
Abstract
Streptomyces sp. CS147 grown on chitin liquid medium incorporating 0.5 % colloidal chitin as a sole carbon source produced an extracellular chitinase (Ch147). The enzyme (Ch147) was purified using Sepharose CL-6B column chromatography and biochemically characterized. The enzymatic reaction products, analyzed using high-performance liquid chromatography and thin layer chromatography clearly indicates the production of N-acetyl-d-glucosamine (GlcNAc) as principal product which can be further hydrolyzed for the production of alcohol, a second generation biofuel. Ch147 hydrolyzed colloidal chitin to 0.278, 0.817, and 1.058 mg/mL of (GlcNAc) as a major product, at retention time of 4.3, respectively, when incubated for 8, 16, and 24 h at 50 °C. GlcNAc is a monosaccharide that usually polymerize linearly through (1, 4) β linkage. The 41 kDa molecular mass chitinase, as determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), has the amino acid sequences DINGGGATLPQKLYL significantly different from other chitinase. Ch147 had K m and V max values of 2.05 ± 5.3 mg/mL and 467.2 ± 2.4 mmol/min, respectively. Further, the purified enzyme (5 U) inhibits the fungal phytopathogens belonging to the genera Fusarium and Aspergillus. We believe that Ch147 is a potential candidate for the conversion of waste materials into simple sugar for productions of biofuels and also can be used as an alternative option for biological control of plant pathogens being unfriendly to chemicals.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Globally, waste materials have become a huge challenge and remain as a center of attraction since they create pollution, unnecessarily occupy space, and require high cost for proper management. On the other hand, these waste materials can be used and utilized for the production of value added products. The hydrolysis of waste materials to sugar and the fermentation of sugar to alcohol (biofuel) offer an alternative option of limited fossil fuels [1], a second generation biofuel. The whole process includes two steps i.e., hydrolysis of lignocellulosic biomass to produce sugars and then fermentation of the produced sugar into ethanol. The first hydrolytic step associated with high processing cost and low yield remained a major challenge. After the first-generation biofuels primarily form food crops and oil seeds had limitation to reach the target for the commercialization of biofuels, an increasing attention has been given to improve this global challenge by pretreating lignocellulosic materials and utilizing more efficient biocatalysts [2]. Despite of other agricultural residue like wheat bran, rice husk, rice straw, etc. every year, approximately 80,000 metric tons of chitins were obtained from the marine waste [3], [4]. Crustacean shells are the most important source of chitin due to its high content and ready availability. Crustaceous which are considered trash or not edible can be used as sources of chitin, which adds more values to the bycatch and represents an economic asset for the fishermen [5]. These huge quantity of wastes can be bioconverted using chitinase, which breaks down the chitin polymer into monomeric units and also can be used in agriculture to control plant pathogens [6]. Chitooligosaccharides are the sugar oligomers derived from chitin. In recent days, the conversion of chitin for the production of chitooligosaccharides and antifungal chitinase for biological control had attracted major research groups. Bearing this in mind, several research groups are exploring the application of chitinase. Chitin and its fragments (chitin oligosaccharides) were found to induce fungal microbe-associated molecular pattern in wide range of plant species [7]. Due to different types of plant disease, 10 % of the total global crop production is lost and most of the plant pathogens are resistant to minor dose of chemicals which has accelerated the environment pollutions and degradation as well [8]. Chitins are white, hard, semi-transparent material, nitrogenous polysaccharide, and inelastic, low chemical reactivity and insoluble in most organic solvents. These natures of chitin made it more feasible for the biological process [7]. Chitin and its deacetylated derivative chitosan have numerous applications in foods, pharmaceuticals, and biotechnological products, cosmetics, textiles, in waste water treatment, and in agriculture [5]. Chitin, a β-(1,4)-linked polymer of N-acetyl-d-glucosa-amine (GlcNAc) is the second most abundant polysaccharide in nature (after cellulose) which functions as a major structural polymer (10–55 % based on dry weight) in many lower life forms including the cell walls of bacteria and fungi, the shell of crustaceans such as crabs and shrimp, squids, oyster, cuttlefish, etc. and the exoskeletons of arthropods such as dust mites and cockroaches [9].
Since the use of chemical fungicides may be lethal to beneficial insects and microorganisms which may enter and affect the food chain, biological control offers an alternative, environmentally friendly strategy for controlling phytopathogens, and the focus on microorganisms producing mycolytic enzymes, especially chitinases, draws prime attention [8]. Chitinase, a chitinolytic enzyme, has been detected in a wide range of organisms such as fungi, crustaceans, and insects as well as from the organisms without chitin in them like archea, bacteria, viruses, higher plants, animals, and humans [10]. Bacterial chitinase has been widely used to inhibit fungal growth, an attractive alternative to synthetic chemicals, which possibly can be effective in controlling plant pathogenic fungal diseases [11]. The reason behind fungal inhibition is the action of chitinase in the fungal cell walls acting as plant protective agents [12]. Chitinase, as promising biocontrol agents, either acting directly on fungal cell walls or initiating increased plants responses against diseases have been already well reported from Streptomyces [10]. Biological control strategies have become an important approach for facilitating sustainable agriculture because of their perceived safety and lower environmental impact. In this work, we describe the isolation and identification of an extracellular chitinase from Streptomyces sp. CS147. Further purification and biochemical characterizations in quest of potential bioindustrial application like oligosaccharide production along with its antifungal activities against Fusarium solani and Aspergillus brasiliensis from this strain were also performed.
Materials and Methods
Materials
Chitin, Glycol chitosan, caucofluor white M2R, and N-acetylglucosamine were purchased from Sigma-Aldrich (St Louis, MO, USA). Thin layer chromatography silica gel plates were from Merck (Darmstadt, Germany). Chitobiose and chitotriose were purchased from Megazyme (Ireland). Sepharose Cl-6B was purchased from Amersham Bioscience (Uppasala, Sweden). F. solani and A. brasiliensis were purchased from Korean Culture Centers of Microorganisms (KCCM). All the reagents used were of the highest grade available.
Bacterial Strain, Growth Conditions, and Screening
For the screening, twenty bacterial strains collected from different provinces of Korea were cultivated in chitin medium (pH 6.5) which was supplemented with (w/v) 0.5 % colloidal chitin, 0.5 % yeast extract, 1 % tryptone, 0.07 % KH2PO4, 0.03 % K2HPO4, 0.4 % NaCl, and 0.05 % MgSO4. These strains were cultivated in 1 L baffled flask containing 250 mL medium at 28 °C and 110 rpm for 3 days. The cultural broths were centrifuged at 10,000×g for 30 min and supernatant was used to determine the enzyme activity. Further, these strains were examined for their ability to produce chitinase. In the chitin plates containing (w/v) 0.5 % colloidal chitin, 0.5 % yeast extract, 1 % tryptone, 0.07 % KH2PO4, 0.03 % K2HPO4, 0.4 % NaCl, 0.05 % MgSO4, and 1.5 % agar, various concentration of enzyme (Unit) were inoculated into spots made in agar plate and incubate at 37 °C for 48 to 72 h [13]. The spot without enzyme was taken as control. The plate was then stained with 0.01 % caucofluor white M2R in 0.5 M Tris/HCl, pH 8.0 for 30 min, and destained with water. The lytic zones were photographed under the UV-transilluminator. Both in liquid culture and agar plate, S. sp. CS147 showed good activity thus was selected for the further study.
Colloidal and Glycol Chitin Preparation
Colloidal chitin was prepared according to the method of Chandrasekaran et al. [14] with some minor modifications. Briefly, 10 g of chitin powder was added slowly into 200 mL of concentrated HCl and left at 4 °C overnight with vigorous stirring. Then, 2 L of ice-cold 95 % ethanol was added to the mixture with rapid stirring and kept overnight at 4 °C. The precipitate was collected by centrifugation at 10,000×g for 30 min at 4 °C. The precipitate was washed with sterile distilled water until the colloidal chitin has neutral pH (7.0). Finally, the volume was raised to 200 mL using buffer (50 mM KCl/NaOH, pH 11) which result in 5 % (w/v) stock solution of colloidal chitin.
Glycol chitin was prepared by the acetylation of glycol chitosan according to the method described by Trudel and Asselin [15] with some minor modification. Briefly, 1 g of glycol chitosan was dissolved in 20 mL of 10 % acetic acid. The viscous solution was allowed to stand overnight at room temperature. Then, 90 mL of methanol was added slowly and the solution was vacuum filtered through a Whatman No.4 filter paper. The filtrate was transferred into a beaker and 1.25 mL of acetic anhydrate was added with magnetic stirring which results in gel formation when stand for 30 min at room temperature. The gel was cut down into pieces and was subjected to Waring Blendor covered with methanol, and homogenized for 4 min at maximum speed. Then, the suspension was centrifuged at 10,000×g for 45 min at 4 °C. The pellet was suspended in distilled water (100 mL) containing 0.02 % (w/v) sodium azide and homogenized which result in 1 % (w/v) stock solution of glycol chitin.
Enzyme Assay and Protein Estimation
The protein concentrations were determined using the Bradford method [16] using bovine serum albumin as standard.
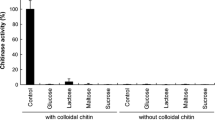
The chitinase activity was determined by measuring the release of reducing sugar using 3,5-dinitrosalicyclic (DNS) method as described by Miller [17]. The reaction mixture contains 0.1 mL enzyme (approximately diluted in 50 mM KCL/NaOH, pH 11.0) with 0.1 mL substrate (2.5 % colloidal chitin prepared in 50 mM KCl/NaOH, pH 11.0) incubated at 60 °C for 30 min. Along with the reaction, a control was run simultaneously with all the reagents in ice-cold water (0 °C). After the incubation, the reaction was stopped by heating in boiling water for 20 min. A standard curve of N-acetylglucosamine was constructed to determine the enzyme activities. One unit of chitinase activity was defined as the amount of enzyme that releases 1 μmol of N-acetylglucosamine per min under standard assay conditions. Additionally, cellulose activities were also evaluated using sigma cell cellulose, carboxymethyl cellulose, avicel and artificial substrate such as paranitrophenyl d-celobioside (pNPC) and paranitrophenyl-β-D glycopyranoside (pNPG).
Enzyme Purification
All the purification procedures were carried out at 0 °C unless stated otherwise. The crude supernatant chitinase was obtained by centrifuging the broth at 10,000×g for 30 min at 4 °C. Then, the supernatant was subjected to 30–80 % saturation with solid ammonium sulfate and left standing overnight at 4 °C. Ammonium sulfate was removed and proteins were recovered by centrifugation at 10,000×g for 45 min at 4 °C, dialyzed against 10 mM Tris/HCl (pH 8.0), and concentrated with an ultra filtration membrane (5 kDa, Millipore Corp). The dialyzed enzyme solution was loaded to Sepharose CL-6B column (85 cm × 1.7 cm) pre-equilibrated with 10 mM Tris/HCl, pH 8.0. Proteins were eluted at 30 mL/h (3 mL/fraction). Chitinase active fractions were pooled, concentrated, and analyzed for the purity. After monitoring the purity using sodium dodecyl sulfate (SDS) and zymography, further characterizations were carried out using the pure enzyme.
Polyacrylamide Gel Electrophoresis
The molecular mass of the enzyme was determined by 12 % (w/v) sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) as describe by Laemmli [18]. The gel was stained with Coomassie Brilliant Blue R-250 and then destained with a solution containing methanol/glacial acetic acid–distilled water = 1:1:8 (by vol). Molecular weight was estimated by comparing with the relative mobility of the reference protein. Protein size marker (MBI, Fermentas) was used for the reference protein. In addition, chitin zymography (activity staining) was performed using 12 % (w/v) gel that contained 0.05 % (w/v) glycol chitin. After electrophoresis, the gel was incubated for 1 h at 60 °C in 50 mM Tris/HCl buffer, pH 8.0 containing 1 % (v/v) Triton X-100. The gel was then stained with 0.01 % caucofluor white M2R in 0.5 M Tris/HCl, pH 8.0 for 30 min, and destained with water. The lytic zones were photographed under the UV-transilluminator.
Effect of pH and Temperature
The effect of pH and temperature on the activity and stability of the enzyme was studied. The pH was optimized at 60 °C using various pH buffer (pH value 2.0–13.6) at 50 mM concentration. The buffers used were citric acid/sodium phosphate (2–7.0), Tris/HCl (7.0–9.5), sodium bicarbonate/NaOH (9.5–11), and KCl/NaOH (11–13.6). To determine the pH stability, the enzyme samples were incubated at 100 mM of various pH buffers (as stated above) at 0 °C for 24 h and the residual activities were measured at 200 mM under standard assay conditions. Further, to determine the optimum temperature, the chitinase activities were examined at various temperatures (50–90 °C) in 50 mM Tris/HCl, pH 11.0. Temperature stability of the chitinase was measured in terms of residual activity under standard assay conditions after incubation of the purified chitinase at various temperatures ranged from 30–90 °C for 60 min.
Effects of Metal Ions and Other Reagents
The effects of different metal ions and other reagents on chitinase activities were measured by the addition of the corresponding ions and other reagents at different concentration to the reaction mixture, and the assay was performed under standard assay conditions. The tested ions were Ca2+, Mg2+, Cu2+, Co2+, Zn2+, K+, Na+, Mn2+, and Fe2+ where as other reagents includes Triton X–100, Tween-20, Tween-80, polyoxylethylene-4-laurylether, deooxycholic acid (DCA), sodium dodecyl sulfate (SDS), 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), hydrogen peroxide, sodium perborate, β-mercaptoethanol, 1,4-Dithiothreitol, ethylenediaminetetraacetic acid (EDTA), and ethylene glycol-bis (β-aminoethyl ether)-N,N,N’,N’-tetraacetic acid (EGTA). The relative activities were determined under standard assay conditions comparing with control without metal ions and other reagents (100 %).
N-terminal Amino Acids
The N-terminal amino acid sequences of Ch147 were determined by Edman degradation using a Procise Model 491 HT protein sequencer (Applied Biosystems, CS, USA).
Substrate Specificity and Kinetic Parameters
The purified chitinase was incubated with various substrates (2.5 %, w/v), such as colloidal chitin, sigma cell cellulose, carboxymethyl cellulose (CMC), avicel, paranitrophenyl d-celobioside (pNPC), and paranitrophenyl-β-D glycopyranoside (pNPG) in 50 mM KCl/NaOH pH 11.0 at 60 °C for 30 min and then the activity was measured under standard assay conditions. For kinetic parameters, five different concentrations of colloidal chitin (6.25–50 mg/mL) with constant enzyme concentration (0.1 mg) were prepared. Assays were performed under standard assay conditions at optimal conditions. The Michaelis-Menten constant (K m) and maximum velocity (V max) were determined from a Lineweaver-Burk plot.
Shelf life Stability
The shelf life stability of Ch147 was studied at room temperature (25 to 30 °C) and under refrigeration conditions (0 to 4 °C) in the presence of enzyme preservatives such as sodium azide (100 μg/mL), sodium metabisulphite (0.1 % w/v), potassium chloride (15 % w/v), and glycerol (30 % v/v). All the treated enzyme samples were kept under aseptic conditions to prevent microbial growth. Enzyme samples were withdrawn at different time intervals for up to 20 weeks to monitor residual chitinase activity under standard assay condition.
Antifungal Activity
Antifungal activity of Ch147 was detected by hyphal extension inhibition method as described by Roberts and Selitrennikoff [19]. F. solani and A. brasiliensis were used as indicator strains. The fungi of actively growing state were streaked on potato dextrose agar plates (3.9 % w/v) and incubated at 28 °C for 3 days. Discs containing purified Ch147 (5, 10, and 20 U) were placed on the plates and incubated at 28 °C. The disc without enzyme was taken as control. The hyphal extension inhibition was observed by the naked eyes after 2 to 3 days.
Chitin Hydrolysis
Thin layer chromatography (TLC) and high-performance liquid chromatography (HPLC) were used to evaluate the enzymatic hydrolysis of Ch147. TLC was analyzed similar to our previous method [20]. Briefly, purified Ch147 (0.12 mg/mL) was incubated with colloidal chitin substrate (250 mg/mL) at 50 mM KCl/NaOH buffer (pH 11.0) at 50 °C. The reaction was stopped by boiling for 20 min and samples were then spotted on the silica gel plates 60F 254 (E. Merck, Germany). The plates were developed with solvent system of chloroform–acetic acid–water (6:10:2, v/v/v) followed by spraying the plates with a methanol–sulfuric acid mixture (95:5, v/v) and heated for a few minutes at 150 °C. In case of HPLC studies, 50 μl (0.2 mg/mL) of purified chitinase was loaded to micro bond pack amino carbohydrate column (4.1 × 300 mm). Elution was done by 70 % acetonitrile at the flow rate of 1 mL/min. The products were detected using refractive index detector. A mixture of chitooligosaccharides consisting N-acetyl-d-glucosamine (GlcNAc), diacetylchitobiose (GlcNAc)2, and triacetlychitotrose (GlcNAc)3 (10 mg/mL) were used as the standard.
Results and Discussion
Bacterial Strain, Growth Conditions, and Screening
All twenty bacterial strains collected from different Korean provinces were cultivated in chitin medium at 28 °C and 110 rpm for 3 days. An alkalophilic and thermophilic bacterium, Streptomyces sp. CS147, was a strong producer (among cultivated strain) of an extracellular thermo stable chitinase and was selected for further study. Based on morphological, biochemical, and 16S rRNA sequences, the strain was identified as S. sp. CS147, as we previously described [20]. 16S rRNA sequence of the strain showed 99.79 % similarity with S. zaomyceticus NBRC 13348(T) (accession no. AB184346), 99.72 % similarity with S. venezuelae JCM 4526(T) (accession no. AB045890), and 99.72 % similarity with S. exfoliates NBRC 13191(T) (accession no. AB184324) (Fig. not shown). Further, when Ch147 (10 to 200 U) was inoculated on the spots of chitin agar plate and incubated at 37 °C for 48 h, the lytic zone increases on increasing the enzyme concentration where the control has no effect (no clear lytic zones), as shown in Fig. 1. Presence of clear lytic zones indicated chitin hydrolysis. In other words, chitin was utilized by bacterial enzymes for chitin degradation.

The plate showing the clear zone of hydrolysis by Ch147. The plates contains 0.5 % (v/v) colloidal chitin, spots inoculated with different concentration of enzyme and incubated at 37 °C for 48 h and then stained with 0.01 % Caucofluor white M2R in 0.5 M Tris/HCl, pH 8.0 for 30 min, and destained with water. The lytic zones were photographed under the UV-transilluminator. The holes contain (A) control, (B) 10 U, (C) 20 U, (D) 50 U, (E) 100 U, and (F) 200 U of purified Ch147
Enzyme Purification and Gel Electrophoresis
After harvesting the crude enzyme, Ch147 was purified through the sequential procedures which involve ammonium sulfate precipitation (30–80 %) and the use of a Sepharose CL-6B gel filtration column (Fig. not shown). Ch147 was purified in a single-step gel chromatography. In the gel filtration step, Ch147 was eluted in 10 mM Tris/HCl, pH 8.0 with a speed of 30 mL/h (3 mL/fraction). Many chitinase from Streptomyces have been purified using multiple column chromatographic steps [21], [22], [23], [24]. Ch147 was purified more efficiently than other available chitinase which shows its novelty. A summary of chitinase purification is illustrated in Table 1. Chitinase was purified to 8.15-fold with a recovery yield of 43.1 %. Furthermore, chitinase activity recovery was greater than those of the chitinase from S. sp. M-20 [21], S. cyaneus SP-27 [22], S. sp J-13-3 [23], S. venezuelae P10 [25], S. sp. DA11 [26], and S. griseus (MTCC 9723) [27]. The purity fold was higher than the chitinase from S. sp. M-20 [21], S. venezuelae P10 [25], and S. sp. DA11 [26].
The purified enzyme was analyzed by SDS-PAGE and activity staining of the gel (Zymography) shown in Fig. 2a, b, respectively. The molecular mass of Ch147was estimated approximately at 41 kDa, and was seen as a single band on gels. In accord with SDS-PAGE results, the pure enzyme showed a clear band by zymography, confirming that it was a chitinase. Its molecular mass was similar to chitinase S. roseolus [24] as shown in Table 2.

12% (w/v) SDS-PAGE (a) and zymograph (b) of purified chitinase from S. sp. CS147. Mr protein molecular weight marker (Fermentas), Crude crude extract, A. Sul 30–80 % ammonium sulfate precipitation fraction, Pure purified chitinase after Sepharose Cl-6B column chromatography
Effect of pH and Temperature on the Activity and Stability of the Purified Chitinase
The effects of pH and temperature on the activity and stability of Ch147 comparing with other chitinase from Streptomyces are shown in Table 2. The effects of pH on the activity and stability of Ch147 were shown in Fig. 3a. The chitinase activity was highest at pH 11.0, and over 60 % activity was detected over a broad range of pH (6–12.5). Although the activity was completely lost in extremely acidic and basic environment, the enzyme was stable in broad range of pH from 4 to 12.0 shown in Fig. 3a. Ch147, being stable in pH 4–12.0, could be stored in acidic to alkaline condition for prolonged period of time without any significant loss of the activity. The alkalophilic nature of some enzymes is very curious. Several physiological and structural assumptions have been made in literature, but the exact reason is yet to be explored. It is most likely that amino acid(s) near the catalytic center of the enzyme are responsible for such uniqueness. To make it clear, we have to perform mutational analysis of the catalytic domain of Ch147 by substitutions of amino acid, which is our future goal to explore this striking feature. Further, the stability on broad range of pH may be due to the reversible denaturation of the protein so that there is no effect on the activity of the enzyme on incubation at different pH. In comparison, Ch147 was found to be more stable in acidic and alkaline media than chitinase from S. cyaneus SP-27 [22], S. roseolus [24], S. venezuelae P10 [25], S. sp. DA11 [26], and S. griseus HUT 6037 [28] as shown in Table 2. Furthermore, Ch147 activity was found optimum at 60 °C and stable at or below 50 °C shown in Fig. 3. The enzyme showed its maximal activity at60 °C despite of stability at this temperature. It may be because of the protective effect of the substrate to Ch147 for optimal time. The temperature optimal and stability of Ch147 was found similar to the chitinase from S. cyaneus SP-27 [22] and S. roseolus [24] shown in Table 2. Many industrial processes are operated at extremes of pH (either acidic or alkaline) and at elevated temperatures thus the enzyme must suit the process requirement and must be capable of withstanding such harsh conditions for prolonged periods or at least during the process time which we can find in Ch147. The reaction rates approximately double for every 10 °C rise in temperature. Therefore, assuming the enzyme is stable at the higher temperature, the amount of enzyme needed can be reduced or the conversion time can be shortened. There are other benefits from operating at higher temperatures carrying out conversions at increased temperatures (above 60–65 °C) significantly reduces microbial infection of the material being processed [29]. In addition, the higher temperatures increase the solubility of polymeric substrates such as carbohydrates, thereby improving their mechanical handling characteristics and rendering them more amenable to enzymatic attack. The activity of Ch147 at high temperature and alkaline pH, stable from acidic to basic environment and high temperature, makes Ch147 a novel among chitinase previously reported from Streptomyces which we believe can be attractive for various biocatalytic applications.

a (filled circle) optimal pH, (filled square) pH stability of chitinase from S. sp. CS147. To determine pH optimum, the activities of chitinase were determined at 60 °C using different pH buffers. To examine chitinase stability, chitinase were incubated with different pH buffers (2–13.6) at 0 °C for 24 h and residual activities were evaluated under standard assay conditions. Each point represents as mean (n = 3). b (filled circle) optimal temperature, (filled square) thermal stability of chitinase from S. sp. CS147. To determine optimum temperature, the reactions were performed at different temperatures at optimum pH (11.0), whereas to determine temperature stability, purified Ch147 was stored at different temperatures for 1 h and residual activities were assayed under standard assay conditions. Each point represents as mean (n = 3)
Effects of Metal Ions and Other Reagents
Ch147 activity was significantly affected by various concentrations of metal ions, detergents, and modulators presented in Table 3. Chitinase activity was inhibited by Triton X-100, Tween-20, Tween-80, Polyoxylethylene-4-laurylether, deooxycholic acid, CHAPS, EDTA, SDS, Fe2+, and Zn2+but enhanced by Mn2+ and Mg2+similar to the reported chitinase from S. roseolus [24] and S. sp. DA11 [26] but against the chitinase from S. sp. M-20 [21], S. roseolus [24], S. griseus (MTCC 9723) [27], and Bacillus thuringiensis subsp. colmeri [30]. In addition, the activity was almost unaffected by K+, Na+, Co2+, and Mg2+ similar to the chitinase from S. sp. DA11 [26] and also modulators like hydrogen peroxide and sodium perborate did not affect the activity of Ch147 but β-mercaptoethanol inhibit the chitinase activity similar to the chitinase from S. viridificans [31] but against the chitinase from S. sp. M-20 [21] and S. griseus (MTCC 9723) [27]. This result reveals what kinds of metal ions and other reagents must be included or excluded for the industrial applications.
N-terminal Amino Acids
The amino acid sequences of the last N-terminal amino acids of Ch147 were found to be DINGGGATLPQKLYL. These sequences were compared with the available sequences in the National Centre for Biotechnology Information (NCBI) protein database using basic local alignment search tool (BLAST; http://blast.ncbi.nlm.nih.gov/Blast.cgi). The BLAST search did not suggest homology with other Streptomyces or chitinase sequences strongly suggesting that it might be the novel chitinase.
Substrate Specificity and Kinetic Parameters
The ability to hydrolyze several substrates is an important criterion for the chitinase potency. To determine the substrate specificity, purified Ch147 was assayed using different substrate (table not shown). It was found that the chitinase had better digestive ability on colloid chitin than other carbohydrates under the same assay condition whereas it did not show any activity towards sigma cell cellulose, carboxymethyl cellulose, avicel, and artificial substrate such as paranitrophenyl d-celobioside (pNPC) and paranitrophenyl-β-D glycopyranoside (pNPG). The kinetic constants of the purified Ch147 were calculated by fitting the data to a linear regression on Lineweaver-Burk double reciprocal plot. The kinetic constants (K m) and (V max) of Ch147were 2.05 ± 5.3 mg/mL and 467.2 ± 2.4 mmol/min mg using different concentrations of colloidal chitin (6.25–50 mg/mL). The K m values were higher than the other chitinase1.41 mg/mL from Enterobacter sp. NRG4 [32], but almost fivefold lower than (12 mg/mL) from Bacillus sp. BG-11 [33], respectively, suggesting that the affinity for the substrate of the enzyme obtained in this study was different from that of chitinase from other microorganisms.
Shelf Life Stability
The chitinase retained 100 % of its activity for up to 18 weeks at 4 °C, and retained about 80 % of its total activity except sodium azide (60 %) for up to 12 weeks in the presence of sodium metabisulphite, potassium chloride, and glycerol (data not shown) which showed efficient stability compared with the chitinase from Bacillus sp. BG-11 [33]. At room temperature (25 °C), the shelf life of chitinase was 14 weeks and it retained almost 100 % of its activity in the presence of sodium metabisulphite, potassium chloride, and glycerol except sodium azide (11 weeks), meanwhile more than 50 % of the total activity has been retained for up to 6 weeks in the case of all four additives. Ch147 activity was found to be less stable in the presence of sodium azide compared with other preservatives. The exact reason behind this phenomenon is yet to be explored but it was reported that sodium azide inhibit the catalytic enzyme activity irreversibly and would therefore render the endogenous enzyme catalytically inactive during the assay [34].
Antifungal Activity
In the present study, Ch147 showed high inhibition level against plant pathogen F. solani and A. brasiliensis shown in Fig. 4a, b. The inhibition was observed on PDA plates with paper disc loaded with purified chitinase of different concentration (5 to 20 U). The highest hyphal inhibition was observed at 20 U and the zone of inhibition had increased on increasing the enzyme concentration where control had no effect. Some previous research had also presented inhibition of hyphal extension growth of A. brasiliensis and F. solani, respectively from S. venezuelae P10, and S. griseus [25], [27]. F. solani is a phytopathogenic fungus which is an important causal agent of several crop diseases, such as root and stem rot of pea, sudden death syndrome of soybean, foot rot of bean, and dry rot of potato. Meanwhile, A. brasiliensis causes disease on certain fruits and vegetables such as grapes, onions, and peanuts, and is a common contaminant of food. Thus, chitinase can be the best option used or added as a supplement along with fungicides or/and pesticides not only to make them potent but also to minimize the concentration of chemicals which are harmful to the environment and health [33]. This result led us to conclude that chitinase produced by endophytic S. sp. CS147 can be used as a promising biocontrol of plant pathogenic fungi. However, a further evaluation of the disease control effectiveness of this strain and the design of a biocontrol formulation and application must be conducted under field conditions. Further, this study also provides a quantitative assessment of Streptomyces used in chitinolytic and antimicrobial activities. In addition, an estimative of the genetic parameters associated with these processes provides a foundation for microorganism screening programs to search for fungal antagonistic agents.

Antifungal activity of Ch147 against F. solani (a) and A. brasiliensis (b). The antifungal activity was assayed by hyphal extension inhibition assay as described in materials and methods. The paper disc contains 0 U (A), 5 U (B), 10 U (C), and 20 U (D) of purified Ch147
Chitin Hydrolysis
The enzyme hydrolyzed products of colloidal chitin were analyzed by both HPLC (not shown) and TLC shown in Fig. 5. According to the results, in the beginning of the enzyme(s) activity on colloidal chitin substrate, the rate of GlcNAc production increased rapidly, suggesting that the high concentration of the substrate and accessibility of nearly all available sites of the decrystallized chitin particles for the enzyme hydrolysis are the main reason for the production of GlcNAc. Chitooligosaccharides were the main oligosaccharides released initially which on further hydrolysis by Ch147 releases N-acetyl-d-glucosamine (GlcNAc), and trace amount of Diacetylchitobiose (GlcNAc)2 whereas (GlcNAc) was the major oligosaccharide. But most bacterial chitinases reported so far produce diacetylchitobiose as a major hydrolyzed product from colloidal chitin. Other end products, GlcNAc and chitooligosaccharides larger than (GlcNAc)3, are either produced in a small amount or not observed at all [28]. This result showed the enzymes’ novelty and attracts the possibility of efficient hydrolyzing the sugar molecules into alcohol, a second generation biofuel. Further, high-performance liquid chromatography (HPLC) analysis using Bond pack (NH2 column) also showed that the hydrolysis products from colloidal chitin was mainly (GlcNAc) similar to the chitinase from Nocardia orientalis [35]. Ch147 hydrolyzed colloidal chitin to 0.278, 0.817, and 1.058 mg/mL of (GlcNAc) as major products, at retention time of 4.3, respectively when incubated for 8, 16, and 24 h at 50 °C. It is reported that use of Bacillus chitinase for the synthesis of chitobiose is by combining GlcNAc and a sugar oxazoline derivative. Diacetylchitobiose has been widely used for the synthesis of biologically active compound as a starting material [36]. The (GlcNAc) as administered by intravenous (iv), intramuscular (im), and even oral routes was reported to be effective as an anti-inflammatory drug useful for the treatment of colitis and other gastrointestinal inflammation disorders [37].

Plot showing chitin degradation after treating with colloidal chitin at 50 °C and pH 11.0 with chitinase from S. sp. CS147. Mr mixture of N-acetyl-d-glucosamine (GlcNAc), diacetylchitobiose (GlcNAc) 2 and triacetlychitotrose (GlcNAc) 3
The mass production of first-generation liquid biofuels primarily from food crops and seed oils has resulted in a series of problems related to food prices, land usage, and carbon emissions [38]; second generation biofuels production are an appealing choice due to its being readily available, cheap, and easy to process using microbial extracellular enzymes. Extensive research has been conducted to investigate the utilization of lignocellulose biomass as an energy feedstock, with applications being developed for the production of biodiesel, bioethanol, and therapeutic fine chemicals.
Conclusions
In this study, we identified a novel chitinase, termed Ch147, from Streptomyces sp. CS147. It was purified by using simple, rapid, and non-expensive chromatographic method. The enzyme derives its novelty from having the following unique features: (i) it is purified in one step column chromatography and is stable in a broad range of pH (4–12.0), may be most alkali-tolerant so far reported from Streptomyces; (ii) its N-terminal amino acid sequence does not show significant homology with that of other chitinase; (iii) it is highly influential in all types of metal ions, modulators, and detergents and particularly in Mn2+; (iv) it produces mainly N-acetyl-d-glucosamine (GlcNAc) and a trace amount of diacetylchitobiose (GlcNAc)2 as hydrolyzed products, and (v) Ch147 shows antifungal activity against F. solani and A. brasiliensis which can be used for the biological control of fungus being unfriendly to chemicals. Further, the production of oligosaccharide like N-acetyl-d-glucosamine (GlcNAc) showed the clear pathway for the utilization of waste materials (chitin sources) for the production of sugar monomers which on further hydrolysis produces alcohol, a second generation biofuel.
References
Bugg, T. D., Ahmad, M., Hardiman, E. M., & Rahmanpour, R. (2011). Natural Product Reports, 28, 1883–18896.
Sun, Y., & Cheng, J. (2002). Bioresource Technology, 83, 1–11.
Patil, R. S., Ghormade, V., & Deshpande, M. V. (2000). Enzyme and Microbial Technology, 26, 473–483.
Makino, A., Ohmae, M., & Kobayashi, S. (2006). Polymer Journel, 38, 1182–1188.
Gortari, M. C., & Hours, R. A. (2013). Electronic Journal of Biotechnology, 16, 14–32.
Karasuda, S., Tanaka, S., Kajihara, H., Yamamoto, Y., & Koga, D. (2003). Bioscience, Biotechnology and Biochemistry, 67, 221–224.
Barikani, M., Oliaei, E., Seddiqi, H., & Honarkar, H. (2014). Iranian Polymer Journal, 23, 307–326.
Brzezinska, M. S., Jankiewicz, U., Burkowska, A., & Walczak, M. (2014). Current Microbiology, 68, 71–81.
Ober, C., & Chupp, G. L. (2009). Current Opinion in Allergy and Clinical Immunology, 9, 401–408.
Bhattacharya, D., Nagpure, A., & Gupta, R. K. (2007). Critical Reviews in Biotechnology, 27, 21–28.
Ordentlich, A. (1988). Phytopathology, 78, 84–88.
Inbar, J., & Chet, I. (1991). Soil Biology and Biochemistry, 23, 973–978.
Chernin, L. S., Winson, M. K., Thompson, J. M., Haran, S., Bycroft, B. W., Chet, I., Williams, P., & Stewart, G. S. (1998). Journal of Bacteriology, 180, 4435–4441.
Chandrasekaran, R., Revathi, K., Nisha, S., Kirubakaran, S. A., Sathish-Narayanan, S., & Senthil-Nathan, S. (2012). Pesticide Biochemistry and Physiology, 104, 65–71.
Trudel, J., & Asselin, A. (1989). Analytical Biochemistry, 178, 362–366.
Bradford, M. M. (1976). Analytical Biochemistry, 72, 248–254.
Miller, G. L. (1959). Analytical Chemistry, 31, 426–428.
Laemmli, U. K. (1970). Nature, 227, 680–685.
Roberts, W. K., & Selitrennikoff, C. P. (1988). Journal of General Microbiology, 134, 169–176.
G.C., P., Choi, Y. H., Choi, Y. S., Seong, C. N., Cho, S. S., Lee, H. J.,Yoo, J. C. (2013). Process Biochemistry, 48, 1188–1196.
Kim, K. J., Yang, Y. J., & Kim, J. G. (2003). Journal of Biochemistry and Molecular Biology, 36, 185–189.
Yano, S., Rattanakit, N., Honda, A., Noda, Y., Wakayama, M., Plikomol, A., & Tachiki, T. (2008). Biosciences, Biotechnology, and Biochemistry, 72, 54–61.
Okazaki, K., Kato, F., Watanabe, N., Yasuda, S., Masui, Y., & Hayakawa, S. (1995). Bioscience, Biotechnology and Biochemistry, 59, 1586–1587.
Xiayun, J., Chen, D., Shenle, H., Wang, W., Chen, S., & Zou, S. (2012). Carbohydrate Polymers, 87, 2409–2415.
Mukherjee, G., & Sen, S. K. (2006). Current Microbiology, 53, 265–269.
Han, Y., Yang, B., Zhang, F., Miao, X., & Li, Z. (2009). Marine Biotechnology, 11, 132–140.
Rabeeth, M., Anitha, A., & Srikanth, G. (2011). Pakistan Journal of Biological Sciences, 14, 788–797.
Tanabe, T., Kawase, T., Watanabe, T., Uchida, Y., & Mitsutomi, M. (2000). Journal of Bioscience and Bioengineering, 89, 27–32.
Ward, O. P., & Moo-Young, M. (1988). Biotechnology Advance, 6, 39–69.
Liu, D., Cai, J., Xie, C-c., Liu, C., Chen, Y-h. (2010). Enzyme and Microbial Technology, 46, 252–256.
Gupta, R., Saxena, R. K., Chaturvedi, P., & Virdi, J. S. (1995). Journal Applied Bacteriology, 78, 378–383.
Dahiya, N., Tewari, R., Tiwari, R. P., & Singh Hoondal, G. (2005). Electronic Journal of Biotechnology, 8, 14–25.
Bhushan, B., & Hoondal, G. S. (1998). Biotechnology Letters, 20, 157–159.
Ohno, Y., & Gallin, J. (1985). Journal of Biological Chemistry., 260, 8438–8446.
Nanjo, F., Katsumi, R., & Sakai, K. (1990). Journal of Biological Chemistry, 265, 10088–10094.
Kobayashi, S., Kiyosada, T., & Shoda, S.-i. (1997). Tetrahedron Letters, 38, 2111–2112.
Friedman, S. J., & Skehan, P. (1980). Proceedings of National Academy of Sciences of United State of America, 77, 1172–1176.
Sims, R. E., Mabee, W., Saddler, J. N., & Taylor, M. (2010). Bioresource Technology, 101, 1570–1580.
Acknowledgment
This work was supported by the research fund from Chosun University, 2014.
Author information
Authors and Affiliations
Corresponding author
Additional information
G. C. Pradeep and Hah Young Yoo contributed equally to this work.
Rights and permissions
About this article

Cite this article
G. C., P., Yoo, H.Y., Cho, S.S. et al. An Extracellular Chitinase from Streptomyces sp. CS147 Releases N-acetyl-d-glucosamine (GlcNAc) as Principal Product. Appl Biochem Biotechnol 175, 372–386 (2015). https://doi.org/10.1007/s12010-014-1267-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-014-1267-6