
Abstract
Purpose of the review
Although calcium pyrophosphate deposition (CPPD) has been known since the 1960s, our understanding of its pathogenesis remains rudimentary. This review aims to illustrate the known mechanisms underlying calcium pyrophosphate (CPP) crystal formation and deposition and explore future directions in research. By examining various perspectives, from basic research to clinical and imaging assessments, as well as new emerging methodologies, we can establish a starting point for a deeper understanding of CPPD pathogenesis.
Recent Findings
Recent years have seen significant advances in CPPD research, particularly in the clinical field with the development of the 2023 ACR/EULAR classification criteria for CPPD disease, and in imaging with the introduction of the OMERACT ultrasonographic definitions and scoring system. However, progress in basic research has been slower. New laboratory approaches, such as Raman spectroscopy and omics sciences, offer promising insights that may help piece together the puzzle of CPPD.
Summary
CPPD is a common yet understudied condition. As the population ages and CPPD becomes more prevalent, there is an urgent need to better understand the disease and the mechanisms involved in crystal formation and deposition, in order to improve diagnosis and therapeutic approaches.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Understanding calcium pyrophosphate (CPP) crystal formation and deposition remains an area ripe for exploration, with numerous aspects awaiting clarification [1,2,3]. Calcium pyrophosphate deposition (CPPD) is prevalent, especially among older people, and encompasses a broad spectrum of clinical presentations, ranging from asymptomatic deposition to acute or chronic arthritis with a significant impact on patients' lives [4].
Our current comprehension of CPP crystal formation derives from studies on cellular and extracellular mechanisms, ion imbalances, and investigations into genetic conditions that predispose to CPPD. Imaging studies, particularly conventional radiography (CR) and ultrasound (US) have primarily elucidated the joints and structures most frequently affected by CPP crystal deposition, along with their association with other conditions and the prevalence of depositions within selected populations. Newer modalities, such as dual-energy conventional tomography (DECT) and Raman spectroscopy are gaining prominence in this field, and hold promise for providing novel insight into this disorder.
This review focuses on illustrating the mechanisms underlying CPP crystal formation and deposition, examining different perspectives that collectively could hopefully enhance a better understanding of CPPD pathogenesis.
Lessons from the Lab
Cell changes in CPPD
Although the exact mechanisms that lead to the formation of CPP crystals are not fully understood, various cell types including chondrocytes, synoviocytes, as well as inflammatory and bone cells are differently involved in the production of crystals and in the inflammatory response. Much of the research on the interaction between joint cells and CPP crystals stems from studies conducted in patients with osteoarthritis (OA) [5].
CPP crystals primarily form within fibrocartilage, hyaline cartilage, or tissue undergoing chondroid metaplasia, with chondrocytes being the principal cells involved in their production [6]. Chondrocytes represent the sole cell type in articular cartilage (comprising both hyaline cartilage and fibrocartilage) and contribute to CPP crystal formation by producing high levels of extracellular inorganic pyrophosphate (PPi) and generating articular cartilage vesicles (ACVs) [7]. It has been reported that ACVs are mainly derived from hypertrophic chondrocytes, however, a recent study indicates that CPP crystal deposition is associated with aging and chondrocyte senescence. Indeed, cellular senescence markers, such as p16 and p21, are increased in CPPD cartilage compared to OA cartilage [8] (Fig. 1).

Main mechanisms involved in CPP crystal formation. Activated osteoclasts in subchondral bone release TGFβ1 into cartilage, which induces an increase in the levels of nucleotide pyrophosphohydrolase (NTPPH) enzymes and PPi production. Chondrocytes and senescent chondrocytes release articular cartilage vesicles (ACVs) in pericellular regions of extracellular matrix that contain NTPPH enzymes, particularly ENPP1. In the same cells, Ank mediates the extracellular efflux of ATP. In ACVs, ENPP1 hydrolyzes ATP in PPi and AMP. Excess PPi complexes with calcium to form amorphous CPP precursors, which are then converted into more stable CPP crystals and grow within or along collagen fibrils. Once formed in the ECM, CPP crystals may shed into the synovial fluid. TGFβ1: transforming growth factor beta 1; ANK: progressive ankylosis protein; ENPP1: ectonucleotide pyrophosphatase/phosphodiesterase 1; ATP: adenosine triphosphate; AMP: Adenosine monophosphate; PPi: inorganic pyrophosphate; CPP: calcium pyrophosphate
While it's intuitive to attribute CPP crystal formation to cartilaginous structures, the presence of these crystals in non-cartilaginous tissues like tendons and ligaments, where chondrocytes are typically absent, raises intriguing questions. This could be explained by the fact that the enthesis of tendons and ligaments presents a fibrocartilaginous layer [9], thus providing a plausible explanation for the preferential deposition of CPP crystals at the insertions of tendons and ligaments. Additionally, changes in extracellular matrix composition and the presence of chondrocyte-like cells, instead of fibroblasts, have been observed around CPP and basic calcium phosphate (BCP) deposits in tendons [10, 11]. The acquisition of chondrocyte phenotype and the consistent formation of calcific deposits in the midsubstance of tendons could be due to an erroneous differentiation of tenocytes into fibrocartilage, or more likely to a fibrochondrogenic differentiation of human tendon-derived stem/progenitor cells [11].
CPP crystals can also be found in the synovial membrane, but it remains uncertain whether they are produced by synoviocytes or derived from synovial fluid (SF) and articular cartilage [5]. Nonetheless, it is recognized that CPP crystal contributes to synovitis by stimulating synovial fibroblast to increase the production of matrix metalloproteinase-8 (MMP-8) and Interleukin 6 (IL-6) [12, 13]. Macrophages are the primary inflammatory cells in the synovial membrane and are implicated in promoting inflammation and contributing to joint damage [5]. In vitro studies have shown that upon stimulation by CPP or BCP crystals, macrophages release pro-inflammatory cytokines and chemokines, exacerbating inflammation and stimulating the production of reactive oxygen species (ROS) and matrix-degrading enzymes by chondrocytes [14, 15]. Furthermore, macrophages' attempted digestion of insoluble crystals often results in inefficient phagocytosis, leading to lysosomal rupture, ROS production, adenosine triphosphate (ATP) release and potassium efflux, activating the NACHT domain-, leucine-rich repeat-, and PYD-containing protein 3 (NLRP3) inflammasome [16], resulting in the production of IL-1β, which perpetuates inflammation through additional upregulation of cytokines and chemokines, neutrophil recruitment, and production of inflammatory mediators [15, 17].
In addition, CPP crystals have been found to trigger a regulated form of cell-death, known as necroptosis, in fibroblasts, epithelial cells, and neutrophils [16]. This process is linked with the formation of neutrophil extracellular traps (NETs), that contribute to inflammation. All these mechanisms, which activate the innate and adaptive immune system, can explain the acute or chronic arthritis phenotypes observed in CPPD, and moreover, low-grade synovitis can be linked to the progression of joint damage.
Bone health and repair processes have been reported to play a key role in CPPD [18]. Osteoclasts in subchondral bone seem to be a new player in the CPP crystal formation scenario, and this is related to an excess of osteoclast activity, which may release transforming growth factor beta (TGFβ) and other factors into cartilage, increasing PPi production [7, 18]. The increased presence of osteoclasts in the subchondral bone of humans and mice with OA, along with aberrant bone structures and reduced mechanical strength, supports this hypothesis. CPP crystals enhance osteoclastogenesis mediated by receptor activator of nuclear factor-κB ligand (RANKL) and macrophage colony-stimulating factor 1 (M-CSF) by promoting the p38 mitogen-activated protein kinase (MAPK) and extracellular-signal-regulated kinase (ERK) pathways [19].
Extracellular Matrix and Synovial Fluid (SF) Changes in CPPD
Histologic and in vitro observations suggest that CPP crystals form extracellularly, initially in ACVs in pericellular regions of extracellular matrix [20,21,22], where excess PPi can accumulate and complex with calcium. ACVs are chondrocyte-derived organelles that contain enzymes involved in both CPP and BCP crystals formation [23,24,25]. Among these enzymes, nucleotide pyrophosphohydrolases (NTPPH) play a key role in extracellular PPi production by hydrolyzing ATP [7]. In particular, the main participant was identified in NTPPH enzyme plasmacell membrane glycoprotein 1 (ENPP1/PC1), a regulator of tissue mineralization. ENPP1 up-regulated expression was detected in calcified degenerative menisci, and associated with intracellular and extracellular PPi (ePPi) [26]. In contrast, ENPP1 downregulation was reported in human and mouse OA cartilage, correlating with increased cartilage calcification [27]. However, direct evidence linking the presence of CPP and ENPP1 levels is lacking in both studies, thus suggesting the need for further investigation into the role of this enzyme and the potential involvement of other factors in these processes. In this context, reduction in tissue-non-specific alkaline phosphatase (TNAP) levels or increase in progressive ankylosis protein homolog (Ank) activity are supposed to regulate ePPi elaboration. TNAP antagonizes ENPP1 by hydrolyzing ePPi to extracellular inorganic phosphate (ePi), thus promoting BCP crystal formation [28, 29]. Ank is a transmembrane transporter detected in high levels in CPPD cartilage [30], whose mutations in the gene encoding are associated with familial CPPD [31, 32]. It has long been debated whether Ank transports PPi or ATP; with recent findings indicating its role in mediating the cellular efflux of ATP rather than PPi [33, 34].
The ePPi levels and enzyme activity are regulated by different factors, such as TGFβ, insulin-like growth factor-1, IL-1β, retinoic acid and thyroid hormone [7]. Usually, increases in ePPi levels correspond to decreases in those of ePi and vice versa, suggesting that Pi/PPi ratio in ACVs is determinant to the type of crystal formed [35]. An imbalance toward PPi production promotes CPP crystal formation, while BCP crystal formation is prevented [36]. Initially, excess PPi complexes with calcium to form amorphous CPP precursors, which are then converted into more stable crystalline forms and grow within or along collagen fibrils [5]. Despite its potential importance, no studies have been conducted on how calcium levels contribute to crystal formation.
CPP crystal formation and growth can also be promoted by other extracellular matrix components, such as osteopontin, transglutaminase and type I collagen [37,38,39]. In contrast, proteoglycans and type II collagen have been shown to inhibit CPP formation [39]. Interestingly, enhanced type X collagen expression has been observed in CPPD cartilage [40].
Once formed in the extracellular matrix, CPP crystals may shed into the SF. Most studies evaluating the role of SF in crystal formation are dated, and updated investigations are needed to better characterize changes in its composition. In these studies, CPPD patients have higher SF NTPPH, ATP and PPi levels than patients with other arthropathies [41,42,43,44]. In addition, it is suggested that high ePPi concentrations in SF are mainly produced by fibrocartilage and hyaline cartilage and are a key factor for CPP crystal formation [32, 45]. However, the effective role and origin of these ions in SF are not yet completely understood and could also derive from the crystal dissolution in this medium. In contrast, a more recent study comparing SF of patients with OA to those with CPPD plus OA, found no significant differences in the concentrations of calcium ions, magnesium ions, Pi and PPi [46].
Lessons from the Clinics
CPPD and Association with Other Diseases
Together with OA, CPPD is the most common abnormality observed in joints in older people. Prevalence of CPPD estimated from radiographs of the appendicular skeleton is up to 13.7% at around 60 years of age, and continuously increases with age-groups to reach 50% among people older than 80 years, with no predilection for either sex [6, 16, 47, 48]. While CPPD disease covers a variety of phenotypes ranging from acute inflammatory episodes, persistent CPP crystal inflammatory arthritis, to chronic non-inflammatory joint pain in the context of CPPD associated with OA [2, 6, 49,50,51], to date, no relationship has been established between the extent and anatomical localization of CPPD and the phenotype(s) of CPPD disease that will eventually develop.
The very specific and rare clinical situations of familial CPPD disease allowed to identify genetic mutations associated with early and severe CPPD, and provided a better understanding of CPP crystal formation in general [32]. Two genes in particular have been linked to these familial phenotypes of CPPD disease: gain-of-function mutations of Ank human gene (ANKH) which codes for a transporter of ATP which will be transformed in ePPi, which will precipitate with calcium ions [34, 52], and TNFRSF11B which encodes osteoprotegerin [53]. The implication of ANKH-associated mutations appears to be responsible for very early onset of CPPD and secondarily of severe joint damage, while people with osteoprotegerin mutations present with a predominant phenotype of early onset of severe and diffuse OA and concomitant CPPD, possibly due to a mechanism involving subchondral bone abnormalities [54]. ANKH mutations have also been suggested to be involved in sporadic CPPD disease [55]. However, the preliminary results of the first attempt to inhibit ANKH in the clinical setting using probenecid, an anion transporter inhibitor [56], did not show a meaningful clinical impact.
The identification of risk factors for CPPD and associated comorbidities also helped out in the understanding of the pathophysiology of CPPD, and on the general balance between CPPD and hydroxyapatite, involved in bone formation. Gitelman’s disease, a renal tubulopathy characterized by severe hypomagnesemia, has been linked with severe and extensive CPPD, including involvement of the axial skeleton [57]. Magnesium is a natural inhibitor of TNAP, which cleaves PPi into Pi, thereby shifting the equilibrium from CPP crystal formation towards hydroxyapatite [34]. While the only four-decade-old trial including 38 patients using magnesium supplementation failed to demonstrate a decrease in the extent of CPPD on radiographs at 6 months, its results suggested that magnesium could have a positive impact on pain relief [58].
The recently proposed association between CPPD disease and osteoporotic fractures may bring new insight into the balance between CPP crystal and hydroxyapatite formation [59]. In a study including 1148 patients with acute CPP crystal arthritis matched to 3730 comparators, the fracture relative risk was found to be twice as high in participants with CPPD (hazard ratio 1.8 [95% confidence interval 1.3–2.3]) [59]. Given that TNAP plays a central role in the balance between PPi and Pi, and sporadic CPPD disease appears to be associated with osteoporosis, it is probable that TNAP regulation needs to be further explored to understand this association. Moreover, this association may be more complex than the sole imbalance in calcium crystal formation and also involves the role of systemic inflammation, suspected to be responsible for the increased risk of cardiovascular events following acute episodes of CPP crystal arthritis [60, 61], as it has been shown in gout [62].
Imaging Contribution in CPPD Understanding
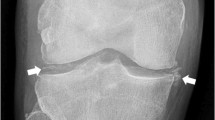
The link between CPPD and imaging extends to 1960, even before CPP crystals identification in SF, when the calcification of articular cartilage, termed “chondrocalcinosis” was first documented by Zitnan and Sitaj [63]. Since then, imaging has contributed to characterizing the disease, assessing the prevalence of calcification in joint structures, elucidating the association of CPPD with other conditions and facilitating differential diagnosis.
Imaging techniques have played a crucial role in defining CPPD as a systemic disease affecting both peripheral and axial regions [49, 64]. They have also allowed us to understand that CPP crystal deposition is not confined to the hyaline cartilage, as initially thought, but extends to other joint and periarticular structures, including fibrocartilage, tendons, ligaments and synovial membrane [65,66,67,68] (Fig. 2). A radiological study conducted at the knee level by Neame et al., demonstrated that the fibrocartilage was the most affected structure in CPPD, observed in 95% of cases, compared to hyaline cartilage in 45% of cases, and joint capsules or the synovial membrane in 30% of cases [69]. These findings were corroborated by a recent systematic literature review (SLR) assessing the prevalence of CPPD across various joint structures using ultrasound and radiography [70] (Table 1). The SLR revealed a higher prevalence in the knee menisci and the triangular fibrocartilage of the wrist, compared to the knee hyaline cartilage. Despite such evidence, much of the basic research has remained narrowly focused on hyaline cartilage. However, shifting perspectives may provide a pathway to delve deeper into understanding this complex disease.

Ultrasonographic appearance of CPPD in various joint structures. CPP deposits (arrows) appear as hyperechoic (white) deposits (similar to the bone cortex) without creating acoustic shadowing. In fibrocartilage, hyaline cartilage, and synovial. membrane, they present as deposits of variable size and shape, while in tendons, they appear as multiple linear deposits parallel to the tendon’s fibres, not continuous with the bone profile. A) CPP deposits (arrows) in femoral hyaline cartilage, B) in fibrocartilage (knee meniscus traced with a white continuous line), C) in tendons (Achilles tendon), and D) in the synovial membrane (synovial hypertrophy traced with a white continuous line). HC: hyaline cartilage, MM: medial meniscus, AT: Achilles tendon, SM: synovial membrane, SF: synovial fluid
Moreover, imaging has shed some light on how CPP crystals deposit within joint structures. The identification of the pseudo double contour sign on ultrasound or cartilage icing on radiography has enhanced our understanding, indicating CPP crystal deposition not only within but also over the cartilage [71,72,73]. Though the exact mechanism remains unclear, a recent study offers an anatomopathological explanation of the pseudo double contour sing, suggesting CPP deposits within the capsule/ligament atop the hyaline cartilage [71]. Thus, dynamic scanning can be useful in distinguishing the pseudo double contour of CPPD from the double contour characteristic of gout [72].
Imaging has also been instrumental in enhancing our understanding of CPPD epidemiology. Radiographic cartilage calcification (formerly known as chondrocalcinosis) has conventionally been considered a surrogate for CPPD prevalence, and radiographic studies have shown CPPD prevalence ranging from 7% to 13.7% at around 60 years of age, increasing with age to 50% among individuals over 80 years old [6, 16, 48, 74]. Ultrasound, with its higher sensitivity, has gone further, revealing CPPD in over 55% of patients presenting at outpatient rheumatologic clinics for the first time with any joint complaint, rising to 66% among those older than 60 and 73% among individuals aged 80 years or older [75]. However, the exact prevalence of CPPD remains undetermined, as current studies have focused solely on symptomatic individuals. Nonetheless, imaging techniques, particularly ultrasound, hold promise for conducting epidemiological studies in the general population.
A lesser-explored aspect of CPPD is its natural history. In this realm, imaging techniques, particularly ultrasound, thanks to the recently developed ultrasonographic scoring system for assessing CPPD extent [76], could provide new insight into the evolution of CPP crystal deposition over time, for example following therapy or after acute attacks, as well as exploring the correlation between CPPD extent and joint damage or OA, and moreover can offer a means of monitoring the efficacy of potential therapies.
The future
Raman Spectroscopy “for Everyone”
Raman spectroscopy is a technique based on the interaction between light and electrons in a material. Atoms in a molecular bond make oscillatory motions, modulating the electronic response with unique frequencies, enabling precise material identification. When irradiated with monochromatic (laser) light, the small energy changes associated with the modulation of the bonds lead to a distinct presentation of higher and lower frequencies in the scattered light. This can be measured as a color shift from the original light source. Analysis of the color spectrum of the scattered light provides a fingerprint of the bonds present in the sample, useful for identifying the material. CPP crystal is identified by several specific Raman features, most notably a strong band which can be attributed to the symmetric P-O stretching mode [77]. The location of this band is dependent on the type of CPP crystal, and is found at 1049 cm−1 for triclinic CPP dihydrate, 1045 cm−1 for monoclinic CPP dihydrate, and 1036 cm−1 for monoclinic CPP tetrahydrate β [78]. These specific spectral characteristics allow for detailed crystal analysis, revealing even subtle differences in structure. This is interesting as different crystal phases may elicit different inflammatory responses [15].
The current gold standard for SF crystal analysis is compensated polarized light microscopy (CPLM), which is significantly less accurate for CPP crystals compared to monosodium urate (MSU) crystals [79, 80]. The birefringence of triclinic CPP is roughly 20 × lower than that of MSU [81], and the varied crystal morphology of CPP further complicates its identification with CPLM. Additionally, BCP crystals are even less birefringent and therefore not identifiable with CPLM. Raman spectroscopy can be used to identify CPP, BCP, but also calcium oxalate crystals in SF and tissue with 100% spectral specificity. Spectroscopic techniques are more objective than operator-dependent microscopic classification and are also well-suited for artificial intelligence-based classification models [82]. In SF analysis, Raman spectroscopy is not performed on bulk material; instead, laser light must be focused directly on the crystals, which can be challenging due to their small size and relative sparsity. Li et al. developed a point-of-care Raman spectroscope capable of isolating and analyzing CPP crystals from SF using filtration and enzymatic digestion [83]. While effective, this method is also time-consuming and destructive to the sample. Raman hyperspectral imaging, however, can analyze crystals directly in SF, but requires manual crystal selection. Adding polarization filters to a hyperspectral Raman microscope has proven helpful and has been successfully implemented [84] (Fig. 3). In a consecutive series of 400 SF samples, this method demonstrated an 86.0% sensitivity and 99.1% specificity for CPP crystal identification according to the 2023 ACR/EULAR CPPD disease classification criteria [85]. Additionally, hyperspectral Raman imaging can be applied to identifying CPPD in tissue slides [86]. With a spatial resolution of around 1 μm, Raman imaging can provide detailed information on the nature and morphology of CPP foci in ex vivo tissue.

A hybrid combination of hyperspectral Raman imaging and polarized light microscopy can identify CPP with high specificity. A) Raman spectroscopic imaging can be performed with unprocessed synovial fluid, using standard microscope slides. B) Ordinary polarized light microscopy is used to locate birefringent crystals such as CPP or MSU. Once located, an area of interest is determined by the operator. C) Using the integrated Raman spectroscope, selected crystals are automatically scanned. D) The result: a collection of Raman spectra of the triclinic CPP crystal shown in panel B)
Future applications of Raman spectroscopy in rheumatology include the study of calcium crystals in OA [87] and the investigation of the role of body-foreign material, such as nanoparticles and microplastics, in inflammatory joint disease [88, 89]. Raman spectroscopic examination of SF from OA patients demonstrated the presence of BCP and CPP crystals, as well as calcium oxalate crystals and a novel crystal type: calcium carbonate [87]. All of these crystals exhibit pro-inflammatory properties, suggesting they might play a role in OA progression. Body foreign materials can also be pro-inflammatory and might cause joint inflammation. Raman spectroscopic identification of the causative particles might provide insights into new disease pathways and has already been demonstrated in clinical practice.
A series of validation studies have shown clear clinical benefits of using Raman spectroscopy for diagnosing CPPD. Depending on the type of Raman spectroscope, analysis takes between 5 to 15 min. While Raman spectroscopes are more expensive than CPLM, typically costing between €100,000 and €400,000, their rapid processing times allow to analyze thousands of SF samples annually. The design of Raman devices dedicated for clinical use will reduce the amount of training required for operating the machine. Overall, the implementation of Raman spectroscopy in CPPD assessment could enhance the specificity, objectivity, and patient stratification.
New Approaches in Basic Science
New laboratory approaches in the study of CPPD, and particularly those related to omics sciences, have seen limited application thus far. The omics path begins with the analysis of the gene sequences and genomics, and proceeds towards the investigation of the transcription profiles, transcriptomics, and the protein expression profile, proteomics, up to the functional results of proteins activity, metabolomics.
In the last twenty years, genetic studies, first, and genomics approaches, subsequently, have identified loci associated with abnormal cartilage calcifications. Familial cases of CPPD appear inherited in an autosomal dominant manner. Monogenic forms have been associated with mutations in genes determining the increase of ePPi as loss-of-function mutations in ANKH gene [32, 90], encoding for Inorganic Pyrophosphate Transport Regulator, PC-1, encoding for a nucleotide pyrophosphate synthetase [32], TNAP, encoding for tissue-non-specific alkaline phosphatase found in matrix vesicles and also associated with hypophosphatasia [91, 92]. However, later studies have not consistently confirmed some of these associations, as in the case of TNAP and ENPP1, encoding for ectonucleotide pyrophosphatase [93]. Associations have also been noted with genetically determined iron and copper metabolism disorders (haemochromatosis, Wilson’s disease), as well as phosphate disorders (X-linked dominant hypophosphataemic rickets due to mutations in the PHEX gene) [91, 92]. A 2017 whole-genome-wide linkage study failed to identify specific gene variants associated with CPPD, including those in ANKH, in ten individuals from five pedigrees [94]. Recently, a whole exosome sequencing of patients with diffuse idiopathic skeletal hyperostosis and CPPD suggests an association with variants of a new gene, PPP2R2D. This gene encodes a serine/threonine protein phosphatase that regulates basal cellular activities by dephosphorylating substrates. [95]. Ectopic calcifications have been observed in mice carrying a gain-of-function mutation in the TNFRSF11B gene, which encodes for osteoprotegerin, an osteoblast-derived inhibitor of the pro-osteoclastogenic RANK-RANKL axis [54, 96]. Further, read through mutation in TNFRSF11B results in the translation of an aberrant protein with additional 19 amino acids at its C-terminus (OPG-XL), leading to excessive fibrosis and mineralization in cartilage [97].
To our knowledge, no transcriptomic studies have been conducted on CPPD. However, in the few published articles analysing the expression profiles of chondrocytes isolated from calcium-containing crystal cartilages, a common finding was the upregulation of the expression of genes involved in mineralization, such as ANKH, PC-1 and TNAP [98]. Interestingly, in human primary articular chondrocytes, in vitro, the histone deacetylase inhibitors (HDACi), trichostatin A and vorinostat, downregulate mRNA and protein expression of ANKH and ENPP1 and upregulate TNAP, limiting levels of ePPi [99].
In addition, there is a notable absence of proteomics studies in this area. In 2020, de Seny et al. attempted to delineate common inflammatory mechanisms among synovial membranes affected by OA, CPPD and rheumatoid arthritis (RA). Among the 4336 proteins identified by mass spectrometry, 51 were selected for their strong correlation with histological scores. Of these, 11 proteins (DNAJB11, CALR, ERP29, GANAB, HSP90B1, HSPA1A, HSPA5, HYOU1, LMAN1, PDIA4, and TXNDC5) are involved in the endoplasmic reticulum stress. S100A8 and S100A9 were significantly elevated in RA compared to OA (both) or CPPD (S100A8 only) and significantly correlated with histological scores [100]. Further, 1871 proteins were associated with histological inflammatory scores and, of these, 10 proteins (LAP3, MANF, LCP1, CTSZ, PTPRC, DNAJB11, EML4, SCARA5, EIF3K, C1orf123) resulted differentially expressed in the synovial membrane of at least one of the three disease groups [101].
To the best of our knowledge, no metabolomics studies are available in the field of CPPD. The synovial joint is a closed system with limited exchange with the external environment, such as the bloodstream. Therefore, besides genomics, omics studies should highlight the similarities in the two environments associated with the disease. Another approach could rely on the spatial analysis of variations in cellular and molecular structures in the target tissue, surely providing a more detailed understanding of the pathophysiology. Spatially resolved biology enables the investigation of cells within the context of their tissue microenvironment. These techniques allow for genomics, transcriptomics, proteomics, and metabolomics analysis of intact tissue sections, associated with precise spatial coordinates [102].
Conclusions
The journey toward a comprehensive understanding of CPPD formation and deposition is ongoing. Progress has been made through basic science research, imaging studies, and investigations into the genetic forms of CPPD, yet significant gaps in knowledge remain. Emerging techniques in both imaging and basic sciences offer promise for deepening our understanding of this complex disease.
This is an exciting time for CPPD research, supported by international initiatives that lay the groundwork for standardized approaches to the disease. Notable advancements include the ACR/EULAR’s first set of classification criteria for CPPD disease [49], the OMERACT US definitions and scoring system [65, 66, 74, 76], the ACR/EULAR recommendations for the use of imaging in crystal-induced arthritis [103], the G-CAN project refining CPPD nomenclature, and the OMERACT project establishing a core outcome domain set for CPPD [104]. With current technological advancements and increasing scientific knowledge, the potential for breakthroughs has never been greater, and this is the perfect time to embark on new research initiatives.
Data Availability
No datasets were generated or analysed during the current study.
References
Abhishek A, Neogi T, Choi H, Doherty M, Rosenthal AK, Terkeltaub R. Review: Unmet Needs and the Path Forward in Joint Disease Associated With Calcium Pyrophosphate Crystal Deposition. Arthritis Rheumatol. 2018;70:1182–91.
Latourte A, Ea H-K, Richette P. Calcium Pyrophosphate and Basic Calcium Phosphate Crystal Arthritis: 2023 in Review. Gout, Urate, and Crystal Deposition Disease. 2024;2:101–7.
McCarthy GM. Calcium Pyrophosphate and Basic Calcium Phosphate Deposition Diseases: The Year in Review 2022. Gout, Urate, and Crystal Deposition Disease. 2023;1:234–42.
Pascart T, Filippou G, Lioté F, Sirotti S, Jauffret C, Abhishek A. Calcium Pyrophosphate Deposition Disease. The Lancet Rheumatology. 2024; in press
Bernabei I, So A, Busso N, Nasi S. Cartilage calcification in osteoarthritis: mechanisms and clinical relevance. Nat Rev Rheumatol. 2023;19:10–27.
Rosenthal AK, Ryan LM, Campion EW. Calcium pyrophosphate deposition disease. N Engl J Med. 2016;374:2575–84.
Williams CJ, Rosenthal AK. Pathogenesis of calcium pyrophosphate deposition disease. Best Pract Res Clin Rheumatol. 2021;35:101718.
Meyer F, Dittmann A, Kornak U, Herbster M, Pap T, Lohmann CH, et al. Chondrocytes From Osteoarthritic and Chondrocalcinosis Cartilage Represent Different Phenotypes. Front Cell Dev Biol. 2021;9:622287.
McGonagle D, Lories RJU, Tan AL, Benjamin M. The concept of a “synovio-entheseal complex” and its implications for understanding joint inflammation and damage in psoriatic arthritis and beyond. Arthritis Rheum. 2007;56:2482–91.
Riley GP, Harrall RL, Constant CR, Cawston TE, Hazleman BL. Prevalence and possible pathological significance of calcium phosphate salt accumulation in tendon matrix degeneration. Ann Rheum Dis. 1996;55:109–15.
Lui PP, Fu S, Chan L, Hung L, Chan K. Chondrocyte phenotype and ectopic ossification in collagenase-induced tendon degeneration. J Histochem Cytochem. 2009;57:91–100.
Reuben PM, Wenger L, Cruz M, Cheung HS. Induction of matrix metalloproteinase-8 in human fibroblasts by basic calcium phosphate and calcium pyrophosphate dihydrate crystals: effect of phosphocitrate. Connect Tissue Res. 2001;42:1–12.
Guerne PA, Terkeltaub R, Zuraw B, Lotz M. Inflammatory microcrystals stimulate interleukin-6 production and secretion by human monocytes and synoviocytes. Arthritis Rheum. 1989;32:1443–52.
Wu C-L, Harasymowicz NS, Klimak MA, Collins KH, Guilak F. The role of macrophages in osteoarthritis and cartilage repair. Osteoarthritis Cartilage. 2020;28:544–54.
Campillo-Gimenez L, Renaudin F, Jalabert M, Gras P, Gosset M, Rey C, et al. Inflammatory Potential of Four Different Phases of Calcium Pyrophosphate Relies on NF-κB Activation and MAPK Pathways. Front Immunol. 2018;9:2248.
McCarthy GM, Dunne A. Calcium crystal deposition diseases - beyond gout. Nat Rev Rheumatol. 2018;14:592–602.
Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–41.
Mitton-Fitzgerald E, Gohr CM, Williams CM, Rosenthal AK. Identification of Common Pathogenic Pathways Involved in Hemochromatosis Arthritis and Calcium Pyrophosphate Deposition Disease: a Review. Curr Rheumatol Rep. 2022;24:40–5.
Chang C-C, Tsai Y-H, Liu Y, Lin S-Y, Liang Y-C. Calcium-containing crystals enhance receptor activator of nuclear factor κB ligand/macrophage colony-stimulating factor-mediated osteoclastogenesis via extracellular-signal-regulated kinase and p38 pathways. Rheumatology (Oxford). 2015;54:1913–22.
Ryan LM, Kurup IV, Derfus BA, Kushnaryov VM. ATP-induced chondrocalcinosis. Arthritis Rheum. 1992;35:1520–5.
Derfus BA, Rachow JW, Mandel NS, Boskey AL, Buday M, Kushnaryov VM, et al. Articular cartilage vesicles generate calcium pyrophosphate dihydrate-like crystals in vitro. Arthritis Rheum. 1992;35:231–40.
Ishikawa K, Masuda I, Ohira T, Yokoyama M. A histological study of calcium pyrophosphate dihydrate crystal-deposition disease. J Bone Joint Surg Am. 1989;71:875–86.
Harmey D, Hessle L, Narisawa S, Johnson KA, Terkeltaub R, Millán JL. Concerted regulation of inorganic pyrophosphate and osteopontin by akp2, enpp1, and ank: an integrated model of the pathogenesis of mineralization disorders. Am J Pathol. 2004;164:1199–209.
Kirsch T, Nah HD, Shapiro IM, Pacifici M. Regulated production of mineralization-competent matrix vesicles in hypertrophic chondrocytes. J Cell Biol. 1997;137:1149–60.
Derfus B, Kranendonk S, Camacho N, Mandel N, Kushnaryov V, Lynch K, et al. Human osteoarthritic cartilage matrix vesicles generate both calcium pyrophosphate dihydrate and apatite in vitro. Calcif Tissue Int. 1998;63:258–62.
Johnson K, Hashimoto S, Lotz M, Pritzker K, Goding J, Terkeltaub R. Up-regulated expression of the phosphodiesterase nucleotide pyrophosphatase family member PC-1 is a marker and pathogenic factor for knee meniscal cartilage matrix calcification. Arthritis Rheum. 2001;44:1071–81.
Bertrand J, Nitschke Y, Fuerst M, Hermann S, Schäfers M, Sherwood J, et al. Decreased levels of nucleotide pyrophosphatase phosphodiesterase 1 are associated with cartilage calcification in osteoarthritis and trigger osteoarthritic changes in mice. Ann Rheum Dis. 2012;71:1249–53.
Garimella R, Bi X, Anderson HC, Camacho NP. Nature of phosphate substrate as a major determinant of mineral type formed in matrix vesicle-mediated in vitro mineralization: An FTIR imaging study. Bone. 2006;38:811–7.
Johnson KA, Hessle L, Vaingankar S, Wennberg C, Mauro S, Narisawa S, et al. Osteoblast tissue-nonspecific alkaline phosphatase antagonizes and regulates PC-1. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1365-1377.
Uzuki M, Sawai T, Ryan LM, Rosenthal AK, Masuda I. Upregulation of ANK protein expression in joint tissue in calcium pyrophosphate dihydrate crystal deposition disease. J Rheumatol. 2014;41:65–74.
Williams CJ. The role of ANKH in pathologic mineralization of cartilage. Curr Opin Rheumatol. 2016;28:145–51.
Abhishek A, Doherty M. Pathophysiology of articular chondrocalcinosis–role of ANKH. Nat Rev Rheumatol. 2011;7:96–104.
Rosenthal AK, Gohr CM, Mitton-Fitzgerald E, Lutz MK, Dubyak GR, Ryan LM. The progressive ankylosis gene product ANK regulates extracellular ATP levels in primary articular chondrocytes. Arthritis Res Ther. 2013;15:R154.
Szeri F, Niaziorimi F, Donnelly S, Fariha N, Tertyshnaia M, Patel D, et al. The Mineralization Regulator ANKH Mediates Cellular Efflux of ATP. Not Pyrophosphate J Bone Miner Res. 2022;37:1024–31.
Thouverey C, Bechkoff G, Pikula S, Buchet R. Inorganic pyrophosphate as a regulator of hydroxyapatite or calcium pyrophosphate dihydrate mineral deposition by matrix vesicles. Osteoarthritis Cartilage. 2009;17:64–72.
Stücker S, Bollmann M, Garbers C, Bertrand J. The role of calcium crystals and their effect on osteoarthritis pathogenesis. Best Pract Res Clin Rheumatol. 2021;35:101722.
Rosenthal AK, Gohr CM, Uzuki M, Masuda I. Osteopontin promotes pathologic mineralization in articular cartilage. Matrix Biol. 2007;26:96–105.
Heinkel D, Gohr CM, Uzuki M, Rosenthal AK. Transglutaminase contributes to CPPD crystal formation in osteoarthritis. Front Biosci. 2004;9:3257–61.
Jubeck B, Gohr C, Fahey M, Muth E, Matthews M, Mattson E, et al. Promotion of articular cartilage matrix vesicle mineralization by type I collagen. Arthritis Rheum. 2008;58:2809–17.
Fuerst M, Bertrand J, Lammers L, Dreier R, Echtermeyer F, Nitschke Y, et al. Calcification of articular cartilage in human osteoarthritis. Arthritis Rheum. 2009;60:2694–703.
Doherty M, Belcher C, Regan M, Jones A, Ledingham J. Association between synovial fluid levels of inorganic pyrophosphate and short term radiographic outcome of knee osteoarthritis. ANN RHEUM DIS. 1996;55:432–6.
Altman RD, Muniz OE, Pita JC, Howell DS. Articular chondrocalcinosis. Microanalysis of pyrophosphate (PPi) in synovial fluid and plasma. Arthritis Rheum. 1973;16:171–8.
Rachow JW, Ryan LM. Adenosine triphosphate pyrophosphohydrolase and neutral inorganic pyrophosphatase in pathologic joint fluids. Elevated pyrophosphohydrolase in calcium pyrophosphate dihydrate crystal deposition disease. Arthritis Rheum. 1985;28:1283–8.
Ryan LM, Rachow JW, McCarty DJ. Synovial fluid ATP: a potential substrate for the production of inorganic pyrophosphate. J Rheumatol. 1991;18:716–20.
Ryan LM, McCarty DJ. Understanding inorganic pyrophosphate metabolism: toward prevention of calcium pyrophosphate dihydrate crystal deposition. Ann Rheum Dis. 1995;54:939–41.
Filippou G, Scanu A, Adinolfi A, Picerno V, Toscano C, Bortoluzzi A, et al. The two faces of the same medal… or maybe not? Comparing osteoarthritis and calcium pyrophosphate deposition disease: a laboratory and ultrasonographic study. Clin Exp Rheumatol. 2021;39:66–72.
Richette P, Bardin T, Doherty M. An update on the epidemiology of calcium pyrophosphate dihydrate crystal deposition disease. Rheumatology (Oxford). 2009;48:711–5.
Abhishek A. Calcium pyrophosphate deposition disease: a review of epidemiologic findings. Curr Opin Rheumatol. 2016;28:133–9.
Abhishek A, Tedeschi SK, Pascart T, Latourte A, Dalbeth N, Neogi T, et al. The 2023 ACR/EULAR classification criteria for calcium pyrophosphate deposition disease. Ann Rheum Dis. 2023;82:1248–57.
Tedeschi SK. A New Era for Calcium Pyrophosphate Deposition Disease Research: The First-Ever Calcium Pyrophosphate Deposition Disease Classification Criteria and Considerations for Measuring Outcomes in Calcium Pyrophosphate Deposition Disease. Gout, Urate, and Crystal Deposition Disease. 2024;2:52–9.
Zhang W, Doherty M, Bardin T, Barskova V, Guerne P-A, Jansen TL, et al. European League Against Rheumatism recommendations for calcium pyrophosphate deposition Part. I: terminology and diagnosis. Ann Rheum Dis. 2011;70:563–70.
Ho AM, Johnson MD, Kingsley DM. Role of the mouse ank gene in control of tissue calcification and arthritis. Science. 2000;289:265–70.
Mitton-Fitzgerald E, Gohr CM, Williams CJ, Ortiz A, Mbalaviele G, Rosenthal AK. Effects of the TNFRSF11B Mutation Associated With Calcium Pyrophosphate Deposition Disease in Osteoclastogenesis in a Murine Model. Arthritis Rheumatol. 2021;73:1543–9.
Williams CJ, Qazi U, Bernstein M, Charniak A, Gohr C, Mitton-Fitzgerald E, et al. Mutations in osteoprotegerin account for the CCAL1 locus in calcium pyrophosphate deposition disease. Osteoarthritis Cartilage. 2018;26:797–806.
Abhishek A, Doherty S, Maciewicz R, Muir K, Zhang W, Doherty M, et al. The association between ANKH promoter polymorphism and chondrocalcinosis is independent of age and osteoarthritis: results of a case-control study. Arthritis Res Ther. 2014;16:R25.
Rosenthal AK, Ryan LM. Probenecid inhibits transforming growth factor-beta 1 induced pyrophosphate elaboration by chondrocytes. J Rheumatol. 1994;21:896–900.
Chotard E, Blanchard A, Ostertag A, Latourte A, Gailly G, Frochot V, et al. Calcium pyrophosphate crystal deposition in a cohort of 57 patients with Gitelman syndrome. Rheumatology (Oxford). 2022;61:2494–503.
Doherty M, Dieppe PA. Double blind, placebo controlled trial of magnesium carbonate in chronic pyrophosphate arthropathy. Ann Rheum Dis. 1983;42:106–7.
Tedeschi SK, Hayashi K, Rosenthal A, Gill M, Marrugo J, Fukui S, et al. Fractures in Patients With Acute Calcium Pyrophosphate Crystal Arthritis Versus Matched Comparators in a Large Cohort Study. Arthritis & Rheumatology. 2024;76(6):936–41. https://doi.org/10.1002/art.42798.
Tedeschi SK, Huang W, Yoshida K, Solomon DH. Risk of cardiovascular events in patients having had acute calcium pyrophosphate crystal arthritis. Ann Rheum Dis. 2022;annrheumdis-2022–222387
Bashir M, Sherman KA, Solomon DH, Rosenthal A, Tedeschi SK. Cardiovascular Disease Risk in Calcium Pyrophosphate Deposition Disease: A Nationwide Study of Veterans. Arthritis Care Res (Hoboken). 2023;75:277–82.
Cipolletta E, Tata LJ, Nakafero G, Avery AJ, Mamas MA, Abhishek A. Association Between Gout Flare and Subsequent Cardiovascular Events Among Patients With Gout. JAMA. 2022;328:440–50.
Zitnan D. Sitaj S [Chondrocalcinosis polyarticularis (familiaris): roentgenological and clinical analysis]. Cesk Rentgenol. 1960;14:27–34.
Filippou G, Sirotti S, Cipolletta E, Filippucci E. Optimizing the Use of Ultrasound in Calcium Pyrophosphate Deposition (CPPD): A Review from the Ground Up. GUCDD. 2024;2:17–33.
Filippou G, Scirè CA, Damjanov N, Adinolfi A, Carrara G, Picerno V, et al. Definition and Reliability Assessment of Elementary Ultrasonographic Findings in Calcium Pyrophosphate Deposition Disease: A Study by the OMERACT Calcium Pyrophosphate Deposition Disease Ultrasound Subtask Force. J Rheumatol. 2017;44:1744–9.
Filippou G, Scirè CA, Adinolfi A, Damjanov NS, Carrara G, Bruyn GAW, et al. Identification of calcium pyrophosphate deposition disease (CPPD) by ultrasound: reliability of the OMERACT definitions in an extended set of joints-an international multiobserver study by the OMERACT Calcium Pyrophosphate Deposition Disease Ultrasound Subtask Force. Ann Rheum Dis. 2018;77:1194–9.
Sirotti S, Becce F, Sconfienza LM, Terslev L, Naredo E, Zufferey P, et al. Reliability and Diagnostic Accuracy of Radiography for the Diagnosis of Calcium Pyrophosphate Deposition: Performance of the Novel Definitions Developed by an International Multidisciplinary Working Group. Arthritis & Rheumatology. 2023;75:630–8.
Tedeschi SK, Becce F, Pascart T, Guermazi A, Budzik J-F, Dalbeth N, et al. Imaging features of calcium pyrophosphate deposition (CPPD) disease: consensus definitions from an international multidisciplinary working group. Arthritis Care Res (Hoboken). 2023;75(4):825–34. https://doi.org/10.1002/acr.24898.
Neame RL, Carr AJ, Muir K, Doherty M. UK community prevalence of knee chondrocalcinosis: evidence that correlation with osteoarthritis is through a shared association with osteophyte. Ann Rheum Dis. 2003;62:513–8.
Adinolfi A, Sirotti S, Sakellariou G, Cipolletta E, Filippucci E, Porta F, et al. Which are the most frequently involved peripheral joints in calcium pyrophosphate crystal deposition at imaging? A systematic literature review and meta-analysis by the OMERACT ultrasound – CPPD subgroup. Frontiers in Medicine [Internet]. 2023 [cited 2023 Mar 11];10. Available from: https://www.frontiersin.org/articles/https://doi.org/10.3389/fmed.2023.1131362
Filippou G, Miguel-Pérez M, Coronel L, Sirotti S, Pacini G, Scanu A, et al. The ultrasonographic pseudo–double contour sign in calcium pyrophosphate deposition disease: an anatomic explanation and how to distinguish it from gout. Arthritis & Rheumatology. 2023;75:639–40.
Cipolletta E, Abhishek A, Di Matteo A, Grassi W, Filippucci E. Dynamic assessment of the double contour sign by ultrasonography helps to distinguish between gout and calcium pyrophosphate deposition disease. RMD Open. 2023;9:e002940.
Falkowski AL, Jacobson JA, Kalia V, Meyer NB, Gandikota G, Yosef M, et al. Cartilage icing and chondrocalcinosis on knee radiographs in the differentiation between gout and calcium pyrophosphate deposition. PLoS One. 2020;15(4):e0231508. https://doi.org/10.1371/journal.pone.0231508.
Filippou G, Scanu A, Adinolfi A, Toscano C, Gambera D, Largo R, et al. Criterion validity of ultrasound in the identification of calcium pyrophosphate crystal deposits at the knee: an OMERACT ultrasound study. Ann Rheum Dis. 2021;80:261–7.
Sirotti S, Madruga-Dias J, Adinolfi A, Sakellariou G, Rozza D, Carrara G, et al. Preliminary descriptive analysis of the RADIAL cohort study about the prevalence and the clinical characteristics of patients with CPPD in daily clinical practice [abstract]. Arthritis Rheumatol. 2023;75(suppl 9).
Sirotti S, Terslev L, Filippucci E, Iagnocco A, Moller I, Naredo E, et al. Development and validation of an OMERACT ultrasound scoring system for the extent of calcium pyrophosphate crystal deposition at the joint level and patient level. The Lancet Rheumatol. 2023;5:e474–82.
Chen K-H, Li M-J, Cheng W-T, Balic-Zunic T, Lin S-Y. Identification of monoclinic calcium pyrophosphate dihydrate and hydroxyapatite in human sclera using Raman microspectroscopy. Int J Exp Pathol. 2009;90:74–8.
Gras P, Rey C, Marsan O, Sarda S, Combes C. Synthesis and Characterisation of Hydrated Calcium Pyrophosphate Phases of Biological Interest. Eur J Inorg Chem. 2013;2013:5886–95.
Berendsen D, Neogi T, Taylor WJ, Dalbeth N, Jansen TL. Crystal identification of synovial fluid aspiration by polarized light microscopy. An online test suggesting that our traditional rheumatologic competence needs renewed attention and training. Clin Rheumatol. 2017;36:641–7.
Bernal J-A, Andrés M, López-Salguero S, Jovaní V, Vela-Casasempere P, Pascual E. Agreement Among Multiple Observers on Crystal Identification by Synovial Fluid Microscopy. Arthritis Care Res (Hoboken). 2022;
Takanabe A, Tanaka M, Taniguchi A, Yamanaka H, Asahi T. Quantitative analysis with advanced compensated polarized light microscopy on wavelength dependence of linear birefringence of single crystals causing arthritis. J Phys D Appl Phys. 2014;47:285402.
Ralbovsky NM, Lednev IK. Towards development of a novel universal medical diagnostic method: Raman spectroscopy and machine learning. Chem Soc Rev. 2020;49:7428–53.
Li B, Singer NG, Yeni YN, Haggins DG, Barnboym E, Oravec D, et al. A Point-of-Care Raman Spectroscopy-Based Device for the Diagnosis of Gout and Pseudogout: Comparison With the Clinical Standard Microscopy. Arthritis Rheumatol. 2016;68:1751–7.
Niessink T, Kuipers C, de Jong BZ, Lenferink ATM, Janssen M, Jansen TL, et al. Raman hyperspectral imaging detects novel and combinations of crystals in synovial fluids of patients with a swollen joint. J Raman Spectrosc. 2023;54:47–53.
Niessink T, Janssen M, Giesen T, Efdé MN, Comarniceanu AC, Otto C, et al. Diagnostic Accuracy of Raman Spectroscopy Integrated With Polarized Light Microscopy for Calcium Pyrophosphate-Associated Arthritis. Arthritis Care Res (Hoboken). 2024;
Buchholz A, Stücker S, Koßlowski F, Lohmann CH, Bertrand J. High-Resolution Imaging Methods for Identification of Calcium Crystal Types in Osteoarthritis. Gout, Urate, and Crystal Deposition Disease. 2023;1:62–82.
Niessink T, Stassen RHMJ, Kischkel B, Vuscan P, Emans PJ, van den Akker GGH, et al. Discovery of calcite as a new pro-inflammatory calcium-containing crystal in human osteoarthritic synovial fluid. Osteoarthr Cartil. 2024. https://doi.org/10.1016/j.joca.2024.05.004.
van der Weele L, Niessink T, Maas M, Don Griot P, Janssen M, Otto C, et al. Clinical image: synovitis of the metacarpophalangeal joints. Ann Rheum Dis. 2023;82:992–3.
Niessink T, Janssen M, Jansen TL, Otto C. The Prevalence of Titanium Dioxide Particles in Synovial Fluid Samples Drops after European Union Ban. Gout, Urate, and Crystal Deposition Disease. 2024;2:45–51.
Gurley KA, Reimer RJ, Kingsley DM. Biochemical and genetic analysis of ANK in arthritis and bone disease. Am J Hum Genet. 2006;79:1017–29.
Zhang Y, Brown MA. Genetic studies of chondrocalcinosis. Curr Opin Rheumatol. 2005;17:330–5.
Zaka R, Williams CJ. Genetics of chondrocalcinosis. Osteoarthritis Cartilage. 2005;13:745–50.
Zhang Y, Brown MA, Peach C, Russell G, Wordsworth BP. Investigation of the role of ENPP1 and TNAP genes in chondrocalcinosis. Rheumatology (Oxford). 2007;46:586–9.
Couto AR, Parreira B, Thomson R, Soares M, Power DM, Stankovich J, et al. Combined approach for finding susceptibility genes in DISH/chondrocalcinosis families: whole-genome-wide linkage and IBS/IBD studies. Hum Genome Var. 2017;4:17041.
Parreira B, Couto AR, Rocha F, Sousa M, Faustino V, Power DM, et al. Whole exome sequencing of patients with diffuse idiopathic skeletal hyperostosis and calcium pyrophosphate crystal chondrocalcinosis. Acta Reumatol Port. 2020;45:116–26.
Ramos YFM, Bos SD, van der Breggen R, Kloppenburg M, Ye K, Lameijer E-WEMW, et al. A gain of function mutation in TNFRSF11B encoding osteoprotegerin causes osteoarthritis with chondrocalcinosis. Ann Rheum Dis. 2015;74:1756–62.
Rodríguez Ruiz A, van Hoolwerff M, Sprangers S, Suchiman E, Schoenmaker T, Dibbets-Schneider P, et al. Mutation in the CCAL1 locus accounts for bidirectional process of human subchondral bone turnover and cartilage mineralization. Rheumatology (Oxford). 2022;keac232
Nguyen C, Bazin D, Daudon M, Chatron-Colliet A, Hannouche D, Bianchi A, et al. Revisiting spatial distribution and biochemical composition of calcium-containing crystals in human osteoarthritic articular cartilage. Arthritis Res Ther. 2013;15:R103.
Chang C-C, Lee K-L, Chan T-S, Chung C-C, Liang Y-C. Histone Deacetylase Inhibitors Downregulate Calcium Pyrophosphate Crystal Formation in Human Articular Chondrocytes. Int J Mol Sci. 2022;23 2604
de Seny D, Bianchi E, Baiwir D, Cobraiville G, Collin C, Deliège M, et al. Proteins involved in the endoplasmic reticulum stress are modulated in synovitis of osteoarthritis, chronic pyrophosphate arthropathy and rheumatoid arthritis, and correlate with the histological inflammatory score. Sci Rep. 2020;10:14159.
de Seny D, Baiwir D, Bianchi E, Cobraiville G, Deroyer C, Poulet C, et al. New Proteins Contributing to Immune Cell Infiltration and Pannus Formation of Synovial Membrane from Arthritis Diseases. Int J Mol Sci. 2021;23 434
Fan X, Sun AR, Young RSE, Afara IO, Hamilton BR, Ong LJY, et al. Spatial analysis of the osteoarthritis microenvironment: techniques, insights, and applications. Bone Res. 2024;12:7.
Mandl P, D’Agostino MA, Navarro-Compán V, Geßl I, Sakellariou G, Abhishek A, et al. 2023 EULAR recommendations on imaging in diagnosis and management of crystal-induced arthropathies in clinical practice. Ann Rheum Dis. 2024;83:752–9.
Cai K, Fuller A, Zhang Y, Hensey O, Grossberg D, Christensen R, et al. Towards development of core domain sets for short term and long term studies of calcium pyrophosphate crystal deposition (CPPD) disease: A framework paper by the OMERACT CPPD working group. Semin Arthritis Rheum. 2021;51:946–50.
Cipolletta E, Moscioni E, Sirotti S, Di Battista J, Abhishek A, Rozza D, et al. Diagnosis of calcium pyrophosphate crystal deposition disease by ultrasonography: how many and which sites should be scanned? Rheumatology. 2023;kead565.
Funding
Open access funding provided by Università degli Studi di Milano within the CRUI-CARE Agreement. No funding was received for the submitted work.
Author information
Authors and Affiliations
Contributions
G.F. designed the work. All authors drafted the work and revised it critically for important intellectual content; approved the version to be published; and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Conflict of Interest
Georgios Filippou has received a research grant from Lilly. Other authors have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sirotti, S., Scanu, A., Pascart, T. et al. Calcium Pyrophosphate Crystal Formation and Deposition: Where Do we Stand and What Does the Future hold?. Curr Rheumatol Rep 26, 354–365 (2024). https://doi.org/10.1007/s11926-024-01161-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11926-024-01161-w