy and protein therapeutics. However, these, by themselves, often do not exert sufficiently adequate anti-tumor responses. Hence, treatment of cancer has witnessed a paradigm shift culminating in the emergence of combination therapy.
Cell signaling pathways are intricately designed processes, where often certain genes/proteins are found to perform multiple roles. As cisplatin and doxorubicin both act by inhibiting the mechanism of DNA synthesis, whereas sFRPs inhibit the Wnt signaling pathway, there seems to be no direct interaction between them. However, cells may have been chemosensitized towards drugs due to inhibition of the Wnt pathway by sFRP1. Stress conditions resulting from cell cycle arrest may render the cells more sensitive towards the chemotherapeutic drugs, resulting in increased cell death. Whether the effect on cells is due to the independent functions of GST-sFRP1 and drugs or whether their actions are somehow intertwined can only be concluded after extensive analysis. One group had reported that Adriamycin upregulated hsFRP protein in a non-cancerous cell line (HBL-100). The study concluded that hsFRP brings about both apoptosis and cell cycle regulation but did not explicitly explain the link between the two networks [11].
Although further explorations into this field are essential to draw definitive conclusions, our experiments demonstrate that this combination therapy may be used for chemosensitization of cancer cells towards conventional anti-cancer drugs.
Conclusions
Targeting the Wnt family, which constitutes a largely uncharted territory, holds great promises in cancer therapy. The current investigation is not only limited to purification, characterization of the purified recombinant sFRP1 protein, but it paves the way for this recombinant protein to be used in combination therapy with conventional chemotherapeutic drugs to augment anti-tumor responses with the complete understanding of molecular processes.
References
Molina, M. A., Codony-Servat, J., Albanell, J., Rojo, F., Arribas, J., & Baselga, J. (2001). Trastuzumab (herceptin), a humanized anti-Her2 receptor monoclonal antibody, inhibits basal and activated Her2 ectodomain cleavage in breast cancer cells. Cancer Research, 61, 4744–4749.
Kabbinavar, F., Hurwitz, H. I., Fehrenbacher, L., Meropol, N. J., Novotny, W. F., Lieberman, G., Griffing, S., & Bergsland, E. (2003). Phase II, randomized trial comparing bevacizumab plus fluorouracil (FU)/leucovorin (LV) with FU/LV alone in patients with metastatic colorectal cancer. Journal of Clinical Oncology : Official Journal of the American Society of Clinical Oncology, 21, 60–65.
Yang, X. D., Jia, X. C., Corvalan, J. R., Wang, P., Davis, C. G., & Jakobovits, A. (1999). Eradication of established tumors by a fully human monoclonal antibody to the epidermal growth factor receptor without concomitant chemotherapy. Cancer Research, 59, 1236–1243.
Giles, R. H., van Es, J. H., & Clevers, H. (2003). Caught up in a Wnt storm: Wnt signaling in cancer. Biochimica et Biophysica Acta, 1653, 1–24.
Lee, A. Y., He, B., You, L., Dadfarmay, S., Xu, Z., Mazieres, J., Mikami, I., McCormick, F., & Jablons, D. M. (2004). Expression of the secreted frizzled-related protein gene family is downregulated in human mesothelioma. Oncogene, 23, 6672–6676.
Suzuki, H., Gabrielson, E., Chen, W., Anbazhagan, R., van Engeland, M., Weijenberg, M. P., Herman, J. G., & Baylin, S. B. (2002). A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nature Genetics, 31, 141–149.
Marsit, C. J., Karagas, M. R., Andrew, A., Liu, M., Danaee, H., Schned, A. R., Nelson, H. H., & Kelsey, K. T. (2005). Epigenetic inactivation of SFRP genes and TP53 alteration act jointly as markers of invasive bladder cancer. Cancer Research, 65, 7081–7085.
Hu, E., Zhu, Y., Fredrickson, T., Barnes, M., Kelsell, D., Beeley, L., & Brooks, D. (1998). Tissue restricted expression of two human Frzbs in preadipocytes and pancreas. Biochemical and Biophysical Research Communications, 247, 287–293.
Finch, P. W., He, X., Kelley, M. J., Uren, A., Varmus, H. E., & Rubin, J. S. (1997). Purification and molecular cloning of a secreted, Frizzled-related antagonist of Wnt action. Proceedings of the National Academy of Sciences of the United States of America, 94(13), 6770–6775.
Nojima, M., Suzuki, H., Toyota, M., Watanabe, Y., Maruyama, R., Sasaki, S., Sasaki, Y., Mita, H., Nishikawa, N., Yamaguchi, K., Hirata, K., Itoh, F., Tokino, T., Mori, M., Imai, K., & Shinomura, Y. (2007). Frequent epigenetic inactivation of SFRP genes and constitutive activation of Wnt signaling in gastric cancer. Oncogene, 26, 4699–4713.
Zhou, Z., Wang, J., Han, X., Zhou, J., & Linder, S. (1998). Up-regulation of human secreted frizzled homolog in apoptosis and its down-regulation in breast tumors. International Journal of Cancer. Journal International du Cancer, 78, 95–99.
Rubin, J. S., Barshishat-Kupper, M., Feroze-Merzoug, F., & Xi, Z. F. (2006). Secreted WNT antagonists as tumor suppressors: pro and con. Frontiers in Bioscience : A Journal and Virtual Library, 11, 2093–2105.
Suzuki, H., Watkins, D. N., Jair, K. W., Schuebel, K. E., Markowitz, S. D., Chen, W. D., Pretlow, T. P., Yang, B., Akiyama, Y., Van Engeland, M., Toyota, M., Tokino, T., Hinoda, Y., Imai, K., Herman, J. G., & Baylin, S. B. (2004). Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nature Genetics, 36, 417–422.
Caldwell, G. M., Jones, C., Gensberg, K., Jan, S., Hardy, R. G., Byrd, P., Chughtai, S., Wallis, Y., Matthews, G. M., & Morton, D. G. (2004). The Wnt antagonist sFRP1 in colorectal tumorigenesis. Cancer Research, 64, 883–888.
Veeck, J., Niederacher, D., An, H., Klopocki, E., Wiesmann, F., Betz, B., Galm, O., Camara, O., Durst, M., Kristiansen, G., Huszka, C., Knuchel, R., & Dahl, E. (2006). Aberrant methylation of the Wnt antagonist SFRP1 in breast cancer is associated with unfavourable prognosis. Oncogene, 25, 3479–3488.
Takada, T., Yagi, Y., Maekita, T., Imura, M., Nakagawa, S., Tsao, S. W., Miyamoto, K., Yoshino, O., Yasugi, T., Taketani, Y., & Ushijima, T. (2004). Methylation-associated silencing of the Wnt antagonist SFRP1 gene in human ovarian cancers. Cancer Science, 95, 741–744.
Fukui, T., Kondo, M., Ito, G., Maeda, O., Sato, N., Yoshioka, H., Yokoi, K., Ueda, Y., Shimokata, K., & Sekido, Y. (2005). Transcriptional silencing of secreted frizzled related protein 1 (SFRP 1) by promoter hypermethylation in non-small-cell lung cancer. Oncogene, 24, 6323–6327.
Joesting, M. S., Perrin, S., Elenbaas, B., Fawell, S. E., Rubin, J. S., Franco, O. E., Hayward, S. W., Cunha, G. R., & Marker, P. C. (2005). Identification of SFRP1 as a candidate mediator of stromal-to-epithelial signaling in prostate cancer. Cancer Research, 65, 10423–10430.
Qu, Y., Ray, P. S., Li, J., Cai, Q., Bagaria, S. P., Moran, C., Sim, M. S., Zhang, J., Turner, R. R., Zhu, Z., Cui, X., & Liu, B. (2013). High levels of secreted frizzled-related protein 1 correlate with poor prognosis and promote tumourigenesis in gastric cancer. European Journal of Cancer (Oxford, England : 1990), 49, 3718–3728.
Yang, Z. Q., Liu, G., Bollig-Fischer, A., Haddad, R., Tarca, A. L., & Ethier, S. P. (2009). Methylation-associated silencing of SFRP1 with an 8p11-12 amplification inhibits canonical and non-canonical WNT pathways in breast cancers. International Journal of Cancer. Journal International du Cancer, 125, 1613–1621.
Shih, Y. L., Hsieh, C. B., Lai, H. C., Yan, M. D., Hsieh, T. Y., Chao, Y. C., & Lin, Y. W. (2007). SFRP1 suppressed hepatoma cells growth through Wnt canonical signaling pathway. International Journal of Cancer. Journal International du Cancer, 121, 1028–1035.
Kelley, L. A., & Sternberg, M. J. (2009). Protein structure prediction on the Web: a case study using the Phyre server. Nature Protocols, 4, 363–371.
Zhang, Y. (2008). I-TASSER server for protein 3D structure prediction. BMC Bioinformatics, 9, 40.
Roy, A., Kucukural, A., & Zhang, Y. (2010). I-TASSER: a unified platform for automated protein structure and function prediction. Nature Protocols, 5, 725–738.
Roy, A., Yang, J. and Zhang, Y. (2012) COFACTOR: an accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Research
Tardito, S., Isella, C., Medico, E., Marchio, L., Bevilacqua, E., Hatzoglou, M., Bussolati, O., & Franchi-Gazzola, R. (2009). The thioxotriazole copper(II) complex A0 induces endoplasmic reticulum stress and paraptotic death in human cancer cells. The Journal of Biological Chemistry, 284, 24306–24319.
Lu, D. Y., Huang, M., Xu, C. H., Yang, W. Y., Hu, C. X., Lin, L. P., Tong, L. J., Li, M. H., Lu, W., Zhang, X. W., & Ding, J. (2005). Anti-proliferative effects, cell cycle G2/M phase arrest and blocking of chromosome segregation by probimane and MST-16 in human tumor cell lines. BMC Pharmacology, 5, 11.
Ferrer-Miralles, N., Domingo-Espin, J., Corchero, J., Vazquez, E., & Villaverde, A. (2009). Microbial factories for recombinant pharmaceuticals. Microbial Cell Factories, 8, 17.
Mercado-Pimentel, M. E., Jordan, N. C., & Aisemberg, G. O. (2002). Affinity purification of GST fusion proteins for immunohistochemical studies of gene expression. Protein Expression and Purification, 26, 260–265.
Schwanke, R. C., Renard, G., Chies, J. M., Campos, M. M., Batista, E. L., Jr., Santos, D. S., & Basso, L. A. (2009). Molecular cloning, expression in Escherichia coli and production of bioactive homogeneous recombinant human granulocyte and macrophage colony stimulating factor. International Journal of Biological Macromolecules, 45, 97–102.
Tsumoto, K., Ejima, D., Kumagai, I., & Arakawa, T. (2003). Practical considerations in refolding proteins from inclusion bodies. Protein Expression and Purification, 28, 1–8.
Park, D. W., Kim, S. S., Nam, M. K., Kim, G. Y., Kim, J., & Rhim, H. (2011). Improved recovery of active GST-fusion proteins from insoluble aggregates: solubilization and purification conditions using PKM2 and HtrA2 as model proteins. BMB Reports, 44, 279–284.
Achilonu, I., Siganunu, T. P., & Dirr, H. W. (2014). Purification and characterisation of recombinant human eukaryotic elongation factor 1 gamma. Protein Expression and Purification, 99C, 70–77.
James, P., Quadroni, M., Carafoli, E., & Gonnet, G. (1993). Protein identification by mass profile fingerprinting. Biochemical and Biophysical Research Communications, 195, 58–64.
Pappin, D. J., Hojrup, P., & Bleasby, A. J. (1993). Rapid identification of proteins by peptide-mass fingerprinting. Current Biology : CB, 3, 327–332.
Fang, H. L., Yu, Z. C., Zhu, H. B., & Jin, Y. T. (2012). Effects of 5-Aza-2-deoxycytidine on DNA methylation of anti-oncogenes in non-small cell lung cancer cells. Zhonghua Zhong Liu Za Zhi [Chinese Journal of Oncology], 34, 658–663.
Saran, U., Arfuso, F., Zeps, N., & Dharmarajan, A. (2012). Secreted frizzled-related protein 4 expression is positively associated with responsiveness to cisplatin of ovarian cancer cell lines in vitro and with lower tumour grade in mucinous ovarian cancers. BMC Cell Biology, 13, 25.
Acknowledgments
This work was supported by the Department of Biotechnology (No. BT/01/NE/PS/08) and the Department of Electronics and Information Technology, Government of India for financial support (No. 5(9)/2012-NANO (Vol. II)). The authors acknowledge the aid of the Central Instruments Facility (CIF) and the Centre for Nanotechnology, IIT Guwahati.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 3459 kb)
Rights and permissions
About this article

Cite this article
Ghoshal, A., Ghosh, S.S. Expression, Purification, and Therapeutic Implications of Recombinant sFRP1. Appl Biochem Biotechnol 175, 2087–2103 (2015). https://doi.org/10.1007/s12010-014-1354-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-014-1354-8