
Abstract
Purpose of Review
We focus on recent advances in diagnosis and therapeutic strategies, as well as on pathogenesis of Schnitzler syndrome.
Recent Findings
New diagnostic criteria were established, and their external validity was assessed in a retrospective cohort study. The cytokine interleukin-1 (IL-1) plays a crucial role in the pathogenesis of the Schnitzler syndrome, and this explains the spectacular efficiency of IL-1 blocking therapies.
Summary
The Schnitzler syndrome is now considered as a late-onset acquired autoinflammatory syndrome in which the cytokine IL-1 plays a crucial role. IL-1 blocking therapies are efficient on the inflammation-linked symptoms but not on the monoclonal component. Therefore, they probably don’t reduce the risk of the development of lymphoproliferative disorders that remains the main prognostic issue. The link between autoinflammation and the monoclonal component needs to be further elucidated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The Schnitzler syndrome is a rare acquired autoinflammatory syndrome, first described in 1972 by Liliane Schnitzler [1]. Its main clinical features are urticarial rash, bone and/or joint pain, enlarged lymph nodes, and fever. It is associated with a monoclonal gammopathy, typically of IgM type, sometimes of IgG type (variant Schnitzler syndrome).
The Schnitzler syndrome’s pathophysiology has not yet been elucidated. Particularly, it is not yet known if there is a link between the monoclonal gammopathy and the clinical signs.
Nevertheless, Schnitzler syndrome is a typical example of a monoclonal gammopathy of cutaneous/clinical significance as recently outlined [2••].
The clinical similarities of the Schnitzler syndrome with other genetically determined autoinflammatory syndromes such as cryopyrin-associated periodic syndromes (CAPS) could be a useful clue to understanding Schnitzler syndrome’s pathogenesis.
The main complications of the Schnitzler syndrome are the development of a lymphoproliferative disorder in about 15 to 20% of cases and rarely AA amyloidosis in untreated patients.
Treatment of the Schnitzler syndrome relies on IL-1 blocking agent that led to a significant improvement in patient care, allowing complete control of clinical symptoms. However, treatment is only suspensive and symptoms always recur when the treatment is stopped.
Over the past few years, advances have been made in the management and the comprehension of the Schnitzler syndrome. Herein, we have reviewed the published literature on Schnitzler syndrome since 2011.
Material and Methods
In April 2017, a PubMed search using the keywords “Schnitzler syndrome” and “Schnitzler’s syndrome” was performed to retrieve all articles. Relevant articles in English and French published since 2011 were analyzed. Only patients that fulfilled the Strasbourg criteria (Table 1) or the Lipsker criteria (Table 2) were included. Hence, if the paraprotein was lacking, the patients were excluded.
Physiopathology
The pathogenesis of the Schnitzler syndrome remains incompletely understood. Many features indicate that the Schnitzler syndrome is an acquired autoinflammatory disorder. It shares many clinical and biological characteristics with genetically determined autoinflammatory syndromes such as CAPS, which is caused by activating mutations in the NLRP3 gene (nucleotide-binding oligomerization domain-leucine-rich repeats containing pyrin domain 3). In both disorders, patients have recurrent fever, urticarial eruption with a neutrophilic infiltrate on biopsy, neutrophilia, and an increase in C-reactive protein (CRP). No germline NLRP3 mutation has been reported in the Schnitzler syndrome, but somatic mosaicism of NLRP3 mutations in the myeloid lineage was reported in two patients with variant (IgG) Schnitzler syndrome [3]. However, the authors of this review rather consider these two cases as authentic mosaics of CAPS.
Increased IL-1β and IL-6 secretion by lipopolysaccharide-stimulated peripheral blood mononuclear cells was reported [4, 5]. Moreover, IL-6 levels in serum seem to be correlated with disease activity. Dermal mast cells could be the source of IL-1β in patients with Schnitzler syndrome [6]. Neutrophils are then recruited and produce IL-17.
Furthermore, IL-1β suppresses IL-10 production by Th17 cells [7]. Recently, it has been demonstrated that in the Schnitzler syndrome, systemic overproduction of IL-1β results in a profound loss of anti-inflammatory Th17 cell functionalities [8]. IL-1β blocking therapy seems to restore IL-10 expression and regulatory properties of Th17. This could have an important implication in the development of new therapeutic strategies.
The role of the IgM paraprotein in the pathogenesis of the Schnitzler syndrome remains unclear. It is still not known whether the paraprotein is the cause or the consequence of the disease process, as it is still not known whether it precedes or follows the very first clinical signs. Delayed development of an IgM paraprotein approximately 4 years after the first clinical symptoms has been reported in one patient [9]. The observation of an association between Waldenström’s macroglobulinemia and mutations in MyD88 [10] raised the question of a potential link between IgM paraprotein and autoinflammation in the Schnitzler syndrome. Indeed, MyD88 is a toll-like receptor signal transduction molecule that acts as an adaptor in IL-1 signaling by interacting with IL-1 receptor complex and IL-1 receptor-associated kinase. Thus, increased IL-1 stimulation could contribute to clonal IgM production.
Epidemiology
The first cases of Schnitzler syndrome were described in Europe, mainly due to a better knowledge of the disease in this continent. By now, there have been about 300 cases reported in literature [11•] all over the world including Japan [12] and Australia [13]. The Schnitzler syndrome does not seem to predominate in certain ethnic subgroups. There is a slight predominance of the disease among males. To our knowledge, no pediatric case has ever been reported.
Diagnostic Criteria
The Schnitzler syndrome is characterized by a recurrent febrile urticarial eruption, joint and/or bone pain, enlarged lymph nodes, hepatomegaly, and/or splenomegaly. The presence of a monoclonal gammopathy, mainly of IgMκ type, is a defining criterion. It can be associated with elevated markers of inflammation such as CRP or leukocytosis. There is no gold standard to diagnose the Schnitzler syndrome, and a number of diseases must be ruled out before considering the diagnosis, especially adult-onset Still’s disease (AOSD), CAPS (including chronic infantile neurologic cutaneous and articular syndrome, Muckle-Wells syndrome), lymphoma, and Waldenström’s disease. In 2001, Lipsker et al. [14] established the first diagnostic criteria (Table 1). To do so, they carefully analyzed all published cases at this time and four new of their own cases. These criteria were revised in 2013 by an expert meeting in Strasbourg [15], and they are known as the “Strasbourg criteria” (Table 2). In 2016, a multicentric study was published [16••], validating for the first time the two sets of criteria. They were applied on patients with Schnitzler syndrome and relevant controls including patients with AOSD, CAPS, and Waldenström’s disease. It appeared that both were reliable and discriminating for the diagnosis of the Schnitzler syndrome, confirming their validity in real-life patients.
Clinical Signs
Onset Manifestations
Median age at clinical onset is 55 years. The first clinical signs are generally urticarial rash, mainly associated with recurrent fever or joint pain. Clinical findings at onset of Schnitzler syndrome can be less specific such as recurrent fever alone, bone and/or joint pain, fatigue, or loss of weight.
Rash
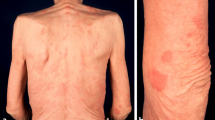
By definition, urticarial recurrent rash is present in all patients, mostly on the trunk and limbs. Most of the time, the eruption consists of rose or red macules or gently raised papules or plaques (Fig. 1). It can be slightly itchy. It lasts less than 24 h and the frequency of the eruption is variable. The eruption can be exacerbated by stress or physical exercise. Sometimes, a halo of vasoconstriction can be observed, as well as dermographism. Angioedema is not common.

Clinical aspect of the urticarial recurrent rash in a patient with a Schnitzler syndrome. Red macules or gently raised papules and plaques of the trunk and limbs
The histopathological findings are important, and skin biopsy should be performed on a recent lesion, as the presence of a neutrophilic dermal infiltrate on skin biopsy is now a minor criterion of the Strasbourg criteria. The most typical skin finding is a neutrophilic urticarial dermatosis as described in 2009 [17], namely a perivascular and interstitial infiltrate of neutrophils with leukocytoclasia but without vasculitis or dermal edema (Fig. 2). Neutrophilic epitheliotropism, especially around sweat glands, is suggestive [18]. A similar rash can be observed in AOSD and lupus erythematosus [17, 19]. Another histopathological finding is vasculitis, reported in up to 20% of cases [11•], but it may be overestimated as reevaluation of several of them revealed the absence of fibrinoid necrosis of the vessel walls [20]. Very subtle histopathological findings, such as a mild mononuclear cell perivascular infiltrate, have been reported [21].

Histopathological aspect of neutrophilic urticarial dermatosis in a patient with Schnitzler syndrome. Perivascular and interstitial infiltrate of neutrophils with leukocytoclasia but without vasculitis or dermal edema
Other Clinical Signs
Recurrent fever is present in a majority of patients [16••], usually simultaneously with rash and joint and/or bone pain. Joint and/or bone pain occurs in about 40% of patients, mostly on the lower limbs. Other common clinical signs are asthenia, loss of weight, myalgia, or headache. Enlarged lymph nodes and splenomegaly and/or hepatomegaly could be more unusual than previously thought [11•].
Biological Signs
By definition, all patients with Schnitzler syndrome have a monoclonal component. Monoclonal IgM gammopathy is predominant (in about 88% of cases [11•, 16••]), mainly associated with a kappa light chain. Monoclonal IgG gammopathy is present in less than 10% of cases but it could be underestimated, as it was not initially included in the definition. Patients with monoclonal IgA gammopathy have also been reported, but in addition to IgMκ gammopathy [22]. The level of the monoclonal component at diagnosis is highly variable; it can only be present at a very low level (trace) or be very high at once (up to 41 g/l) [16••]. A high level (> 10 g/l) should always raise suspicion of Waldenström’s disease.
The other biological hallmark is the elevation of inflammatory markers such as ESR, CRP, or increased neutrophils count. These are relevant findings, as CRP and neutrophils count are usually not elevated in autoimmune syndromes. Inflammatory anemia or thrombocytosis can also be observed [12]. Finally, markers of abnormal bone remodeling and an increase in VEGF are newly individualized biological findings (see next section).
Musculoskeletal Involvement
Skeletal examination is crucial in the Schnitzler syndrome, since bone pain is an important clinical finding and as it is part of both diagnostic criteria systems. It should be done even in the absence of bone and/or joint pain. Bone involvement can also lead to the diagnosis in some patients [23]. The iliac bone, the tibia, and the femur are the most commonly involved, but other localizations have been reported including axial skeleton and peripheral bones [24]. None of the imaging findings are specific, as they can be observed in other diseases, in particular infiltrative diseases (such as systemic mastocytosis or Erdheim-Chester disease), or dysplastic diseases (such as melorheostosis or Camurati-Engelmann disease) [25]. Standard radiographies typically show sclerotic lesions, but lytic lesions have also been described [26]. Bone scintigraphy is considered as the examination of choice to demonstrate bone damage [24], which results in focally increased radiotracer uptake. MRI can also be performed [24].
The possible discriminating value of the dosage of VEGF and markers of bone formation, namely osteocalcin and bone-specific alkaline phosphatase (bALP), has been suggested by Terpos et al. [27] and needs to be further evaluated. High uptake on technetium bone scintigraphy suggests increased osteoblast function. This is confirmed by the increase of bALP and osteocalcin, which are directly produced by osteoblasts. On the other hand, no increase in bone resorption markers (namely sRANKL and CTX (collagen type I cross-linked C-telopeptide)) was detected. It is not well elucidated how IL-1β overproduction leads to increased bone formation as IL-1 and IL-6 are known to stimulate osteoclasts functions. However, angiogenesis is known to enhance osteogenesis and VEGF could therefore participate in bone formation in Schnitzler syndrome, as it is elevated in untreated patients. In successfully treated patients, the VEGF level was significantly reduced.
Treatment
Currently, there is no approved treatment for the Schnitzler syndrome.
Before the emergence of interleukin-1 blocking therapies in 2005 [28], numerous treatments had been used unsuccessfully to treat the Schnitzler syndrome [29]. Colchicine, pefloxacine, interferon-alpha, or corticosteroids were only moderately effective and could lead to serious side effects.
IL-1 blocking agents are the most effective therapies. This is especially true for the IL-1 receptor antagonist anakinra, since clinical signs resolve within hours following the first injection. However, the effect is only suspensive and it does not cure the disease.
Anakinra, an IL-1 receptor antagonist, is the most commonly used agent to treat Schnitzler syndrome. During a meeting in 2012, the leading experts in the field recommended anakinra as the treatment of choice of Schnitzler syndrome [13].
The first reported case of its efficiency was published in 2005 [28], and its long-term effectiveness was subsequently confirmed by a multicentric retrospective cohort study [30]. It relieves all clinical symptoms within the hours that follow the first injection. If the patient forgets an injection, symptoms recur between 36 and 48 h. It is delivered subcutaneously at a dose of 100 mg/day. The major side effect is an erythematous reaction at the injection site. Neutropenia can also occur, but anakinra has been successfully reintroduced after normalization of the neutrophil count [31].
The efficiency of another IL-1 blocking therapy, namely canakinumab, a fully human IL-1β-specific antibody has also been reported [32,33,34]. More recently, a phase II randomized placebo-controlled multicenter study was conducted to assess the effects of canakinumab [35]. A unique dose of 150 mg of canakinumab was given subcutaneously. Then, the other injections were given according to individual clinical and laboratory responses. Fourteen of 20 patients needed further canakinumab injections within the first 16 weeks of the study. They received one to three further canakinumab injections at the dose of 150 or 300 mg. This study showed that canakinumab was significantly more effective than the placebo in reducing clinical signs and in lowering the level of inflammation markers in the blood. The main adverse events were infections (respiratory and urinary tract). Injection-site reactions, abdominal pain, vertigo, and neutropenia can also occur.
Rilonacept is a recombinant fusion protein comprising the extracellular ligand-binding domains of human IL-1 type I receptor and IL-1 receptor accessory protein, fused to the Fc portion of human IgG1. After a loading dose of 320 mg, injections of 160 mg are administrated once weekly subcutaneously. It has shown to result in a rapid clinical response, with reduction of markers of inflammation [36]. The main side effects are injection-site reactions, upper airway infections, headache, and neutropenia.
The level of complete remission in patients treated with IL-1 blocking therapies is about 83% [30]. The remaining patients have partial remission, mainly with the persistence of joint pain. However, some patients may not respond at all to anti-IL-1 [37]. In this particular case, diagnosis should be reconsidered. If the diagnosis of the Schnitzler syndrome remains certain, an anti-IL-6 treatment such as tocilizumab can be effective [37]. In our experience, 100% of patients treated with anakinra are in complete remission.
Evolution
The course of Schnitzler syndrome is long-standing. Only one case of spontaneous remission has been reported so far [38].
The overall prognosis of Schnitzler syndrome depends on the potential evolution into a lymphoproliferative disorder and the occurrence of AA amyloidosis. Waldenström’s disease is the most common complication and occurs in about 15% of cases after 10 to 20 years of evolution.
In Schnitzler syndrome, continuous inflammation is known to be associated with an increased risk of AA amyloidosis [39]. Treatment with IL-1 blocking therapies probably minimizes this risk by significantly reducing the level of inflammation. Therefore, in case of persistent inflammation, treatment is indicated even in pauci-symptomatic patients.
However, IL-1 blocking therapies do not seem to be effective on the monoclonal component [30] and probably do not prevent the development of lymphoproliferation such as Waldenström’s disease [40]. Moreover, recently, a patient with an 18-year story of Schnitzler syndrome developed rheumatoid arthritis while being successfully treated with anakinra [41] for 8 years. This case raises the question of the impact of long-term IL-1 blockade and its implication in the pathogenesis of rheumatoid arthritis.
Conclusion
The Schnitzler syndrome is now considered as the paradigm of late-onset acquired autoinflammatory syndrome. Cutaneous symptoms, in particular urticarial eruption and the presence of a neutrophilic infiltrate in skin biopsy, are part of the spectrum of autoinflammatory disorders and practitioners should be able to recognize it. Bone involvement is a characteristic of the Schnitzler syndrome and should be systematically investigated.
The link between the IgM paraprotein and autoinflammation needs to be investigated further as it is so far not understood. Inflammation-linked symptoms can be completely reversed by IL-1 blocking therapy. Nevertheless, the development of hematological disorder, in particular Waldenström’s disease, is still an issue and careful monitoring should be carried out in every patient.
References
Recently published papers of particular interest have been highlighted as: • Of importance •• Of major importance
Schnitzler L. Lésions urticariennes chroniques permanentes (érythème pétaloïde?). Journées Dermatologiques d’Angers. 1972; Cas Clinique. N°46b.
•• Lipsker D. Monoclonal gammopathy of cutaneous significance: review of a relevant concept. J Eur Acad Dermatol Venereol. 2017;31:45–52. This article introduces the concept of monoclonal gammopathy of cutaneous significance and reviews the main entities that fit into it.
De Koning HD, van Gijn ME, Stoffels M, Jongekrijg J, Zeeuwen PLJM, Elferink MG, et al. Myeloid lineage-restricted somatic mosaicism of NLRP3 mutations in patients with variant Schnitzler syndrome. J Allergy Clin Immunol. 2015;135:561–4.
Ryan JG, de Koning HD, Beck LA, Booty MG, Kastner DL, Simon A. IL-1 blockade in Schnitzler syndrome: ex vivo findings correlate with clinical remission. J Allergy Clin Immunol. 2008;121:260–2.
De Koning HD, Schalkwijk J, Stoffels M, Jongekrijg J, Jacobs JFM, Verwiel E, et al. The role of interleukin-1 beta in the pathophysiology of Schnitzler’s syndrome. Arthritis Res Ther. 2015;17:187.
De Koning HD, van Vlijmen-Willems IMJJ, Rodijk-Olthuis D, van der Meer JWM, Zeeuwen PLJM, Simon A, et al. Mast-cell interleukin-1beta, neutrophil interleukin-17 and epidermal antimicrobial proteins in the neutrophilic urticarial dermatosis in Schnitzler’s syndrome. Br J Dermatol. 2015;173:448–56.
Zielinski CE, Mele F, Aschenbrenner D, Jarrossay D, Ronchi F, Gattorno M, et al. Pathogen-induced human TH17 cells produce IFN-gamma or IL-10 and are regulated by. Nature. 2012;484:514–8.
Noster R, de Koning HD, Maier E, Prelog M, Lainka E, Zielinski CE. Dysregulation of proinflammatory versus anti-inflammatory human TH17 cell functionalities in the autoinflammatory Schnitzler syndrome. J Allergy Clin Immunol. 2016;138:1161–9.
Mulla E, Neame R. Delayed development of the IgM paraprotein in Schnitzler’s syndrome. Scand J Rheumatol. 2015;44:521–2.
Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. MYD88 L265P somatic mutation in Waldenstrom’s macroglobulinemia. N Engl J Med. 2012;367:826–33.
• De Koning HD. Schnitzler’s syndrome: lessons from 281 cases. Clin Transl Allergy. 2014;4:41. This study summarizes the clinical, biological, histological, and morphological data of all the patients with a Schnitzler syndrome reported to date.
Akimoto R, Yoshida M, Matsuda R, Miyasaka K, Itoh M. Schnitzler’s syndrome with IgG kappa gammopathy. J Dermatol. 2002;29:735–8.
Welsh B, Tate B. Schnitzler’s syndrome: report of a case with progression to Waldenstrom’s macroglobulinaemia. Australas J Dermatol. 1999;40:201–3.
Lipsker D, Veran Y, Grunenberger F, Cribier B, Heid E, Grosshans E. The Schnitzler syndrome: four new cases and review of the literature. Medicine (Baltimore). 2001;80:37–44.
Simon A, Asli B, Braun-Falco M, De Koning H, Fermand J-P, Grattan C, et al. Schnitzler’s syndrome: diagnosis, treatment, and follow-up. Allergy. 2013;68:562–8.
•• Gusdorf L, Asli B, Barbarot S, Neel A, Masseau A, Puechal X, et al. Schnitzler syndrome: validation and applicability of diagnostic criteria in real-life patients. Allergy. 2017;72:177–82. This study confirms the applicability and the validity of the existing diagnostic criteria in real-life patients.
Kieffer C, Cribier B, Lipsker D. Neutrophilic urticarial dermatosis: a variant of neutrophilic urticaria strongly associated with systemic disease. Report of 9 new cases and review of the literature. Medicine (Baltimore). 2009;88:23–31.
Broekaert SMC, Boer-Auer A, Kerl K, Herrgott I, Schulz X, Bonsmann G, et al. Neutrophilic epitheliotropism is a histopathological clue to neutrophilic urticarial dermatosis. Am J Dermatopathol. 2016;38:39–49.
Gusdorf L, Bessis D, Lipsker D. Lupus erythematosus and neutrophilic urticarial dermatosis: a retrospective study of 7 patients. Medicine (Baltimore). 2014;93:e351.
Lipsker D. The Schnitzler syndrome. Orphanet J Rare Dis. 2010;5:38.
Sokumbi O, Drage LA, Peters MS. Clinical and histopathologic review of Schnitzler syndrome: the Mayo Clinic experience (1972-2011). J Am Acad Dermatol. 2012;67:1289–95.
Carlesimo M, Abruzzese C, Narcisi A, La Verde G, De Marco G, Verga E, et al. Chronic vasculitis urticaria associated to a monoclonal gammopathy of IgM and IgA type, a Schnitzler syndrome? Eur J Dermatol. 2010;20:838–9.
Ricci M, Bettazzi LG, Varenna M, Marchesoni A. Atypical bone sclerosis. Arthritis Rheumatol Hoboken NJ. 2016;68:816.
Niederhauser BD, Dingli D, Kyle RA, Ringler MD. Imaging findings in 22 cases of Schnitzler syndrome: characteristic para-articular osteosclerosis, and the “hot knees” sign differential diagnosis. Skelet Radiol. 2014;43:905–15.
Willekens I, Walgraeve N, Goethals L, De Geeter F. Correlative bone imaging in a case of Schnitzler’s syndrome and brief review of the literature. Hell J Nucl Med. 2015;18:71–3.
Ferrando FJ, Pujol J, Hortells JL, Navarro M, Pinol J, Carapeto FJ. Schnitzler’s syndrome: report of a case with bone osteolysis. J Investig Allergol Clin Immunol. 1994;4:203–5.
Terpos E, Asli B, Christoulas D, Brouet J-C, Kastritis E, Rybojad M, et al. Increased angiogenesis and enhanced bone formation in patients with IgM monoclonal gammopathy and urticarial skin rash: new insight into the biology of Schnitzler syndrome. Haematologica. 2012;97:1699–703.
Martinez-Taboada VM, Fontalba A, Blanco R, Fernandez-Luna JL. Successful treatment of refractory Schnitzler syndrome with anakinra: comment on the article by Hawkins et al. Arthritis Rheum. 2005;52:2226–7.
Lipsker D, Lenormand C. Management of Schnitzler’s syndrome. Expert Opin Orphan Drugs. 2014;2:947–55.
Neel A, Henry B, Barbarot S, Masseau A, Perrin F, Bernier C, et al. Long-term effectiveness and safety of interleukin-1 receptor antagonist (anakinra) in Schnitzler’s syndrome: a French multicenter study. Autoimmun Rev. 2014;13:1035–41.
Perrin F, Neel A, Graveleau J, Ruellan A-L, Masseau A, Hamidou M. Two cases of anakinra-induced neutropenia during auto-inflammatory diseases: drug reintroduction can be successful. Presse Med. 2014;43:319–21.
De Koning HD, Schalkwijk J, van der Ven-Jongekrijg J, Stoffels M, van der Meer JWM, Simon A. Sustained efficacy of the monoclonal anti-interleukin-1 beta antibody canakinumab in a 9-month trial in Schnitzler’s syndrome. Ann Rheum Dis. 2013;72:1634–8.
Vanderschueren S, Knockaert D. Canakinumab in Schnitzler syndrome. Semin Arthritis Rheum. 2013;42:413–6.
Pesek R, Fox R. Successful treatment of Schnitzler syndrome with canakinumab. Cutis. 2014;94:E11–2.
Krause K, Tsianakas A, Wagner N, Fischer J, Weller K, Metz M, et al. Efficacy and safety of canakinumab in Schnitzler syndrome: a multicenter randomized placebo-controlled study. J Allergy Clin Immunol. 2017;139:1311–20.
Krause K, Weller K, Stefaniak R, Wittkowski H, Altrichter S, Siebenhaar F, et al. Efficacy and safety of the interleukin-1 antagonist rilonacept in Schnitzler syndrome: an open-label study. Allergy. 2012;67:943–50.
Krause K, Feist E, Fiene M, Kallinich T, Maurer M. Complete remission in 3 of 3 anti-IL-6-treated patients with Schnitzler syndrome. J Allergy Clin Immunol. 2012;129:848–50.
Asli B, Brouet JC, Fermand JP. Spontaneous remission of Schnitzler syndrome. Ann Allergy Asthma Immunol Off Publ Am Coll Allergy Asthma Immunol. 2011;107:87–8.
Conlon NP, Hayden P, Barnes L, Doran M, O’Shea F, Feighery C. Schnitzler’s syndrome: a case highlighting the complications of long-standing acquired autoinflammation. Eur J Dermatol. 2014;24:405–6.
Gameiro A, Gouveia M, Pereira M, Tellechea O, Goncalo M. Clinical characterization and long-term follow-up of Schnitzler syndrome. Clin Exp Dermatol. 2016;41:461–7.
Margerin F, Gottenberg JE, Lipsker D. Occurrence of rheumatoid arthritis in a patient treated with anakinra for Schnitzler syndrome: a case report. J Rheumatol. 2016;43:1447.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Orphan Diseases
Rights and permissions
About this article

Cite this article
Gusdorf, L., Lipsker, D. Schnitzler Syndrome: a Review. Curr Rheumatol Rep 19, 46 (2017). https://doi.org/10.1007/s11926-017-0673-5
Published:
DOI: https://doi.org/10.1007/s11926-017-0673-5