
Abstract
An α-l-arabinofuranosidase gene, abf51S9, was cloned from Streptomyces sp. S9 and successfully expressed in Escherichia coli BL21 (DE3). The full-length gene consisted of 1,506 bp and encoded 501 amino acids with a calculated mass of 55.2 kDa. The deduced amino acid sequence was highly homologous with the α-l-arabinofuranosidases belonging to family 51 of the glycoside hydrolases. The recombinant protein was purified to electrophoretic homogeneity by Ni-NTA affinity chromatography and subsequently characterized. The optimal pH and temperature for the recombinant enzyme were 6.0 and 60∼65 °C, respectively. The enzyme showed a broad pH range of stability, retaining over 75% of the maximum activity at pH 5.0 to 11.0. The specific activity, K m, and V max with p-nitrophenyl-α-l-arabinofuranoside as substrate were 60.0 U mg−1, 1.45 mM, and 221 μmol min−1 mg−1, respectively. Abf51S9 showed a mild but significant synergistic effect in combination with xylanase on the degradation of oat-spelt xylan and soluble wheat arabinoxylan substrates with a 1.19- and 1.21-fold increase in the amount of reducing sugar released, respectively. These favorable properties make Abf51S9 a good candidate in various industrial applications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Xylan is the main hemicellulosic component of plant cell walls and the second most abundant renewable polysaccharide in nature [1, 2]. It is a complex heteropolysaccharide consisting of a β-1,4-linked xylose backbone with substitution on different side chains with l-arabinose, d-galactose, acetyl, feruloyl, and glucuronic acids residues [3, 4]. Due to the structural complexity of xylan, its complete degradation requires the cooperative action of a complex array of enzymes. Among xylanolytic enzymes, endoxylanases and β-xylosidases are the backbone degrading enzymes, and accessory enzymes such as α-glucuronidase, acetyl esterase, and α-l-arabinofuranosidase (AFase) are responsible for the cleavage of side chains [5, 6].
AFases, one of the most important xylan-debranching enzymes, generally catalyze the hydrolysis of terminal α-l-1,5-arabinofuranosidic linkages from arabinoxylan, arabinan, and arabinogalactan [7]. Based on their amino acid sequence similarities, AFases have been classified into glycoside hydrolase (GH) families 43, 51, 54, and 62 [8, 9]. A number of AFases have been purified and characterized from bacteria including Clostridium stercorarium [10], Bacillus pumilus [11], Geobacillus stearothermophilus [12], Bifidobacterium longum [13], and Streptomyces spp. [14–16].
In our previous studies on xylanolytic enzymes from Streptomyces sp. S9, a new xylanase (XynAS9) with broad temperature adaptability [17] has been reported. Here, we describe the cloning of a new AFase gene from Streptomyces sp. S9. The gene was expressed in Escherichia coli, and the purified recombinant enzyme was characterized. Additionally, the synergistic effects of AFase and xylanase during the enzymatic degradation of oat-spelt xylan and soluble wheat arabinoxylan were also determined.
Experimental
Strains and Plasmids
Streptomyces sp. S9 ACCC 41168 was obtained from the Agricultural Culture Collection of China. The strains E. coli JM109 (Promega, USA) and BL21 (DE3) (Novagen, USA) were used for recombinant plasmid amplification and gene expression, respectively. The plasmid pUC19 (TaKaRa, Japan), pGEM-T Easy vector (Promega, USA), and pET-30a(+) (Novagen) were used for plasmid preparations, gene cloning, and expression, respectively.
Substrates and Materials
The synthetic substrate p-nitrophenyl-α-l-arabinofuranoside (pNPA) and oat-spelt xylan were purchased from Sigma (USA). Soluble wheat arabinoxylan, sugar beet arabinan, azurine cross-linked (AZCL)-arabinan (debranched), arabinooligosaccharides (arabinobiose, arabinotriose, arabinotetraose, arabinopentaose, arabinohexaose), and xylooligosaccharides (xylobiose, xylotriose, xylotetraose, xylopentose, xylohexose) were obtained from Megazyme (Australia). The DNA purification kit, restriction endonucleases, T4 DNA ligase, and LA Taq DNA polymerase with GC buffer were purchased from TaKaRa (Japan). Recombinant enterokinase was purchased from Novagen (USA). Other reagents were of analytical grade and commercially available.
Cloning the AFase-Encoding Gene abf51S9
The core region of the AFase-encoding gene abf51S9 was obtained using polymerase chain reaction (PCR) with degenerate primers Abf51F (5′-CGCTACCCNGGNGGNAAYTT-3′) and Abf51R (5′-GTACCANACRTTCCAYTCRTC-3′) designed on the basis of two conserved amino acid sequences RYPGGNF and DEWNVW among GH 51 AFases from bacteria (http://www.cazy.org/fam/GH51.html). The genomic DNA of Streptomyces sp. S9 was used as the template. The amplified fragment was purified and ligated into pGEM-T Easy vector for sequencing and Basic Local Alignment Search Tool analysis.
A genomic library was constructed as described previously [18]. A colony PCR method [19] was used for the target gene isolation with two specific synthetic primers SF (5′-GTCTCCAACTACCGCTGGGA-3′) and SR (5′-GTGCAGGGAGACGTAGTCGA-3′). The clone carrying the candidate fragment was obtained, and the recombinant plasmid was isolated and sequenced.
Sequence Analysis
The sequence assembly was performed using the Vector NTI Suite 7.0 software, and the nucleotide sequence was analyzed using the National Center for Biotechnology Information Open Reading Frame (ORF) Finder tool (http://www.ncbi.nlm.nih.gov/gorf/gorf.html). Signal peptide in the deduced amino acid sequence was predicted by SignalP 3.0 server (http://www.cbs.dtu.dk/services/SignalP/). The DNA and protein sequence alignments were carried out using the blastn and blastp programs, respectively (http://www.ncbi.nlm.nih.gov/BLAST/). Multiple alignments of protein sequences were performed using the CLUSTALW program (http://www.ebi.ac.uk/clustalW/).
Construction of Expression Plasmids
For expression of abf51S9 in E. coli, the structural abf51S9 gene was amplified by PCR using primers S9BF (5′-TACCATGGATATGCCCACCGCCCGTCTCACC-3′) (NcoI site underlined) and S9BR (5′-TTAAAGCTTTCAGCGGGTGGCGAGGCGGATCA-3′; HindIII site underlined). The PCR product was gel-purified, digested with NcoI and HindIII, and cloned into the corresponding sites of pET-30a(+) vector. The recombinant pET-abf51S9 with a His-Tag at the N terminus was transformed into E. coli BL21 (DE3) competent cells and confirmed by sequencing.
Enzyme Assays
The arabinofuranosidase activity was assayed according to the method of Manin et al. [14] with slight modifications. The standard reaction contained 0.1 ml appropriately diluted enzyme and 0.1 ml McIlvaine buffer (pH 6.0) containing 2 mM pNPA. After incubation at 60 °C for 10 min, the reaction was stopped by addition of 0.6 ml of 1.0 M Na2CO3. The released p-nitrophenol was quantitatively determined spectrophotometrically by reading the absorbance at 405 nm. One unit of arabinofuranosidase activity was defined as the amount of enzyme that liberated 1.0 μmol of p-nitrophenol per min. Xylanase activity was determined by measuring the release of reducing sugars from xylan as previously described [17].
Expression and Purification of the Recombinant Proteins
The positive transformant harboring pET-abf51S9 was picked from a single colony and grown overnight at 37 °C in Luria–Bertani (LB) medium supplemented with 50 µg ml−1 kanamycin. The culture was then transferred into fresh LB medium (1:100 dilution) containing kanamycin and grown aerobically at 37 °C to an A 600 of ∼0.8. The subculture was grown at 30 °C for 6 h with agitation at 180 rpm after induction with 0.8 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG).
To purify the recombinant protein, cells were harvested by centrifugation at 12,000×g for 5 min at 4 °C and then resuspended in sonication buffer A (20 mM Na2HPO4/0.1 M citric acid, pH 6.0) using about 5 ml buffer per gram wet weight. The cell suspension was sonicated on ice with 50 short bursts of 10 s followed by intervals of 20 s for cooling. The cell debris was removed by centrifugation, and the concentrated supernatant (crude enzyme, 5 ml) was applied to a 1-ml Ni-NTA chelating column (Qiagen, Germany) that had been previously equilibrated with buffer B (20 mM Tris-HCl, 500 mM NaCl, pH 7.6). The protein was eluted using a step gradient of imidazole (0, 20, 40, 60, 80, 100, 200, and 300 mM) in buffer B containing 10% (w/v) glycerol. Fractions were tested for enzyme activity using the standard method described above.
The purified enzyme was evaluated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), using a 12% running gel [20]. The low molecular weight calibration kit for SDS electrophoresis (GE Healthcare) was used as a standard. The proteins were visualized by staining with Coomassie brilliant blue R-250. To determine the native molecular weight of recombinant Abf51S9, the purified protein was subjected to nondenaturing gradient (4–12%) PAGE analysis and stained with Coomassie brilliant blue. The protein concentration was determined using the Bradford method [21] with bovine serum albumin as the standard.
To identify the effects of His tag on enzyme folding and activity, the N-terminal His tag was cleaved by incubation of recombinant Abf51S9 with enterokinase at room temperature for 16 h. Using the Enterokinase Cleavage Capture Kit (Novagen), enterokinase was removed using EKapture agarose, and the truncated protein was recovered by spin filtration and subjected to enzyme characterization.
Biochemical Characterization
The pH optimum of purified recombinant Abf51S9 activity was determined at various pH values ranging from 4.0 to 9.0. The pH stability was estimated by pre-incubating the purified enzyme solution with different buffers of pH 4.0–12.0 at 37 °C for 1 h and measuring the remaining enzyme activity under standard condition. The buffers used (0.1 M each) were McIlvaine buffer (pH 4.0−8.0), Tris-HCl buffer (pH 8.0−9.0), and glycine–NaOH (pH 9.0−12.0).
The optimal temperature for arabinofuranosidase activity was determined in McIlvaine buffer at the optimal pH (pH 6.0) from 30 °C to 80 °C. Thermostability of the purified recombinant enzyme was evaluated by pre-incubating the enzyme for various periods in McIlvaine buffer (pH 6.0) at 60 °C and 70 °C without substrate. Residual enzyme activity in each case was then assayed under standard assay condition.
The effect of different metal ions and some chemical reagents on the recombinant enzyme activity was assessed in McIlvaine buffer (pH 6.0) at 60 °C. The reactions contained 1 or 10 mM of NaCl, KCl, CaCl2, LiCl, CoCl2, NiSO4, CuSO4, MgSO4, FeCl3, ZnSO4, Pb(CH3COO)2, FeSO4, AgNO3, HgCl2, SDS, EDTA, or β-mercaptoethanol. The assay system without any additive was used as a control.
The K m, V max, and k cat values for the purified recombinant Abf51S9 were analyzed by the Lineweaver–Burk method. The enzyme activity was assayed at 60 °C in McIlvaine buffer (pH 6.0) using pNPA as substrate at concentrations ranging from 0.1 to 1.0 mM.
Substrate Specificity
To determine whether Abf51S9 can hydrolyze oligosaccharides and polysaccharides, the substrate specificity of the enzyme was tested using the following substrates: oat-spelt xylan, soluble wheat arabinoxylan, sugar beet arabinan, AZCL-arabinan (debranched), arabinooligosaccharides (arabinobiose, arabinotriose, arabinotetraose, arabinopentaose, arabinohexaose), and xylooligosaccharides (xylobiose, xylotriose, xylotetraose, xylopentose, xylohexose). The reaction mixture contained 0.5 ml of 1% (w/v) substrate and 0.1 ml of enzyme (1 U) solution in 50 mM McIlvaine buffer (pH 6.0). After incubation at 37 °C for 24 h, the reaction was stopped by boiling for 5 min, and the released reducing sugars in the supernatant were measured as arabinose equivalents, and the reaction without enzyme addition was treated as control.
Enzymatic Hydrolysis—Synergistic Interactions
To determine the effects of Abf51S9 in combination with xylanase on the hydrolysis of oat-spelt xylan and soluble wheat arabinoxylan, different enzyme preparations were tested as described by Raweesri et al. [22] with some modifications. The simultaneous interaction system (1 ml) contained 10-mg oat-spelt xylan or soluble wheat arabinoxylan, 50 mM McIlvaine buffer (pH 6.0), and 0.5 U ml−1 of each individual enzyme. After incubation at 37 °C for 48 h, the mixtures were boiled for 20 min, and the released reducing sugars in the supernatant were measured as xylose equivalents.
Sequential interactions were performed in two-step reactions. The reaction mixture and conditions were the same as the simultaneous reactions described above. The two enzyme preparations were used as follows: a xylan-backbone degrading enzyme XynAS9 (0.5 U ml−1 each) and an accessory enzyme Abf51S9 (0.5 U ml−1 each). The first reaction contained either XynAS9 or Abf51S9 and was incubated at 37 °C for 24 h. The mixture was boiled for 20 min to inactivate the first added enzyme. Then, the second reaction was carried out by adding the other enzyme and incubated at 37 °C for another 24 h. Finally, the released reducing sugars were measured in the supernatant.
Nucleotide Sequence Accession Number
The nucleotide sequence for the Streptomyces sp. S9 α-l-arabinofuranosidase gene (abf51S9) was deposited in GenBank database under accession number GQ359824.
Results
Gene Cloning and Sequence Analysis
A 693-bp DNA fragment was amplified from Streptomyces sp. S9 by PCR using degenerate primers Abf51F and Abf51R derived from the conserved amino acid sequences of the bacteria GH 51 arabinofuranosidases. The sequence of this fragment exhibited 75% identity with the arabinofuranosidase from Streptomyces sviceus ATCC 29083 (GenBank accession no. EDY57062), suggesting that the fragment obtained was a partial arabinofuranosidase-encoding gene sequence. Primers SF and SR were synthesized based on the partially identified sequence and used for screening of the Streptomyces sp. S27 genomic library. Among the 2,500 transformants screened, one clone (PS2201) contained the gene abf51S9. The insert of the plasmid DNA from PS2201 was about 5 kb, containing one complete ORF of 1,506 bp. The ORF had about 72.2% G + C content, started with an ATG, and terminated with a TGA codon. No signal peptide was predicted using SignalP. The mature polypeptide contained 501 residues with a calculated mass of 55.2 kDa.
The deduced amino acid sequence of Abf51S9 was compared with family 51 arabinofuranosidase sequences available from the GenBank. The highest identity was 67.1% with the putative α-l-arabinofuranosidase from Streptosporangium roseum DSM 43021 (GenBank accession no. EEP18193), followed by the α-l-arabinofuranosidase (64.3%, GenBank accession no. AAA61708) from Streptomyces lividans 66. Alignment of Abf51S9 with the other GH 51 AFases predicted that Abf51S9 had two domains: a (β/a)8-barrel (Asp18∼Ala381) and a 12-stranded β sandwich with jelly-roll topology (Met1∼Val17 and Arg382∼Arg501). Two putative catalytic residues, Glu173 (acid/base) and Glu292 (nucleophile), are located in the catalytic domain (Fig. 1).

Amino acid sequence alignment of Abf51S9 from Streptomyces sp. S9 with other α-l-arabinofuranosidases of GH 51. The abbreviations, sources, and GenBank accession nos. of the proteins are Abf51S9, Streptomyces sp. S9, GQ359824; S.ros, Streptosporangium roseum DSM 43021, EEP18193; S.ave, Streptomyces avermitilis MA-4680, BAC73455; S.liv, Streptomyces lividans, P53627; K.fla, Kribbella flavida DSM 17836, EEJ18245; C.aci, Catenulispora acidiphila DSM 44928, EEN29837. Residues that are conserved among all six sequences are highlighted in black. Two putative catalytic site residues are marked (asterisk). Cysteine residues in Abf51S9 are marked (C)
Enzyme Expression and Purification
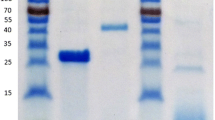
The recombinant gene encoding Abf51S9 containing a N-terminal His-tag was cloned into the vector pET-30a(+) and then transformed into E. coli BL21 (DE3). After induction with IPTG, the crude enzyme activity in cell lysate was 1.2 U ml−1. The recombinant Abf51S9 was purified using a one-step affinity chromatography on a Ni-NTA column. The resulting specific activity of the purified Abf51S9 was 60.0 U mg−1 after 113-fold purification, with a final activity yield of 18.1%. The purified enzyme was a single band with a molecular mass of ∼60.0 kDa as determined by SDS-PAGE (Fig. 2). After digestion with enterokinase, 43 amino acid residues were removed from the N terminus of recombinant Abf51S9 based on sequence analysis, and the truncated protein showed one band of ∼55 kDa, which was essentially identical to the calculated value. The truncated protein showed the same arabinofuranosidase activity as recombinant fusion protein did. Thus, His tag did not influence the folding or activity of the enzyme. Native gradient gel electrophoresis of the purified Abf51S9 demonstrated that the purified protein migrated as one band of ∼240 kDa, suggesting that native Abf51S9 might be a tetramer.

SDS-PAGE analysis of the purification of recombinant Abf51S9 expressed in E. coli BL21 (DE3). Lanes M low molecular weight marker, 1 culture supernatant of the induced transformant harboring empty pET-30a(+), 2 culture supernatant of the induced transformant harboring pET-abf51S9, 3 purified recombinant Abf51S9 after Ni-NTA affinity chromatography
Properties of Purified Recombinant Enzyme
The optimal pH of purified recombinant Abf51S9 was 6.0, and over 80% of the maximum activity remained between pH 5.5 and 7.0 (Fig. 3a). The pH stability assay showed that more than 75% of the initial activity was retained after pre-incubation in buffers ranging from pH 5.0 to 11.0 (Fig. 3b). The optimal temperature for enzyme activity was 60∼65 °C at pH 6.0 (Fig. 3c). After incubation at 60 °C for 1 h, the enzyme retained ∼40% of the initial activity, but lost all activity within 20 min at 70 °C (Fig. 3d).

Properties of the purified recombinant Abf51S9. a Effect of pH on enzyme activity. The assay was performed at 60 °C in buffers with pH ranging from 4.0 to 9.0. b pH stability of Abf51S9. After incubating the enzyme at 37 °C for 1 h in buffers of pH 4.0–12.0, the activity was measured in McIlvaine buffer (pH 6.0) at 60 °C. c Effect of temperature on enzyme activity measured in McIlvaine buffer (pH 6.0). d Temperature stability of Abf51S9. The enzyme was pre-incubated at 60 °C (diamond) and 70 °C (square) in McIlvaine buffer (pH 6.0), and aliquots were removed at specific time points for the measurement of residual activity at 60 °C
The enzyme activity of the recombinant Abf51S9 in the presence of 1 and 10 mM of different metal ions and chemical reagents is shown in Table 1. The activity was absolutely inhibited by Ag+ and strongly inhibited by Hg2+ and Cu2+ even at 1 mM concentration. In the presence of 10 mM Zn2+, SDS, Cr3+, and Ca2+, the enzyme activity was reduced to less than 50%. Fe3+ at 1 and 10 mM enhanced the activity about 1.16- and 1.45-fold, respectively. The addition of other reagents had little or no effect on the activity. Based on the Lineweaver–Burk plot, the K m, V max, and k cat values using pNPA as substrate were 1.45 mM, 221 μmol min−1 mg−1, and 204 s−1, respectively.
Substrate Specificity
Substrate specificity of Abf51S9 was examined by including oligosaccharides and polysaccharides with various linkages. The enzyme did not display activity against oat-spelt xylan, soluble wheat arabinoxylan, sugar beet arabinan, AZCL-arabinan (debranched), and xylooligosaccharides (xylobiose, xylotriose, xylotetraose, xylopentose, xylohexose), but it exhibited activity on α-1,5-linked arabinooligosaccharides (arabinobiose, arabinotriose, arabinotetraose, arabinopentaose, arabinohexaose), with the released reducing sugars of 4.24, 6.87, 6.32, 5.25, and 7.93 mg–1 ml–1, respectively.
Interactions of Abf51S9 and XynAS9 in the Hydrolysis of Xylan
To determine the mechanism of synergistic effects, the application of arabinofuranosidase in combination with xylanase was studied using the hydrolysis of oat-spelt xylan (containing 10% arabinose, 75% xylose, and 15% glucose) and soluble wheat arabinoxylan (containing 37% arabinose, 61% xylose, and 2% other sugars) [13, 23]. Simultaneous interaction-reaction results showed that addition of Abf51S9 preparation to XynAS9 significantly enhanced 19% and 21% of the action of XynAS9 on the oat-spelt xylan or wheat arabinoxylan (Table 2).
The results of sequential interaction reactions showed that the amounts of released reducing sugars were enhanced 1.14- and 1.19-fold for the substrate oat-spelt xylan and wheat arabinoxylan, respectively, when XynAS9 was added in the first reaction. When Abf51S9 was added in the first reaction, no significant enhancement of released reducing sugars was observed.
Discussion
In this study, we cloned, expressed, and characterized an AFase from the xylanase-producing Streptomyces sp. S9 [17]. Several Streptomyces AFases have been reported and showed similar optimal pH and temperature. For example, Abf51S9 under study showed optimal activity at pH 6.0 and 60∼65 °C, ABF from S. lividans 66 was optimally active at pH 6.0 and 60 °C [14], AbfB from S. lividans [24] and AFaseI from Streptomyces chartreusis GS901 [25] exhibited maximal activity at pH 6.0 and 55 °C, and AFase from Streptomyces sp. PC22 [22] was optimal at pH 6.0∼6.5 and 65 °C. However, Abf51S9 showed some distinct enzymatic properties compared with other AFases from Streptomyces. Abf51S9 was highly pH stable over a wide pH range, retaining over 75% of the activity after treatment in buffers ranging from pH 5.0 to 11.0 at 37 °C for 1 h. Comparing with Abf51S9, most AFases from Streptomyces were not stable at alkaline pH. For example, AFase from Streptomyces sp. PC22 at pH 9.0 retained only ∼20% activity [22]. This superior property makes Abf51S9 more potential in research and industrial applications.
A characteristic shared by most AFases from Streptomyces is their dependence on divalent metal ions for activity. The addition of Hg2+, Cu2+, and Zn2+ inhibited the activity of Abf51S9 significantly, suggesting that Abf51S9 is a thiol-sensitive enzyme because these heavy metal ions bind free mercapto groups (–SH) in cysteine residues. Moreover, sensitivity to Hg2+ indicates that Abf51S9 has an active-site thiol group. Based on the predicted structure of Abf51S9 using SWISS-MODEL (http://swissmodel.expasy.org//SWISS-MODEL.html), there are four cysteine residues in Abf51S9, and three of them, Cys169, Cys211, and Cys346, are near the catalytic residues and two glutamate residues, Glu173 and Glu292, are located at the active sites (Fig. 1). Fe3+ stimulated Abf51S9 activity about 1.16- and 1.45-fold at 1 and 10 mM concentration, respectively. The reason might be that Fe3+ may influence the active site and lead to some conformational changes.
Synergistic interactions between AFase and xylanase in the hydrolysis of arabinoxylan have been reported previously [22, 26]. Under the tested conditions, significant synergistic effect occurred in the simultaneous interaction reaction containing Abf51S9 and XynAS9. Sequential reaction results indicated that Abf51S9 had no ability to catalyze the hydrolysis of oat-spelt xylan. This result has also been found in other GH 51 AFases [14, 24]. When added Abf51S9 after XynAS9 hydrolysis, the amount of reducing sugars liberated was similar with that of simultaneous interaction reaction. Abf51S9 exhibited activity only against α-1,5-linked arabinooligosaccharides, indicating that Abf51S9 is an exo-α-l-arabinosyl hydrolase that has no activity on polymers. Some GH 51 enzymes exhibited very little or no activity with arabinose-containing polysaccharides. For example, ArfB from C. stercorarium [10] and AFaseI from S. chartreusis GS901 [25] are active on arabinogalactan; AbfB from S. lividans cannot hydrolyze oat spelt xylan, whereas AFase from Streptomyces sp. PC22 [24] and AraF from a novel thermophilic bacterium PRI-1686 [27] have this ability. Therefore, Abf51S9 may play a role in the assimilation of arabinose moieties from arabinose-containing xylooligosaccharides generated by xylanase. In the future, accessory enzymes such as AFases combining with xylanase might be widely applied in animal feed to convert xylan to simple sugars effectively and enhance feed digestibility.
References
Prade, R. A. (1996). Biotechnology and Genetic Engineering Reviews, 13, 101–131.
Polizeli, M., Rizzatti, A., Monti, R., Terenzi, H., Jorge, J., & Amorim, D. (2005). Applied Microbiology and Biotechnology, 67, 577–591.
Coughlan, M. P., & Hazlewood, G. P. (1993). Biotechnology and Applied Biochemistry, 17, 259–289.
Bocchini, D. A., Alves-Prado, H. F., Baida, L. C., Roberto, I. C., Gomes, E., & Da-Silva, R. (2002). Process Biochemistry, 38, 727–731.
Kebir, H., Dupont, C., & Morosoli, R. (2000). Biochimica et Biophysica Acta, 1491, 177–184.
Saha, B. C. (2000). Biotechnology Advances, 18, 403–423.
Saha, B. C., Dien, B. S., & Bothast, R. J. (1998). Applied Biochemistry and Biotechnology, 72, 115–125.
Henrissat, B., & Davies, G. (1997). Current Opinion in Structural Biology, 7, 637–644.
Fritz, M., Ravanal, M. C., Braet, C., & Eyzaguirre, J. (2008). Mycological Research, 112, 933–942.
Schwarz, W., Bronnenmeier, K., Krause, B., Lottspeich, F., & Staudenbauer, W. (1995). Applied Microbiology and Biotechnology, 43, 856–860.
Degrassi, G., Vindigni, A., & Venturi, V. (2003). Journal of Biotechnology, 101, 69–79.
Shallom, D., Belakhov, V., Solomon, D., Gilead-Gropper, S., Baasov, T., Shoham, G., et al. (2002). FEBS Letters, 514, 163–167.
Margolles, A., & de los Reyes-Gavilán, C. G. (2003). Applied and Environmental Microbiology, 69, 5096–5103.
Manin, C., Shareek, F., Morosoli, R., & Kluepfel, D. (1994). The Biochemical journal, 302, 443–449.
Tajana, E., Fiechter, A., & Zimmermann, W. (1992). Applied and Environmental Microbiology, 58, 1447–1450.
Tsujibo, H., Takada, C., Wakamatsu, Y., Kosaka, M., Tsuji, A., Miyamoto, K., et al. (2002). Bioscience, Biotechnology, and Biochemistry, 66, 434–438.
Li, N., Meng, K., Wang, Y., Shi, P., Luo, H., Bai, Y., et al. (2008). Applied Microbiology and Biotechnology, 80, 231–240.
Li, N., Yang, P., Wang, Y., Luo, H., Meng, K., Wu, N., et al. (2008). Journal of Microbiology and Biotechnology, 18, 410–416.
Bottoli, A. P., Kertesz-Chaloupková, K., Boulianne, R. P., Granado, J. D., Aebi, M., & Kües, U. (1999). Journal of Microbiological Methods, 35, 129–141.
Laemmli, U. (1970). Nature, 227, 680–685.
Bradford, M. (1976). Analytical Biochemistry, 72, 248–254.
Raweesri, P., Riangrungrojana, P., & Pinphanichakarn, P. (2008). Bioresource Technology, 99, 8981–8986.
Rahman, A. K. M. S., Sugitani, N., Hatsu, M., & Takamizawa, K. (2003). Canadian Journal of Microbiology, 49, 58–64.
Vincent, P., Shareck, F., Dupont, C., Morosoli, R., & Kluepfel, D. (1997). The Biochemical Journal, 322, 845–852.
Matsuo, N., Kaneko, S., Kuno, A., Kobyashi, H., & Kusakabe, I. (2000). The Biochemical Journal, 346, 9–15.
Sørensen, H. R., Pedersen, S., Viks-Nielsen, A., & Meyer, A. S. (2005). Enzyme and Microbial Technology, 36, 773–784.
Birgisson, H., Fridjonsson, O., Bahrani-Mougeot, F. K., Hreggvidsson, G. O., Kristjansson, J. K., & Mattiasson, B. (2004). Biotechnology Letters, 26, 1347–1351.
Acknowledgments
This work was supported by the Chinese National High Technology Research and Development Program (863 Program, Grant No. 2007AA100601) and the Chinese Agricultural Microorganism Collection and Share Program (No. 2005DKA21201).
Author information
Authors and Affiliations
Corresponding author
Additional information
Pengjun Shi and Ning Li contributed equally to this work.
Rights and permissions
About this article
Cite this article
Shi, P., Li, N., Yang, P. et al. Gene Cloning, Expression, and Characterization of a Family 51 α-l-Arabinofuranosidase from Streptomyces sp. S9. Appl Biochem Biotechnol 162, 707–718 (2010). https://doi.org/10.1007/s12010-009-8816-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-009-8816-4