
Abstract
The development of cellulase production technology has greatly contributed to the successful use of cellulosic materials as renewable carbon sources. In this study, a putative endoglucanase IV (EG IV) complementary DNA was cloned from the mycelium of a strain of the filamentous fungus Trichoderma viride using a PCR-based exon-splicing method and expressed in both a silkworm BmN cell line and in silkworm larvae. Western blot analysis detected a band of 42 kDa in BmN cells after infection with a recombinant mBacmid/BmNPV/EG IV baculovirus. Sequence alignment analysis of the T. viride EG IV gene showed two domains that were highly conserved with glycosyl hydrolases and a funga-type cellulose-binding domain. Analysis of variance showed that silkworms infected with recombinant baculoviruses exhibited significantly higher enzyme activity that was 48.84% higher than silkworms infected with blank baculoviruses and 46.61% higher than normal silkworms. The expressed bioactive EG IV was also stable at the pH range from 5.0 to 10.0. The availability of large quantities of bioactive EG IV in silkworm provided a possibility to produce cellulase transgenic silkworm, which express bioactive cellulase specially in its digestive tract and improve its metabolism efficiency of mulberry leaves. Its application in the sericulture industry may be very promising.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
With the growth of serious energy and environmental problems, worldwide interest in alternative sources of energy such as agricultural biomass has increased [1–3]. Today, however, nearly all fuel ethanol is produced from corn glucose and sucrose [4], which consumes food materials, making renewable sources a better alternative. Cellulose is a linear polymer composed of β-1,4-linked d-glucopyranosyl units. It is the major component of the rigid cell walls of plants and is one of the most abundant organic compounds in the biosphere. With the current global warming crisis caused by the increasing use of fossil fuels, resulting in significant carbon dioxide emissions, the importance of cellulose hydrolysis in the conversion of plant biomass to fuels and chemicals is widely recognized [5]. Cellulose has great promise as a renewable energy source and has attracted the interest of biotechnologists who are developing its use as a source of fuel and chemicals. Demand is increasing for large quantities of cellulase with high bioactivity that can hydrolyze cellulose in biomass with high efficiency.
The cellulose-converting systems of many cellulolytic microorganisms may provide information about how to use plant cellulosic biomass as a sustainable energy source. Cellulolytic fungi are recognized as having the most promising ability to degrade cellulose. Of the cellulolytic fungi, species of the filamentous fungus Trichoderma are one of the most effective cellulase producers. These species have the strongest cellulose-degrading activity, and their cellulases have been widely investigated [6–8].
Based on enzymatic mode of action and substrate specificity, cellulases are classified into three different types: exo-β-1,4-glucanases (exoglucanases, EC 3.2.1.91), endo-β-1,4-glucanases (endoglucanases, EC 3.2.1.4), and β-1,4-glucosidase (β-glucosidases, EC 3.2.1.21) [9]. Endo-β-1,4-glucanases randomly cleave amorphous sites of cellulose chains, exo-β-1,4-glucanases act on the nonreducing or reducing termini of cellulose fibers to release either cellobiose or glucose, and β-1,4-glucosidases hydrolyze cellobiose or cello-oligomers to glucose from the nonreducing ends [10]. These enzymes act synergistically to facilitate the breakdown of cellulose and its conversion to an utilizable glucose [11].
Recently, many cellulase genes have been cloned with the goal of using the tremendous resource of cellulose waste that is currently available [12–14]. Trichoderma viride secretes at least five types of endoglucanases (EGI, EGII, EGIII, EGIV, and EGV), and the genes for these cellulases might be overexpressed in heterologous microbial hosts, such as Escherichia coli, yeast, or other fungi. However, isolation of the EG IV gene of T. viride has not yet been reported. For cellulase expression, it is difficult to express and purify a recombinant cellulase from microorganisms such as E. coli or yeast [15]. In E. coli, most produced proteins are retained in the periplasm, and only a few can be secreted into the culture medium. In addition, cellulases expressed in E. coli do not receive glycosylation or acetylation, and their expression levels are often too high, requiring renaturation techniques to refold them and restore their activities. In yeast, cellulases can be glycosylated, acetylated, and phosphorylated, but inaccurate glycosylation like mannotetrose glycosylation easily occurs, and glycosylation is often not complete [16]. Moreover, although the content of proteins secreted into the culture medium can be 30 g/L or higher, this is still lower than what can be achieved with filamentous fungi expression systems. In filamentous fungi, cellulases can be produced to a higher yield, with an accurate conformation, but it is still difficult to purify the target cellulase because crude cellulase is a complex system of cellulase proteins.
The advantages of silkworms as bioreactors include high expression efficiency, low feeding cost, natural activity for the expressed products, ease of purification, and nontoxicity [17, 18]. The Bac-to-Bac/Bombyx mori nucleopolyhedrovirus (BmNPV) baculovirus expression system is a preferred eukaryotic expression tool [19] because it greatly simplified technology with a fast experimental protocol. This makes the use of silkworm as a vector for industrial large-scale mass production promising [20].
In this study, a putative EG IV complementary DNA (cDNA) of a filamentous fungus T. viride strain was cloned using a PCR-based exon-splicing method. The gene was sequenced, and the results submitted to the GenBank database. The gene was expressed both in silkworm cell lines and in silkworm larvae using a mutant BmNPV baculovirus (mBacmid) lacking the virus-encoded chitinase (chiA) and cathepsin (v-cath) genes that was recently developed for the purpose of increasing foreign protein expression. The activity of the recombinant EG IV enzyme was characterized. The availability of large quantities of bioactive EG IV in silkworm may greatly contribute to its future application of cellulase transgenic silkworm in sericulture industry.
Materials and methods
Strains and plasmids
T. viride strain AS 3.3711 was from the China General Microbiological Culture Collection Center (CGMCC, Beijing). Culture conditions for T. viride were as described [21]. E. coli TG1 was used as a host for plasmid preparation. The DH10Bac E. coli mutant strain was used as the host of the donor plasmid pFastBacHT A, which contained an mBacmid lacking the chiA and v-cath genes of BmNPV.
Cell lines and silkworms
B. mori cell line BmN (from our lab) was cultured in T-25 flasks at 27 °C with Grace’s insect cell culture medium (Invitrogen, USA), supplemented with 10% fetal bovine serum. Hybrid silkworm larvae (commercial name, Qingsong X Haoyue) were reared conventionally and used in this experiment.
Cloning of putative EG IV cDNA
A PCR-based exon splicing method was used to clone the putative EG IV cDNA with primers in Table 1. Using the sequence of T. reesei EG IV (GenBank number Y11113), EG IV P1 and EG IV P4 were designed to PCR amplify EG IV genomic DNA from T. viride DNA. Isolation of genomic DNA was by urea and phenol/chloroform/isoamyl alcohol extraction.
PCR with EG IV P1-EG IV P4 was 94 °C for 2 min, 32 cycles of 94 °C for 1 min, 60 °C for 1 min and 72 °C for 72 s, followed by 72 °C for 10 min. Purified PCR product was cloned into pMD18-T vector (Takara) for sequencing.
Primer pairs EG IV P1–EG IV P2 and EG IV P3–EG IV P4 were used to clone EG IV exons using the previous PCR product as a template, using PCR as above. The purified products were used as templates in the next PCR reaction with primers EG IV P1–EG IV P4 as above. The product of EG IV exon splicing DNA that was the putative EG IV cDNA was purified and cloned into pMD18-T, named pMD18-T/EG IV, sequenced and submitted to the GenBank database. Properties of the putative EG IV cDNA were analyzed by DNAstar software. The signal peptide sequence of its deduced protein was analyzed by SignalP 3.0 Server (http://www.cbs.dtu.dk/services/SignalP/). Multiple sequence alignment was analyzed using ClustalW software (http://www.ebi.ac.uk/Tools/clustalw2/).
Generation of recombinant donor plasmid
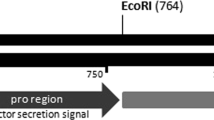
The EG IV gene was PCR-amplified from pMD18-T/EG IV using forward primer (5′-GCCGAATTCCATGGACATATTAATGACATTGTCATC-3′) and reverse primer (5′-TTAAAGCTTCTAGTTAAGGCACTGGGCG-3′) and restriction enzymes EcoRI and HindIII (underlined). PCR was as above. A purified product of the expected 990 bp was cloned into the corresponding sites of pFastBacHT A, yielding the recombinant donor plasmid pFast A/EG IV, and sequenced.
Construction of recombinant mBacmid and baculovirus
The recombinant donor plasmid pFast A/EG IV was transformed into DH10Bac mutant E. coli competent cells and propagated for 48 h on Luria–Bertani medium containing antibiotics (50 μg/ml kanamycin, 7 μg/ml gentamicin, and 10 μg/ml tetracycline), 100 μg/ml X-gal, and the inducer, 40 μg/ml isopropyl β-d-1-thiogalactopyranoside. DNA of the large recombinant mBacmid baculovirus (mBacmid/BmNPV/EG IV) was isolated from white (Lac Z−) colonies by alkaline lysis method and analyzed by PCR with the M13 forward and reverse primers.
The identified recombinant mBacmid was transfected into BmN cells using FuGENE 6 transfection reagent (Roche). The recombinant P1 viral solution was collected at 120 h post-transfection and used to further infect cells to generate the high-titer second generation viral solution for recombinant protein expression.
Expression of EG IV gene in silkworm
At 48 h post-infection by recombinant virus, BmN cells (2 × 106 cells/flask) were collected for Western blotting. For expression of the EG IV gene in silkworm larvae, the recombinant baculovirus was injected into first day larvae of the fifth instar at about 4 × 105 particles per worm in 10 μl before feeding. Enzyme activity analysis used 50 silkworms randomly sampled after 84 h.
Western blotting
After sodium dodecyl sulfate polyacrylamide gel electrophoresis with a 10% separating gel, proteins were transferred onto a nitrocellulose membrane at 110 mA for 1.5 h followed by blocking for 2 h at room temperature. The membrane was incubated with 6 × His antibody (GenScript, USA) for 1 h at room temperature, followed by washing with phosphate- buffered saline Tween and 1 h incubation of horseradish peroxidase (HRP)-labeled second antibody (GenScript, USA). After washing, HRP was detected with TMB solution (Promega).
Bioassay of recombinant EG IV
At 84 h post-infection by a second generation recombinant baculovirus, 50 silkworm larvae were randomly collected and homogenized in 100 ml of citrate buffer solution (pH 5.0), followed by centrifugation at 10,000 rpm at 4 °C for 10 min. Clarified supernatant was used as the source of enzymes, and activity was determined by the 3,5-dinitrosalicylic acid method with CMC-Na as a substrate. The international unit (IU) of activity was defined as the amount of enzyme that liberated 1 μmol of glucose per minute in a standard assay. For enzyme characterization, pH 3–10 and 40 °C-90 °C were used. For statistical analysis, analysis of variance (ANOVA) and Tukey’s honestly significant difference (HSD) tests were used. All experiments were performed in triplicate.
Results
Synthesis and analysis of putative EG IV cDNA from T. viride
A PCR-based exon splicing method was used instead of the traditional method of total RNA isolation followed by reverse transcription PCR (RT-PCR), to avoid contamination by heterogeneous nuclear RNA (hnRNA), which also has poly A structures and introns. The method also allowed easy synthesis of the putative EG IV cDNA from T. viride. The PCR product from EG IV genomic DNA with the primer pair EG IV P1–EG IV P2 is shown in Fig. 1a. Sequence analysis showed that it was 1,089 bp with a possible 54-bp intron.

Synthesis of the EG IV gene from T. viride using the exon splicing method. a PCR product of EG IV genomic DNA; b PCR products of EG IV exons from genomic DNA. Lane 1 DL2000 DNA Marker (TaKaRa); lane 2 first exon; lane 3 second exon; c lane 1, DNA marker G (BBI); lane 2 EG IV exon splicing product; d, schematic diagram of EG IV exon splicing method. Black arrow represents primer EG IV P1, orange arrow represents primer EG IV P2, red arrow represents primer EG IV P3, and blue arrow represents primer EG IV P4. EG IV P1–EG IV P2 were used for exon 1 amplification and EG IV P3–EG IV P4 for exon 2. Primer pair EG IV P1–EG IV P4 was used to synthesize EG IV exon splicing DNA
After exon splicing, the 1,035-bp EG IV gene was obtained (Fig. 1c) and its sequence registered in the GenBank database (GenBank accession number HM222525). This gene codes for 344 amino acids, with 21 amino acids in the N-terminus that could serve as a signal peptide. Sequence analysis by DNAstar software showed that its deduced protein contains 13 strongly basic amino acids, 20 strongly acidic amino acids, 113 hydrophobic amino acids, and 120 polar amino acids, with a predicted molecular weight (MW) of 35.6 kDa and an isoelectric point (pI) of 5.64.
Using the BLAST algorithm (http://www.ncbi.nlm.nih.gov), the putative EG IV cDNA sequence was found to be highly similar to others in GenBank, specifically the genes from T. reesei endoglucanase IV (Y11113, 99% identity), Trichoderma saturnisporum type IV endoglucanase (GU290062, 86% identity), and Neosartorya fischeri NRRL 181 endoglucanase (putative) (XM 001267235, 81% identity).
Relatively high amino acid sequence identities were observed to the known cellulase sequences when the translated amino acid sequence of the T. viride putative EG IV cDNA was compared to Neurospora crassa (67% protein sequence identity), N. fischeri (67%), Chaetomium globosum (64%), and Aspergillus terreus (62%) cellulase, all of which belong to the glycosyl hydrolase superfamily. These sequences were used for multiple sequence alignment using ClustalW software (http://www.ebi.ac.uk/Tools/clustalw2/), and the results are in Fig. 2.

Comparison of amino acid sequences of T. viride EG IV with other reported cellulases. Sequences were aligned using ClustalW software; identical and similar residues are highlighted in different colors. Marked regions in the multiple sequence alignment represent conserved domains of the glycosyl hydrolases-61 superfamily (A) and fungal type CBD (B). Asterisk denotes identity with T. viride sequence, while dashes represent gaps introduced for alignment
Construction of recombinant baculovirus
A 972-bp EG IV fragment encoding the mature peptide (Fig. 3a) was successfully cloned into a pFastBacHTA vector. After transformation of the purified recombinant donor plasmid pFastA/EG IV into DH10Bac E. coli, with a transposition rate of about 10% using a helper plasmid, the large recombinant DNA mBacmid/BmNPV/EG IV was isolated from white (Lac−) colonies for PCR analysis (Fig. 3b, c). Recombinant DNA mBacmid/BmNPV/EG IV was successfully constructed.

The EG IV mature gene fragment synthesized by PCR. a Lane 1 PCR product of EG IV mature gene fragment containing 18 bp of restriction enzyme sites; lane 2 DNA marker G (BBI). b Lane 1 λDNA/EcoRI + HindIII Marker, 3 (MBI); lane 2 PCR products of a white colony, theoretically concordant with ∼2,430 bp plus target EG IV gene (972 bp); c lane 1 DL2000 DNA Marker (TaKaRa); lane 2 PCR product of a blue colony
BmN cells transfected with the recombinant mBacmid/BmNPV/EG IV DNA, displayed obvious differences to uninfected cells, such as an increase in cell diameter and nuclei, and growth suspension after 120 h. Second generation recombinant baculovirus from the supernatant of transfected cell cultures was used to infect silkworm larvae by subcutaneous injection.
Expression of putative EG IV cDNA in silkworm
BmN cells expressed the putative EG IV cDNA by propagating the recombinant baculovirus in vivo. A Western blot of cell lysates showed a 42-kDa protein (Fig. 4), indicating that recombinant baculovirus containing the putative EG IV cDNA was successfully expressed in BmN cells.

Western blot of silkworm BmN cells infected with recombinant mBacmid/BmNPV/EG IV baculovirus. M FineMarker prestained protein ladder (GeneDireX); 1 recombinant EG IV protein expressed in silkworm BmN cells. 2 negative control infected by blank BmNPV
After silkworm larvae were subcutaneously injected with the recombinant baculovirus or blank baculovirus, the worms showed no obvious differences from normal silkworm larvae during the first 3 days, but presented abnormal phenotypes such as loss of appetite, elevation of intersegmental membranes, and swelling of body segments at 72 h post-injection, and died about 120 h after infection.
Empirically, the recombinant proteins were found to be at the highest expression level at 84 h post-infection, so 50 silkworms were randomly sampled at this time for enzyme activity assays.
Analysis of enzyme activity
Significant differences in enzyme activities were seen at different pHs and different temperatures (ANOVA, p < 0.01). The enzyme activity from recombinant baculovirus-infected silkworms was significantly higher than from blank baculovirus-infected silkworms or from normal silkworms (ANOVA, p < 0.01). Significantly higher enzyme activity by Tukey’s HSD test (p < 0.01) was seen at pH 6.0 and 50 °C (Fig. 5a, b), and this was stable from pH 5.0 to 10.0 (Fig. 5a) and from 40 to 60 °C (Fig. 5b). This was an increase of 48.84% compared to blank baculovirus-infected silkworms and 46.61% compared to normal silkworms, respectively.

Analysis of enzyme activity. a Optimum pH for enzyme activity. The pH profile was determined at pH 3.0–10.0. b Optimum temperature for enzyme activity. Temperature profile was determined at reaction temperatures of 40–90 °C. c Comparison of enzyme activities between recombinant baculovirus-infected silkworms, blank baculovirus-infected silkworms and normal silkworms. Data are mean ± SD (n = 6)
Discussion
Nowadays, nearly all fuel ethanol is produced from corn glucose and sucrose [4], which consumes too many food materials; thus, it is better to exploit other renewable sources. Cellulose is the major component in the rigid cell walls of plants and one of the most abundant organic compounds in the biosphere. It has vast prospective as a renewable source of energy and has attracted the interest of biotechnologists who wish to use it as a source of fuels and chemicals. Since the problems of energy and environment are more and more serious, there has been an increasing worldwide interest in alternative sources of energy [1–3], such as agricultural biomass. It is a great demand to exploit large quantities of cellulase with high bioactivity, which can hydrolyze cellulose in biomass with high efficiency.
Recently, more and more cellulase genes were cloned in order to use the tremendous resource of cellulose waste that is currently available [12–14, 22]. In the present study, to avoid hnRNA contamination, which also has poly A structures and introns, and to easily synthesize the EG IV gene from T. viride, we used a PCR-based exon splicing method, rather than traditional method of total RNA isolation followed by RT-PCR. The putative EG IV cDNA cloned from T. viride was registered in GenBank database (GenBank accession number HM222525). Among the known cellulase sequences, relatively high amino acid sequence identities were observed when the T. viride EG IV was compared with N. crassa (67% protein sequence identity), N. fischeri (67%), C. globosum (64%), and A. terreus (62%) cellulase. The sequence alignment of the T. viride putative EG IV cDNA with the reported cellulases showed two highly conserved domains of glycosyl hydrolases and fungal type cellulose-binding domain (CBD) (Fig. 2). Glycosyl hydrolases are a group of enzymes that hydrolyze the glycosidic bond between the carbohydrates. Based on the sequence similarity, glycosyl hydrolases have been classified into 85 different families [23, 24]. The CBD fold is composed of a three-stranded antiparallel β-sheet [25]. Many fungal CBDs composed of 36 amino acid residues are located at the N- or C-terminal extremity of the cellulases [26], and there are four conserved cysteines in this domain (Fig. 2), all involved in disulfide bonds. In terms of 3D protein structure, the T. reesei celllulases have been well characterized and used as a model for other cellulase gene studies [27] . Most fungal cellulases usually have a two-domain structure with one catalytic domain and one cellulose binding domain, which are connected by a short linker sequence rich in proline and/or hydroxy-amino acids [28]. This structure allows the enzyme to diffuse two-dimensionally on an insoluble substrate in a caterpillar way and thus work well.
The advantages of silkworm as a bioreactor include high expression efficiency, low feeding cost, natural activity for its expressed products, and safety for mankind [17, 18]. The Bac-to-Bac/BmNPV baculovirus expression system is one of the best eukaryotic expression tools [19] because it greatly simplifies the technical progress and shortens the experimental period. It is very promising to use the silkworm as vector for industrial large-scale mass production [20]. In this study, we used a recently developed mutant BmNPV bacmid (mBacmid), lacking the virus-encoded chitinase (chiA) and cathepsin (v-cath) genes of B. mori nucleopolyhedrovirus (BmNPV) for the purpose of increasing the expression of foreign proteins, to express the EG IV protein of T.viride in both B. mori cell lines and silkworms. As a result, the recombinant baculovirus with bioactive endoglucanase IV of T. viride was successfully constructed by integrating this gene into the transposition site of host genome and then transfecting the recombinant DNA into the silkworm BmN cells, which was confirmed by Western blotting and enzyme activity analysis. The approach established in this paper is probably one of the most economical and efficient ways of producing EG IV. Because silkworms can be used as feed for other animals, for it is nutritious and the recombinant baculovirus is non-infectious to animals, it is possible to be used as a feed additive for animals [29].
Furthermore, the purpose of this study was to test whether bioactive cellulase can be produced in silkworm for the future application of cellulase transgenic silkworm in sericulture industry. The pH in the normal silkworm’s digestive tract is about 9.2–9.8, which is not suitable for most cellulase activities. We used the Bac-to-Bac/BmNPV expression system to quickly determine whether cellulase protein can be bioactively expressed in silkworm larvae, especially under the alkaline condition. Because silkworm does not have any cellulase gene in its genome, and additionally the result showed the putative recombinant EG IV is stably bioactive at silkworm larvae body’s alkaline contiditon, it is possible and attemptable to produce cellulase transgenic silkworm, which can express bioactive cellulase specially in its digestive tract. It can improve the utilization efficiency of mulberry leaves, and its application in the sericulture industry may be very promising. Right now, we have construct a piggyBac/EG IV transgenic vector, which is confirmed successfully in silkworm BmN cell line (data not shown), and will further to construct a cellulase transgenic silkworm to test this possibility.
In summary, a putative EG IV cDNA was cloned from T. viride and expressed in silkworm. The availability of bioactive EG IV that the silkworm provides might greatly facilitate the future research and testing of this protein for potential application in industries. It provides a proof that producing cellulase transgenic silkworm is possible for the development of sericulture industry. Further studies will be also underway to purify the EG IV protein, elucidate the complete structure and post-translation modification of EG IV expressed in silkworm, and further produce cellulase transgenic silkworm.
References
Aristidou, A., & Penttila, M. (2000). Metabolic engineering applications to renewable resource utilization. Current Opinion in Biotechnology, 11, 187–198.
Jeffries, T. W., & Jin, Y. S. (2002). Ethanol and thermotolerance in the bioconversion of xylose by yeasts. Advances in Applied Microbiology, 47, 221–268.
Zaldivar, J., Nielsen, J., & Olsson, L. (2001). Fuel ethanol production from lignocellulose: A challenge for metabolic engineering and process integration. Applied Microbiology and Biotechnology, 56, 17–34.
Lin, Y., & Tanaka, S. (2006). Ethanol fermentation from biomass resources: Current state and prospects. Applied Microbiology and Biotechnology, 69, 627–642.
Himmel, M. E., Ruth, M. F., & Wyman, C. E. (1999). Cellulase for commodity products from cellulosic biomass. Current Opinion in Biotechnology, 10, 358–364.
Miettinen-Oinonen, A., & Suominen, P. (2002). Enhanced production of Trichoderma reesei endoglucanases and use of the new cellulase preparations in producing the stonewashed effect on denim fabric. Applied and Environmental Microbiology, 68, 3956–3964.
Penttila, M., Lehtovaara, P., Nevalainen, H., Bhikhabhai, R., & Knowles, J. (1986). Homology between cellulase genes of Trichoderma reesei: complete nucleotide sequence of the endoglucanase I gene. Gene, 45, 253–263.
Tomme, P., Van Tilbeurgh, H., Pettersson, G., Van Damme, J., Vandekerckhove, J., Knowles, J., et al. (1988). Studies of the cellulolytic system of Trichoderma reesei QM 9414. Analysis of domain function in two cellobiohydrolases by limited proteolysis. European Journal of Biochemistry, 170, 575–581.
Wilson, D. B., & Irwin, D. C. (1999). Genetics and properties of cellulases. Advances in Biochemical Engineering/Biotechnology: Recent Progress in Bioconversion, 65, 1–21.
Watanabe, H., & Tokuda, G. (2010). Cellulolytic systems in insects. Annual Review of Entomology, 55, 609–632.
Beguin, P., & Aubert, J. P. (1984). The biological degradation of cellulose. FEMS Microbiology Reviews, 13, 25–58.
Murray, P. G., Collins, C. M., Grassick, A., & Tuohy, M. G. (2003). Molecular cloning, transcriptional, and expression analysis of the first cellulase gene (cbh2), encoding cellobiohydrolase II, from the moderately thermophilic fungus Talaromyces emersonii and structure prediction of the gene product. Biochemical and Biophysical Research Communications, 301, 280–286.
Saloheimo, M., Nakari-Setala, T., Tenkanen, M., & Penttila, M. (1997). cDNA cloning of a Trichoderma reesei cellulase and demonstration of endoglucanase activity by expression in yeast. European Journal of Biochemistry, 249, 584–591.
Wang, T. H., Liu, T., Wu, Z. H., Liu, S. L., Lu, Y., & Qu, Y. B. (2004). Novel cellulase profile of Trichoderma reesei strains constructed by cbh1 gene replacement with eg3 gene expression cassette. Acta Biochim Biophys Sin (Shanghai), 36, 667–672.
Okumura, F., Kameda, H., Ojima, T., & Hatakeyama, S. (2010). Expression of recombinant sea urchin cellulase SnEG54 using mammalian cell lines. Biochemical and Biophysical Research Communications, 395, 352–355.
van Peij, N. N., Gielkens, M. M., de Vries, R. P., Visser, J., & de Graaff, L. H. (1998). The transcriptional activator XlnR regulates both xylanolytic and endoglucanase gene expression in Aspergillus niger. Applied and Environmental Microbiology, 64, 3615–3619.
Jeefrey, L. C., & Charles, S. C. (1996) Insect cell expression technology. In: J. L. Cleland et al. (Eds.), Protein engineering, chapter 7 (pp. 183–218). New York: Wiley-Liss Press.
Maeda, S. (1994) Expression of foreign genes in insect cells using baculovirus vectors. In: K. Maramorosch, A. H. McIntosh (Ed.), Insect cell biotechnology (pp. 1–31). Boca Raton: CRC Press.
Motohashi, T., Shimojima, T., Fukagawa, T., Maenaka, K., & Park, E. Y. (2005). Efficient large-scale protein production of larvae and pupae of silkworm by Bombyx mori nuclear polyhedrosis virus bacmid system. Molecular Cell Biology Research Communications, 326, 564–569.
Choudary, P. V., Kamita, S. G., & Maeda, S. (1995). Expression of foreign genes in Bombyx mori larvae using baculovirus vectors. In C. D. Richardson (Ed.), Methods in molecular biology. Baculovirus expression protocols, vol. 39. New Jersey: Humana.
Li, X. H., Wang, D., Zhou, F., Yang, H. J., Bhaskar, R., Hu, J. B., et al. (2010). Cloning and expression of a cellulase gene in the silkworm. Bombyx mori by improved Bac-to-Bac/BmNPV baculovirus expression system. Molecular Biology Reports, 37, 3721–3728. doi:10.1007/s11033-010-0025-2.
Tang, B., Pan, H., Zhang, Q., & Ding, L. (2009). Cloning and expression of cellulase gene EG1 from Rhizopus stolonifer var. reflexus TP-02 in Escherichia coli. Bioresource Technology, 100(23), 6129–6132.
Davies, G., & Henrissat, B. (1995). Structures and mechanisms of glycosyl hydrolases. Structure, 3, 853–859.
Henrissat, B., Callebaut, I., Fabrega, S., Lehn, P., Mornon, J. P., & Davies, G. (1995). Conserved catalytic machinery and the prediction of a common fold for several families of glycosyl hydrolases. Proc Natl Acad Sci USA, 92, 7090–7094.
Mattinen, M. L., Kontteli, M., Kerovuo, J., Linder, M., Annila, A., Lindeberg, G., et al. (1997). Three-dimensional structures of three engineered cellulose-binding domains of cellobiohydrolase I from Trichoderma reesei. Protein Science, 6, 294–303.
Tomme, P., Warren, R. A., & Gilkes, N. R. (1995). Cellulose hydrolysis by bacteria and fungi. Advances in Microbial Physiology, 37, 1–81.
Zhou, X., Smith, J. A., Oi, F. M., Koehler, P. G., Bennett, G. W., & Scharf, M. E. (2007). Correlation of cellulase gene expression and cellulolytic activity throughout the gut of the termite Reticulitermes flavipes. Gene, 395, 29–39.
Shen, H., Schmuck, M., Pilz, I., Gilkes, N. R., Kilburn, D. G., Miller, R. C., et al. (1991). Deletion of the linker connecting the catalytic and cellulose-binding domains of endoglucanase A (CenA) of Cellulomonas fimi alters its conformation and catalytic activity. Journal of Biological Chemistry, 266, 11335–11340.
Zhou, L., Wu, X., Lan, L., & Liu, J. (2010). Expression of Trichoderma reesei endo-beta-glucanase II in silkworm. Bombyx mori L. by using BmNPV/Bac-to-Bac expression system and its bioactivity assay. Biotechnology Letters, 32, 67–72.
Acknowledgments
The work was supported by the National Basic Research Program of China under grand number 2012CB114601 and the National Natural Science Foundation of China (number 30972141/C120110), the Key project of Zhejiang Government (number 2011C14006), and Chinese Universities Scientific Fund.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, Xh., Zhang, P., Liang, S. et al. Molecular Cloning and Characterization of a Putative cDNA Encoding Endoglucanase IV from Trichoderma Viride and its Expression in Bombyx Mori . Appl Biochem Biotechnol 166, 309–320 (2012). https://doi.org/10.1007/s12010-011-9426-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-011-9426-5