
Abstract
Purpose of Review
The aim of this review is to aid in choosing safe options when assessing potential risks of acute migraine treatments based on known mechanisms of action and anticipated safety concerns.
Recent Findings
Part 1 highlights safety issues associated with commonly used medications to treat acute migraine attacks. Strategies to mitigate cardiovascular and gastrointestinal risks of nonsteroidal anti-inflammatory drugs, evaluation of cardiovascular risks of triptan and ergot alkaloids, and precautions with use of antiemetics and the novel drugs gepants and ditans are discussed to help practitioners in clinical decision-making. When available, we included recommendations from professional societies and data from pharmacovigilance systems.
Summary
While guidelines on efficacy are available, one must also consider the possible risks and adverse effects of a drug when creating treatment plans.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Assessing the risks and benefits of medications is a complex and multidimensional task. While potential benefits of certain medications are usually apparent, assessment of possible risks is not as straightforward as these are not always captured in randomized clinical trials [1]. There are multiple reasons for this: (1) study design includes participants that do not represent the population that will be exposed to the drug; (2) exposure to medication in clinical trials may be limited, and delayed toxicity may not be evident; (3) accepted level of risk may be skewed, both in public health terms and at the individual level.
Adverse health effects can be related to mechanism of action directly or to off-target effects of a certain drug. Clinicians also deal with potential drug interactions that can result in loss of efficacy or unintended toxicity, especially in polypharmacy, which is common in headache disorders. Nearly 29% of patients with episodic headache have polypharmacy, taking five or more medications [2]. Patients take multiple combinations to treat headache when medications with different mechanisms of action provide additive or synergistic effects. Medications can be prescribed by multiple physicians, or patients self-medicate with over-the-counter drugs. Patients may take medications to treat other disorders. This can lead to adverse effects that require additional treatment, further escalating polypharmacy. Finally, individual genetic traits can increase side effects or have an impact on treatment response of medications that undergo enzymatic transformation or are substrates of membrane transporters [3].
When considering risk, it is prudent to keep in mind that inaction can have its own consequences. Taking unnecessary precautions promotes waste of medical resources, but withholding treatment for fear of side effects also comes with the risks of increasing disability from undertreated migraine and chronification of migraine. When assessing potential risks of acute migraine treatments based on known mechanisms of action, clinicians should consider the risks of alternative options, including withholding treatment.
Medications with established efficacy in the acute treatment of migraine include triptans, ergotamine derivatives, nonsteroidal anti-inflammatory drugs, and opioids such as butorphanol [4••, 5••]. While butorphanol has established efficacy in acute migraine management, opioids are not a recommended treatment option for migraine and will not be reviewed in this paper. The treatment of migraine during pregnancy and lactation was recently reviewed by R. Burch, and we recommend readers to reference that article for more information on those specific safety concerns [6]. In addition, the acute treatment of migraine in children and adolescents has been recently addressed in an updated American Academy of Neurology and American Headache Society practice guideline [7].
Nonsteroidal Anti-inflammatory Drugs (NSAIDs)
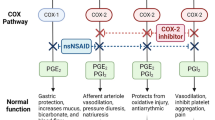
NSAIDs such as acetylsalicylic acid, ibuprofen, naproxen, and diclofenac have established (level A) efficacy in acute treatment of migraine. Their analgesic effect is mainly attributed to inhibition of two isoforms of cyclooxygenase enzymes (COX-1 and COX-2) responsible for the conversion of arachidonic acid to several bioactive lipids and prostaglandins. The COX-1 isoform regulates platelet aggregation, thrombosis, gastric cytoprotection, and renal function, explaining why inhibition of COX-1 leads to an increased risk of gastrointestinal complications and bleeding [8]. COX-2 is induced at the site of inflammation, which is believed to be the basis of the anti-inflammatory effect of NSAIDs. This isoform produces prostaglandin I2 in the vascular endothelium, especially at the time of endothelial injury, which prevents platelet aggregation and aids vasodilation. This mechanism is the basis of cardiovascular side effects of NSAIDs, because inhibition of COX-2 impairs the defense mechanism against endothelial injury [9].
Acetylsalicylic acid (ASA) binds irreversibly to both COX enzyme isoforms and inhibits platelet aggregation for the duration of the platelet life. Non-salicylate NSAIDs inhibit both COX isoforms competitively and reversibly but with varying degrees of selectivity [9]. Higher doses (1000 mg) of ASA are required for analgesic effect on headache [10, 11]. Excessively high doses (6–8 g per day) of acetylsalicylic acid can result in neurotoxicity [12], and chronic use can increase the risk of major bleeding, with relative risk ranging from 1.3 for intracranial hemorrhage to 1.58 for gastrointestinal bleeding [13].
Celecoxib is the only selective COX-2 inhibitor available in the USA. Oral solution of low-dose celecoxib (120 mg) was FDA-approved in 2020 for the treatment of migraine with or without aura in adults and is a promising option for patients with high risk for gastrointestinal (GI) complications, although it does carry a warning about cardiovascular and gastrointestinal risks [14,15,16].
Due to some degree of COX-2 inhibition, all NSAIDs now carry black box warnings emphasizing an increased risk of cardiovascular events, although naproxen at higher dose (1000 mg daily) showed lower vascular risk compared to other NSAIDs [8, 17]. In a large Danish population-based study, diclofenac was associated with a 50% increase in major cardiovascular events in patients with prior cardiovascular risk factors [18]. Use of NSAIDs was also associated with an increased risk of hospital admission for heart failure in a large study in four European countries, with odds ratio highest for ketorolac [19]. Addition of aspirin as an adjunct to other NSAIDs to reduce risk of thrombotic events may result in an increased unfavorable interaction and remains debatable; it also further increases risk of GI side effects [20].
Medications with relatively higher COX-1 inhibition include indomethacin, ibuprofen, naproxen, and ketorolac. These are associated with higher risk of gastrointestinal adverse events with highest risk reported for ketorolac [21]. Several strategies have been proposed to reduce risk of GI side effects of NSAIDs with best advice to use medication at the lowest dose for the shortest period [8]. Upper GI bleeding can be prevented with an addition of proton pump inhibitors (PPI) [22, 23]. NSAID-induced lower enteropathy is not related to acid secretion; addition of PPI does not protect against it and may in fact worsen enteropathy by disrupting the intestinal microbiota. Probiotics can reduce the risk of lower gastrointestinal injury with NSAIDs, but there are insufficient data to recommend specific probiotic strains [24].
Nephrotoxicity of NSAIDs is a long-standing concern and a debated topic in nephrology. Risk factors previously associated with nephrotoxicity from NSAIDs included higher doses, long duration of therapy, concomitant use of renin-angiotensin system inhibitors or diuretics, preexisting chronic kidney disease, and advanced age [25]. Avoidance of NSAIDs may lead to use of alternative treatments which can have harmful consequences as well. Opioid use in patients with chronic kidney disease was associated with higher risk of death. Impaired excretion of gabapentinoids in patients with reduced glomerular filtration can lead to encephalopathy, generalized weakness, ataxia, and myoclonus. Use of acetaminophen may also increase risk of renal impairment [26,27,28].
While liver toxicity of acetaminophen and aspirin is well known, hepatotoxicity of NSAIDs is commonly overlooked. The mechanism of liver toxicity associated with typical NSAIDs is likely idiosyncratic rather than due to direct intrinsic toxicity, unlike toxicity associated with aspirin and acetaminophen. The most common pattern of toxicity is hepatocellular presenting with fever, fatigue, and marked elevation of aminotransferases. Less frequent cholestatic injury presents with itching, jaundice, and elevation in alkaline phosphatase and bilirubin levels [29].
In a systematic review by Sriuttha et al., diclofenac was the most common NSAID causing liver toxicity, followed by celecoxib [30]. Ibuprofen was the most common NSAID causing drug-induced liver injury, likely due to higher doses available without prescription [31].
Drug-induced aseptic meningitis (DIAM) is a rare condition observed in association with NSAID use, especially ibuprofen, naproxen, diclofenac, and sulindac. NSAIDs were the second leading cause of DIAM after intravenous immunoglobulins in a French pharmacovigilance database analysis of 329 cases [32]. The likely mechanism of DIAM with oral NSAIDs is an immunological hypersensitivity reaction, which may explain why patients with systemic lupus erythematosus are more susceptible to this condition [33]. Rapid evolution of symptoms and polymorphonuclear pleocytosis in cerebrospinal fluid is more characteristic for DIAM and may help to differentiate it from central nervous lupus activity [34, 35]. Patients who developed DIAM after ingesting one NSAID can usually take another NSAID, as cross-intolerance between NSAIDs in this condition is rare [36].
There is no evidence-based guidance for NSAID use in patients at risk for side effects, but practical recommendations by experts are available. It is advised that prior to the initiation of NSAIDs, the cardiovascular, gastrointestinal, and renal risk profiles of the patient are assessed. Patients with stage 1 to 3 chronic kidney disease have risk of NSAID-associated nephrotoxicity similar to the general population, but in more advanced stages, hypovolemic states, or when used with concurrent renin-angiotensin system inhibitors or diuretics, NSAIDs should be avoided and alternatives should be used instead [37]. Risk of nephrotoxicity is similar between COX-2-selective and non-selective NSAIDs.
In patients with high cardiovascular risk and low GI risk, naproxen, or ibuprofen with PPI for gastric protection, or lower dose (200 mg) celecoxib may be used. Patients with high risk for both gastrointestinal and cardiovascular complications can use low-dose celecoxib with gastric protection, but other NSAIDs are not recommended [38, 39•].
Triptans
Triptans are selective serotonin (5HT) agonists specific to 5HT-1B/D receptors; some triptans also have activity at the 5HT-1F receptor. Cardiovascular safety concerns regarding triptans stem from their vasoconstrictive effect mediated by 5HT-1B receptors. This mechanism of action led to the contraindication for the use of triptans in patients with cardiovascular risks; however, triptans themselves do not increase the risk of cardiovascular events in healthy individuals. In magnetic resonance angiography studies of cranial arteries, sumatriptan prevents migraine-related dilatation and significantly constricts extracerebral middle meningeal arteries, but not cerebral arteries such as middle cerebral, internal carotid, and basilar arteries [40,41,42]. No link between triptan use and risk of stroke was found in epidemiological studies from a UnitedHealthcare database and from a General Practice Research database [43, 44]. A notable point is that population-based studies can be biased, because patients with cardiovascular risk factors are less likely to be prescribed triptans [45]. A number of angiographic studies, and studies on isolated human coronary artery from patients with normal coronary arteries, showed that triptans have minimal vasoconstrictive effect on coronary arteries and are unlikely to cause myocardial infarction in healthy individuals [46, 47]. A population-based study of adults from Southern California also showed no association between triptan use and an increased risk of myocardial infarction, heart failure, or death [48].
An expert panel from the American Headache Society concluded that triptans have an overall favorable cardiovascular risk–benefit profile in the absence of contraindications [49••]. However, in patients with cardiovascular disease, triptans are contraindicated because a small risk of triptan-induced cardiovascular events cannot be excluded. It has been disputed that this warning does not consider the mechanism of action of triptans or pathophysiology of stroke [50•]. Studies of triptan use after a stroke are lacking, and in the current medico-legal environment, prescription of triptans in patients with prior stroke takes a great deal of deliberation despite reassuring literature [51].
Assessment of vascular risk factors may help to decide if additional cardiac workup is necessary to assure that a patient does not have cardiac disease that precludes use of triptans [52, 53]. Tools for risk calculation are available online [54, 55]. In patients with a history of angina, myocardial infarction, and ischemic stroke, alternative therapies for acute treatment of headache should be considered.
Use of triptans in migraine with brainstem aura (formerly known as basilar migraine) is another topic of debate. Basilar migraine was initially described by Bickerstaff as basilar artery migraine due to vascular spasm [56]. Given the lack of evidence of vasospasm in migraine aura, this form of migraine is now called migraine with brainstem aura [57]. Observations of patients with basilar migraine, hemiplegic migraine, or migraine with prolonged aura did not identify any ischemic vascular events [58]. There are no convincing studies to support the belief that migraine with brainstem aura should be treated differently from migraine with typical aura [59].
Triptans have been reported to precipitate reversible cerebral vasoconstriction syndrome; however, patients in case reports had other risk factors [60, 61]. Cases of ischemic colitis associated with triptans were observed, and a high level of suspicion for ischemic colitis should be maintained when a patient taking triptans develops acute abdominal pain and bloody diarrhea in the absence of a known inflammatory bowel disorder [62].
There is a common hesitancy to use triptans and ergot derivatives in patients with cerebral aneurysms because of vasoconstrictive effects. One retrospective review found no complications in 10 pre-coiling and post-coiling patients with cerebral aneurysms who used triptans, suggesting that triptans can be used in this category of patients [63].
Triptans have an FDA warning about potential life-threatening serotonin syndrome when coadministered with selective serotonin reuptake inhibitors (SSRI) and selective norepinephrine reuptake inhibitors (SNRI). This warning was based on 29 case reports, but none of them met validated Hunter criteria for serotonin toxicity [64]. Serotonin syndrome is a drug-induced and dose-dependent toxidrome due to an excess of serotonin in the synaptic cleft [65, 66•, 67]. In experimental studies, serotonin toxicity was mediated by 5HT-2A receptors with some involvement of 5HT-1A receptors, while triptans are agonists with high affinity at 5HT-1B/5HT-1D/5HT-1F and lower affinity to 5HT-1A receptors. Thus, triptans are unlikely to induce this syndrome [68]. In our population-based study of patients who were co-prescribed SSRI or SNRI antidepressants and triptans, we estimated the risk of serotonin toxicity as very low at 2.3 cases per 10,000 person-years of exposure [69]. Another retrospective study of clinical outcomes after intentional triptan or ergotamine overdoses found no definitive cases of serotonin toxicity [70]. Because of low risk of serotonin toxicity, the American Headache Society does not support withholding triptans to treat migraine in patients who take SSRI or SNRI antidepressants [71].
Notable interactions of triptans include eletriptan with CYP3A4 inhibitors, frovatriptan with CYP1A2 inhibitors, sumatriptan and rizatriptan with monoamine oxidase inhibitors, and rizatriptan with propranolol use, which are all listed in product labeling [72].
Ditans
Ditans are selective agonists of 5HT-1F receptors expressed on trigeminal neurons, allowing for the treatment of migraine attacks via a neural mechanism and without the vasoconstrictive effects typically seen with triptans [73]. Lasmiditan is first in class and currently the only ditan available in the USA. It penetrates the blood–brain barrier and can cause CNS-related side effects such as dizziness, fatigue, paresthesia, and sedation [74]. Lasmiditan can impair driving performance despite subjective perception of the ability to drive safely [75]. It is currently classified as a schedule V controlled substance [76].
Since lasmiditan acts on serotonin receptors, there is a theoretical risk of serotonin syndrome, regardless of the presence of other serotonergic medications. In two phase 3 single-migraine-attack studies of lasmiditan, five cases of possible serotonin syndrome were identified, but none met validated Hunter criteria [77]. This is not surprising, as serotonin receptor 1F has not been implicated in the development of serotonin syndrome.
Lasmiditan can interact with a number of medications, including alcohol (resulting in an additive effect) and substrates of P-glycoprotein, such as calcium-channel blockers, cyclosporine, dabigatran etexilate, digoxin, erythromycin, loperamide, protease inhibitors, and tacrolimus [78], but no clinical studies are available to assess clinical significance of these interactions [79].
Lasmiditan can potentiate bradycardia when used concomitantly with heart rate-lowering medications. This was observed in healthy individuals receiving propranolol 80 mg twice daily after one 200-mg dose of lasmiditan, but exact mechanism of interaction remains unclear [80,81,82].
Risk of medication overuse headache (MOH) with lasmiditan remains unknown. One of the hypothesized mechanisms of MOH is related to desensitization and downregulation of the receptors after prolonged exposure to agonists, which includes ditans and triptans [83]. In a pre-clinical rat model of medication overuse headache, lasmiditan induced acute transient cutaneous allodynia in a similar way to sumatriptan, which suggests that lasmiditan may have the capacity to induce MOH [80, 84]
Ergot Alkaloids
Of more than 80 ergot alkaloids, three are used to treat migraine. Ergotamine and dihydroergotamine (DHE) are used for acute treatment, and methysergide is used for prophylaxis [85]. Like triptans, the anti-migraine properties of ergot alkaloids are due to agonism at 5HT-1B and 5HT-1D serotonin receptors. In addition, ergot alkaloids also have high affinity to 5HT-1A and 5HT-2A receptors, dopamine receptor D, and α1/α2-adrenergic receptors, which may contribute to the potential side effects on the one hand, and more clinical efficacy in those with inadequate response to triptans on the other hand [86, 87].
Compared to triptans, ergot alkaloids have more potent vasoconstrictive effects on peripheral arteries, including pulmonary, cerebral, temporal, and coronary arteries. Ergot alkaloids can transiently increase systemic blood pressure for 3 h after parenteral use and should be avoided in patients with uncontrolled hypertension [88]. Ergot alkaloids are also contraindicated in patients with coronary, cerebral, and peripheral vascular disease; arteriosclerosis; in those with clinical symptoms of coronary vasospasm including Prinzmetal’s variant angina; sepsis; and following vascular surgery; and in those with severely impaired hepatic or renal function. Because of oxytocic properties and the ability to cause developmental toxicity, ergot alkaloids are contraindicated in pregnancy and should not be used by nursing mothers [89].
Although both ergotamine and DHE are potent constrictors of venous capacitance vessels, ergotamine is a more potent arterial vasoconstrictor. It has stronger uterotonic effects and causes nausea more often. DHE is better tolerated and is less likely to cause nausea and vomiting, although intravenous DHE can be associated with diarrhea.
Prolonged use of ergotamine can lead to medication overuse headache and can also result in overt ergotism with gangrene, peroneal nerve ischemic neuropathy, anorectal ulcers with chronic use of suppositories, and retroperitoneal, pulmonary, pleural, pericardial, or heart valve fibrosis [90]. Cases of ergotism have been reported when ergotamine was administered with strong CYP3A4 inhibitors, such as macrolide antibiotics; protease inhibitors; and antifungals such as ketoconazole, because both ergotamine and DHE are believed to be a substrate to CYP3A4 [91, 92].
Unlike ergotamine, DHE has minimal risk of MOH and can be used to treat refractory migraine and status migrainosus via continuous infusions over 1–3 days or as repeated intermittent boluses [93, 94]. The bioavailability of ergotamine and DHE highly depends on route of administration because of significant first-pass metabolism. Due to very low oral bioavailability (less than 1%), oral administration of DHE is not useful for acute treatment of migraine [94]. Parenteral formulations of DHE for intravenous and intramuscular administration have 100% bioavailability, and intranasal delivery of DHE using the nasal pump has 40% bioavailability.
The INP104 (DHE administered via Impel’s Proprietary Precision Olfactory Delivery Technology) is another formulation for intranasal use and was approved by the FDA in September 2021 [95] backed by reassuring safety and tolerability study results [96]. Other formulations of DHE for intranasal or orally inhaled formulations are in development with data reassuring for cardiovascular safety of DHE. Orally inhaled DHE MAP0004 was efficacious and well tolerated, but MAP0004 was not approved because of manufacturing issues with the delivery system [87]. A trial of STS101 (dihydroergotamine nasal) powder did not achieve statistical significance on the co-primary endpoints of freedom from pain and freedom from most bothersome symptoms, but a new phase 3 efficacy trial started in 2021 with estimated completion in 2022 [97].
Antiemetics
Antiemetics comprise a diverse group of medications which includes dopamine receptors antagonists, antihistamines, anticholinergic agents, 5HT-3 antagonists, cannabinoids, benzodiazepines, corticosteroids, and neurokinin-1 receptor antagonists [98].
Dopamine antagonists are the most commonly used antiemetics in migraine and are well studied. They include chlorpromazine, prochlorperazine, promethazine, haloperidol, droperidol, and metoclopramide. The antiemetic effect of these medications is mediated by both peripheral (enteric) and central (area postrema) D2 dopamine receptors. Dopamine antagonists can also be used to prevent a migraine attack in patients who have clear prodromal symptoms such as yawning, mood changes, irritability, and fatigue. The atypical neuroleptics olanzapine and quetiapine are used for both acute treatment of prolonged migraine and prophylaxis; however, weight gain and sedation limit their long-term use [99].
These dopamine antagonists are associated with various drug-induced movement disorders. Acute dystonia and acute akathisia can emerge within hours to days of starting a dopamine antagonist. Diphenhydramine reduces risk of akathisia when used with prochlorperazine or high dose (20 mg) metoclopramide [100]. Drug-induced parkinsonism can develop days to weeks after exposure to dopamine antagonists, more commonly with chronic use, with two-thirds of patients recovering within weeks after discontinuation of the offending drug [101]. Tardive syndromes, such as classic tardive dyskinesia, tardive dystonia, tardive akathisia, tardive tremor, and tardive tics, typically appear after months or years of exposure; however, it can be seen with just over 3 months of metoclopramide use [102]. Older women and patients with diabetes, liver or kidney failure, or concomitant antipsychotic drug therapy are at increased risk for this condition [103, 104]. Although development of tardive dyskinesia after an isolated dose of antidopaminergic drug is unlikely, the risk with intermittent use remains unknown and it has been suggested that their use should be limited to 2 days a week [105].
Neuroleptic malignant syndrome is a feared life-threatening reaction to dopamine-blocking drugs reported after exposure to metoclopramide, prochlorperazine, and other dopamine antagonists [101]. This idiosyncratic reaction to therapeutic doses of dopamine antagonists presents with cogwheel rigidity, hyperthermia, autonomic dysfunction, and mental status change typically associated with an elevated creatine phosphokinase and requires an intensive level of care as a neurological emergency [106]. Although there are similarities between neuroleptic malignant syndrome and serotonin toxicity, hyperkinesia and clonus in serotonin syndrome can be distinguished from bradykinesia and lead-pipe rigidity in neuroleptic malignant syndrome [107].
When dopaminergic antiemetics are contraindicated or poorly tolerated, 5HT-3 receptor antagonists can be used. Of the four such antiemetics currently available in the USA, ondansetron is most used. There is a lack of research on this group of medications in migraine, and they are not included in current guidelines for acute migraine treatment.
Prolonged QT is a potential side effect of both dopamine receptor antagonists and serotonergic antiemetics, among many other medications. It increases risk of torsades de pointes (TdP) and sudden cardiac death. Cases are reported after intravenous administration of ondansetron, although arrhythmia was not observed after a single dose of oral ondansetron in healthy individuals [108, 109]. QTc prolongation of more than 500 ms is a contraindication for some neuroleptics, including chlorpromazine, droperidol, and haloperidol [99]. Monitoring of QTc intervals to ensure they remain below 500 ms during treatment is recommended.
Many medications commonly prescribed in patients with migraine can increase risk of QTc prolongation. These include citalopram/escitalopram, venlafaxine, nortriptyline, amitriptyline, imipramine, and tizanidine, among others. An extensive list of medications that prolong QT and induce TdP can be found at the CredibleMeds.org website [110].
CGRP Blocking Agents
Two classes of calcitonin gene-related peptide (CGRP) blocking agents are used to treat migraine: CGRP receptor blocking small molecules (gepants) and CGRP monoclonal antibodies (mAbs) that block either the CGRP receptor (erenumab) or ligand (galcanezumab, fremanezumab, eptinezumab) [111]. Although central activity of gepants was suggested, the blood–brain barrier permeability of gepants and CGRP mAbs is very low, implicating a peripheral site of action for these medications, most likely at the level of the trigeminal ganglion or dura mater and meninges outside the blood–brain barrier [112].
CGRP belongs to a family of neuropeptides and exists in two forms. α-CGRP, most relevant to migraine, is the principal form found in the central and peripheral nervous systems. The β-CGRP isoform is mainly found in the gut; it is formed by a different gene but has 90% homology with α-CGRP. CGRP is contained in perivascular nerves, providing the link with the cardiovascular system [113]. CGRP receptors belong to a group of family B G-protein-coupled receptors that also share structural homology. They are located at multiple sites involved in the pathophysiology of migraine, in both the central and peripheral nervous systems [112].
At this time, two orally administered gepants (ubrogepant and rimegepant) are approved for acute migraine treatment, and intranasal zavegepant (formerly known as vazegepant) is in late-stage development [114]. Another member of this class, atogepant, was approved by the FDA in September 2021 only for the preventive treatment of episodic migraine based on promising results from clinical trials [115]. Rimegepant gained FDA approval for the preventive treatment of episodic migraine treatment as well, making it the first CGRP-specific drug with both acute and prophylactic indications for migraine [116]. CGRP mAbs are currently approved for prevention of migraine, and we further discuss safety of prolonged CGRP antagonists in Part 2: Preventive Treatments.
Concerns for hepatotoxicity halted development of some small‐molecule CGRP receptor antagonists, but both ubrogepant and rimegepant are well tolerated without concerns for hepatotoxicity in healthy individuals, even when used at a high frequency [117, 118]. Rimegepant as a single dose was also well tolerated in healthy individuals, as well as in those with various degrees of liver function impairment. However, maximum observed plasma concentration was increased twofold in persons with severe liver impairment compared to matched healthy individuals [119].
In a long‐term safety evaluation trial, ubrogepant was also well tolerated. Of 1230 participants receiving ubrogepant 50 mg, 100 mg, or usual care, there were only three cases of alanine aminotransferase (ALT) or aspartate aminotransferase (AST) elevations of ≥ 3 times the upper limit of normal that were either possibly or probably related to treatment [120]. Every other day administration of rimegepant for 12 weeks in a phase 2/3 randomized, double-blind placebo-controlled trial showed tolerability similar to placebo with low rates of increased enzymes in both treatment groups [121]. In patients with severe hepatic impairment (Child–Pugh C), rimegepant and atogepant should be avoided; ubrogepant can be used at a reduced dose. In patients with end-stage renal disease (creatinine clearance, CLcr < 15 mL/min) rimegepant and ubrogepant should be avoided; atogepant can be used at the lowest dose 10 mg. Ubrogepant can be used at reduced dose in those with severe renal impairment (CLcr 15–29 mL/min).
The cardiovascular safety of gepants deserves special attention. Gepants were well tolerated in regulatory clinical trials, and this class of drugs has become a welcome addition to the current migraine management armamentarium. These do not have direct vasoconstrictive effects, and neither ubrogepant nor rimegepant has any cardiovascular contraindications listed on the product label. A trial of ubrogepant in study participants with cardiovascular risk factors had no serious cardiovascular adverse events after treatment of a single attack [122]. In long-term studies that included individuals with cardiovascular risk factors, both rimegepant and ubrogepant, used intermittently for up to 1 year, were well tolerated, and there were no cardiovascular safety issues [120, 123]. It is important to note, however, that clinical trials of both rimegepant and ubrogepant excluded patients that had cardiovascular events within 6 months prior to enrollment [122, 124] and use of these medications in patients with recent stroke or myocardial infarction has not been studied.
In animal studies by Mulder et al. [125], olcegepant (not used clinically) and rimegepant worsened ischemic stroke in mice via collateral dysfunction. While these data cannot be directly extrapolated to clinical practice, they raise the question of whether these drugs can worsen an outcome of coincidental stroke or myocardial infarction.
Given that gepants are marketed as medications without cardiovascular concerns, this can lead to the perception that they are safer alternatives to triptans in patients with migraine who experience a recent acute ischemic event. More data on safety with long-term use are needed, but we believe it is reasonable to avoid gepants for at least 6 months after an acute stroke or myocardial infarction, as well as those with unstable disease (such individuals likewise were excluded from clinical trials). Although arbitrary, this time frame seems to be reasonable to assume recovery from an acute vascular event and allow time for risk factors to be adequately addressed prior to initiation of treatment with gepants in this category of patients.
All currently available gepants are subject to multiple drug interactions when coadministered with CYP34A inhibitors or inducers [126, 127]. Medications that may decrease levels of gepants with loss of efficacy include strong CYP3A4 inducers such as phenytoin, barbiturates (including butalbital), rifampin, St. John’s wort, and carbamazepine [92].
Unlike inducers, CYP3A inhibitors increase the exposure to gepants and have the potential to lead to high serum levels. Coadministration of ubrogepant and rimegepant with strong CYP3A4 inhibitors such as ketoconazole, itraconazole, clarithromycin, and protease inhibitors should be avoided. Atogepant can be used with strong CYP3A4 inhibitors at the lowest dose 10 mg daily; no dose adjustment is necessary when used with moderate and weak inhibitors [128]. Limiting ubrogepant dose to 50 mg is advised with concomitant use of moderate CYP3A4 inhibitors such as cyclosporine, ciprofloxacin, fluconazole, fluvoxamine, grapefruit juice, and verapamil. When rimegepant is coadministered with moderate CYP3A4 inhibitors, avoidance of the next dose of rimegepant within 48 h is advised.
Dose reduction of ubrogepant or avoidance of a second dose of rimegepant within 48 h is recommended with concomitant use of inhibitors of P-glycoprotein (verapamil, carvedilol, curcumin, amiodarone, cyclosporine, lapatinib, quinidine, ranolazine, eltrombopag) or breast cancer resistance protein (BCRP) efflux transporter inhibitors (tyrosine kinase inhibitors imatinib, anti-HIV protease inhibitors nelfinavir and ritonavir, antifungal azoles, tamoxifen) [129,130,131].
Dose reduction to 10 mg or 30 mg is recommended for atogepant when used concomitantly with organic anion transporting polypeptide (OATP) inhibitors. OATPs are membrane influx transporters that participate in enzyme-based detoxification. Many drugs can inhibit OATPs causing drug-drug interactions, including gemfibrozil, cyclosporine, leflunomide, teriflunomide, clarithromycin, erythromycin, rifampicin, and anti-HIV protease inhibitors [132].
Conclusion
The acute management of migraine is both fascinating and complex. Clinicians should take into consideration not only adverse health effects directly related to known mechanisms of action, off-target effects, and drug interactions, but also risks associated with withholding the treatment for fear of side effects. With the introduction of migraine-specific therapies such as gepants and ditans, physicians and other healthcare providers have many options to choose from when considering acute treatment for their patients Table 1.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Curtin F, Schulz P. Assessing the benefit: risk ratio of a drug–randomized and naturalistic evidence. Dialogues Clin Neurosci. 2011;13:183–90.
Ferrari A, Baraldi C, Licata M, Rustichelli C. Polypharmacy Among Headache Patients: A Cross-Sectional Study. CNS Drugs. 2018;32:567–78.
Pomes LM, Guglielmetti M, Bertamino E, Simmaco M, Borro M, Martelletti P. Optimising migraine treatment: from drug-drug interactions to personalized medicine. J Headache Pain. 2019;20:56.
•• Marmura MJ, Silberstein SD, Schwedt TJ. The acute treatment of migraine in adults: the American Headache Society evidence assessment of migraine pharmacotherapies. Headache. 2015;55:3–20. AHS guidelines for an acute treatment of migraine.
•• American Headache Society. The American Headache Society Position Statement On Integrating New Migraine Treatments Into Clinical Practice. Headache. 2019;59:1–18. Position statement with recommendations for acute and preventive treatment, including novel drugs.
Burch R. Epidemiology and Treatment of Menstrual Migraine and Migraine During Pregnancy and Lactation: A Narrative Review. Headache. 2020;60:200–16.
Oskoui M, Pringsheim T, Holler-Managan Y, Potrebic S, Billinghurst L, Gloss D, et al. Practice guideline update summary: Acute treatment of migraine in children and adolescents: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the American Headache Society. Neurology. 2019;93:487–99.
Antman EM, Bennett JS. Daugherty Alan, Furberg Curt, Roberts Harold, Taubert Kathryn A. Use of Nonsteroidal Antiinflammatory Drugs Circulation. 2007;115:1634–42.
Meek IL, Van de Laar MAFJ, E Vonkeman H. Non-Steroidal Anti-Inflammatory Drugs: An Overview of Cardiovascular Risks. Pharmaceuticals. 2010;3:2146–2162.
Lange R, Schwarz JA, Hohn M. Acetylsalicylic acid effervescent 1000 mg (Aspirin) in acute migraine attacks; a multicentre, randomized, double-blind, single-dose, placebo-controlled parallel group study. Cephalalgia. 2000;20:663–7.
Diener HC, Bussone G, de Liano H, Eikermann A, Englert R, Floeter T, et al. Placebo-controlled comparison of effervescent acetylsalicylic acid, sumatriptan and ibuprofen in the treatment of migraine attacks. Cephalalgia. 2004;24:947–54.
Sheppard A, Hayes SH, Chen G-D, Ralli M, Salvi R. Review of salicylate-induced hearing loss, neurotoxicity, tinnitus and neuropathophysiology. Acta Otorhinolaryngol Ital. 2014;34:79–93.
Xie W, Luo Y, Liang X, Lin Z, Wang Z, Liu M. The Efficacy And Safety Of Aspirin As The Primary Prevention Of Cardiovascular Disease: An Updated Meta-Analysis. Ther Clin Risk Manag. 2019;15:1129–40.
Lipton RB, Munjal S, Brand-Schieber E, Tepper SJ, Dodick DW. Efficacy, tolerability, and safety of DFN-15 (celecoxib oral solution, 25 mg/mL) in the acute treatment of episodic migraine: A randomized, double-blind, placebo-controlled study. Headache. 2020;60:58–70.
BRIEF-FDA Approves Dr. Reddy’s Laboratories’ Elyxyb Oral Solution For Acute Treatment Of Migraine With Or Without Aura In Adults. 5 May 2020 [cited 20 Apr 2021]. Available: https://www.reuters.com/article/brief-fda-approves-dr-reddys-laboratorie-idUSFWN2CN10I.
Center for Drug Evaluation, Research. COX-2 Selective (includes Bextra, Celebrex, and Vioxx). In: U.S. Food & Drug Administration [Internet]. 2 Jan 2021 [cited 19 May 2021]. Available: https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/cox-2-selective-includes-bextra-celebrex-and-vioxx-and-non-selective-non-steroidal-anti-inflammatory.
Coxib and traditional NSAID Trialists’ (CNT) Collaboration, Bhala N, Emberson J, Merhi A, Abramson S, Arber N, et al. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet. 2013;382: 769–779.
Schmidt M, Sørensen HT, Pedersen L. Diclofenac use and cardiovascular risks: series of nationwide cohort studies. BMJ. 2018;362: k3426.
Arfè A, Scotti L, Varas-Lorenzo C, Nicotra F, Zambon A, Kollhorst B, et al. Non-steroidal anti-inflammatory drugs and risk of heart failure in four European countries: nested case-control study. BMJ. 2016;354: i4857.
Antman EM. The Aspirin-NSAID Interaction. J Am Coll Cardiol. 2018;71:1752–4.
David Waterbury L, Silliman D, Jolas T. Comparison of cyclooxygenase inhibitory activity and ocular anti-inflammatory effects of ketorolac tromethamine and bromfenac sodium. Curr Med Res Opin. 2006;22:1133–40.
Scally B, Emberson JR, Spata E, Reith C, Davies K, Halls H, et al. Effects of gastroprotectant drugs for the prevention and treatment of peptic ulcer disease and its complications: a meta-analysis of randomised trials. Lancet Gastroenterol Hepatol. 2018;3:231–41.
Gwee KA, Goh V, Lima G, Setia S. Coprescribing proton-pump inhibitors with nonsteroidal anti-inflammatory drugs: risks versus benefits. J Pain Res. 2018;11:361–74.
Lué A, Lanas A. Protons pump inhibitor treatment and lower gastrointestinal bleeding: Balancing risks and benefits. World J Gastroenterol. 2016;22:10477–81.
Dreischulte T, Morales DR, Bell S, Guthrie B. Combined use of nonsteroidal anti-inflammatory drugs with diuretics and/or renin-angiotensin system inhibitors in the community increases the risk of acute kidney injury. Kidney Int. 2015;88:396–403.
Novick TK, Surapaneni A, Shin J-I, Alexander GC, Inker LA, Wright EA, et al. Associations of Opioid Prescriptions with Death and Hospitalization across the Spectrum of Estimated GFR. Clin J Am Soc Nephrol. 2019;14:1581–9.
Zand L, McKian KP, Qian Q. Gabapentin toxicity in patients with chronic kidney disease: a preventable cause of morbidity. Am J Med. 2010;123:367–73.
Kanchanasurakit S, Arsu A, Siriplabpla W, Duangjai A, Saokaew S. Acetaminophen use and risk of renal impairment: A systematic review and meta-analysis. Kidney Res Clin Pract. 2020;39:81–92.
Nonsteroidal Antiinflammatory Drugs (NSAIDs). LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases 2020.
Sriuttha P, Sirichanchuen B, Permsuwan U. Hepatotoxicity of Nonsteroidal Anti-Inflammatory Drugs: A Systematic Review of Randomized Controlled Trials. Int J Hepatol. 2018;2018:5253623.
Zoubek ME, González-Jimenez A, Medina-Cáliz I, Robles-Díaz M, Hernandez N, Romero-Gómez M, et al. High Prevalence of Ibuprofen Drug-Induced Liver Injury in Spanish and Latin-American Registries. Clin Gastroenterol Hepatol. 2018;16:292–4.
Bihan K, Weiss N, Théophile H, Funck-Brentano C, Lebrun-Vignes B. Drug-induced aseptic meningitis: 329 cases from the French pharmacovigilance database analysis. Br J Clin Pharmacol. 2019;85:2540–6.
Yelehe-Okouma M, Czmil-Garon J, Pape E, Petitpain N, Gillet P. Drug-induced aseptic meningitis: a mini-review. Fundam Clin Pharmacol. 2018;32:252–60.
Marinac JS. Drug- and chemical-induced aseptic meningitis: a review of the literature. Ann Pharmacother. 1992;26:813–22.
Schwartz N, Stock AD, Putterman C. Neuropsychiatric lupus: new mechanistic insights and future treatment directions. Nat Rev Rheumatol. 2019;15:137–52.
Morgan A, Clark D. CNS Adverse Effects of Nonsteroidal Anti-Inflammatory Drugs : Therapeutic Implications. CNS Drugs. 1998;9:281–90.
Barreto EF, Feely MA. Can NSAIDs Be Used Safely for Analgesia in Patients with CKD?: PRO. Kidney360. 2020;1: 1184–1188.
Schjerning A-M, McGettigan P, Gislason G. Cardiovascular effects and safety of (non-aspirin) NSAIDs. Nat Rev Cardiol. 2020;17:574–84.
• Ho KY, Cardosa MS, Chaiamnuay S, Hidayat R, Ho HQT, Kamil O, et al. Practice Advisory on the Appropriate Use of NSAIDs in Primary Care. J Pain Res. 2020;13:1925–39. Practical guidance for the use of NSAIDs in cardiovascular and GI risk patients.
Asghar MS, Hansen AE, Kapijimpanga T, van der Geest RJ, van der Koning P, Larsson HBW, et al. Dilation by CGRP of middle meningeal artery and reversal by sumatriptan in normal volunteers. Neurology. 2010;75:1520–6.
Amin FM, Asghar MS, Ravneberg JW, de Koning PJH, Larsson HBW, Olesen J, et al. The effect of sumatriptan on cephalic arteries: A 3T MR-angiography study in healthy volunteers. Cephalalgia. 2013;33:1009–16.
Khan S, Amin FM, Christensen CE, Ghanizada H, Younis S, Olinger ACR, et al. Meningeal contribution to migraine pain: a magnetic resonance angiography study. Brain. 2019;142:93–102.
Velentgas P, Cole JA, Mo J, Sikes CR, Walker AM. Severe vascular events in migraine patients. Headache. 2004;44:642–51.
Hall GC, Brown MM, Mo J, MacRae KD. Triptans in migraine: the risks of stroke, cardiovascular disease, and death in practice. Neurology. 2004;62:563–8.
Bigal ME, Golden W, Buse D, Chen Y-T, Lipton RB. Triptan use as a function of cardiovascular risk. A population-based study Headache. 2010;50:256–63.
Razzaque Z, Pickard JD, Ma Q-P, Shaw D, Morrison K, Wang T, et al. 5-HT1B-receptors and vascular reactivity in human isolated blood vessels: assessment of the potential craniovascular selectivity of sumatriptan. Br J Clin Pharmacol. 2002;53:266–74.
Maassen Van Den Brink A, Saxena PR. Coronary vasoconstrictor potential of triptans: a review of in vitro pharmacologic data. Headache. 2004;44 Suppl 1: S13–9.
Ghanshani S, Chen C, Lin B, Duan L, Shen Y-JA, Lee M-S. Risk of Acute Myocardial Infarction, Heart Failure, and Death in Migraine Patients Treated with Triptans. Headache. 2020;60: 2166–2175.
•• Dodick D, Lipton RB, Martin V, Papademetriou V, Rosamond W, MaassenVanDenBrink A, et al. Consensus statement: cardiovascular safety profile of triptans (5-HT agonists) in the acute treatment of migraine. Headache. 2004;44:414–25. Comprehensive review of cardiovascular effects and safety of triptans.
• Diener H-C. The Risks or Lack Thereof of Migraine Treatments in Vascular Disease. Headache. 2020;60:649–53. Thought-provoking opinion statement about use of triptans in patients with cardiovascular disorders.
Leroux E, Rothrock J. Triptans for migraine patients with vascular risks: New insights, new options. Headache. 2019;59:1589–96.
Loder E, Burch R. What can data mining teach us about triptan safety that we don’t already know? Cephalalgia: an international journal of headache. 2014. pp. 3–4.
Evans RW, Martin V. Assessing cardiac risk prior to use of triptans. J Headache Pain. 2000. Available: https://headachejournal.onlinelibrary.wiley.com/doi/abs/; https://doi.org/10.1046/j.1526-4610.2000.00094.x?casa_token=0mMV-5fbIKwAAAAA:SpYV7ROJxWNxoX8CBP7ml7lFL06LvDVdtwEAGPYTezFY48CKv3fRkhLoV6LBt7Ll-6Pbce2ygJxAjQM.
ASCVD Risk Estimator +. In: American College of Cardiology [Internet]. [cited 31 May 2021]. Available: https://tools.acc.org/ascvd-risk-estimator-plus/#!/calculate/estimate/.
ACC/AHA CV Risk Calculator (2013). In: Medscape [Internet]. [cited 31 May 2021]. Available: https://reference.medscape.com/calculator/37/acc-aha-cv-risk-calculator-2013.
Bickerstaff E. BASILAR ARTERY MIGRAINE. Lancet. 1961;277:15–7.
Headache Classification Committee of the International Headache Society (IHS) The International Classification of Headache Disorders, 3rd edition. Cephalalgia. 2018;38: 1–211.
Klapper J, Mathew N, Nett R. Triptans in the treatment of basilar migraine and migraine with prolonged aura. Headache. 2001;41:981–4.
Di Stefano V, Rispoli MG, Pellegrino N, Graziosi A, Rotondo E, Napoli C, et al. Diagnostic and therapeutic aspects of hemiplegic migraine. J Neurol Neurosurg Psychiatry. 2020;91:764–71.
Ducros A. Reversible cerebral vasoconstriction syndrome. Lancet Neurol. 2012;11:906–17.
Kato Y, Hayashi T, Mizuno S, Horiuchi Y, Ohira M, Tanahashi N, et al. Triptan-induced Reversible Cerebral Vasoconstriction Syndrome: Two Case Reports with a Literature Review. Intern Med. 2016;55:3525–8.
Nguyen TQ, Lewis JH. Sumatriptan-associated ischemic colitis: case report and review of the literature and FAERS. Drug Saf. 2014;37:109–21.
Baron EP. Headache, cerebral aneurysms, and the use of triptans and ergot derivatives. Headache. 2015;55:739–47.
Evans RW. The FDA alert on serotonin syndrome with combined use of SSRIs or SNRIs and Triptans: an analysis of the 29 case reports. MedGenMed. 2007;9:48.
Isbister GK, Buckley NA, Whyte IM. Serotonin toxicity: a practical approach to diagnosis and treatment. Medical Journal of Australia. 2007. pp. 361–365. https://doi.org/10.5694/j.1326-5377.2007.tb01282.x.
• Gillman PK. Triptans, serotonin agonists, and serotonin syndrome (serotonin toxicity): a review. Headache. 2010;50:264–72. Review of mechanisms of serotonin toxicity that addresses common misconceptions.
Pilgrim JL, Gerostamoulos D, Drummer OH. Deaths involving serotonergic drugs. Forensic Sci Int. 2010;198:110–7.
Isbister GK, Buckley NA. The pathophysiology of serotonin toxicity in animals and humans: implications for diagnosis and treatment. Clin Neuropharmacol. 2005;28:205–14.
Orlova Y, Rizzoli P, Loder E. Association of Coprescription of Triptan Antimigraine Drugs and Selective Serotonin Reuptake Inhibitor or Selective Norepinephrine Reuptake Inhibitor Antidepressants With Serotonin Syndrome. JAMA Neurol. 2018;75:566–72.
Robblee JV, Butterfield RJ, Kang AM, Smith JH. Triptan and ergotamine overdoses in the United States: Analysis of the National Poison Data System. Neurology. 2020;94:e1460–9.
Evans RW, Tepper SJ, Shapiro RE, Sun-Edelstein C, Tietjen GE. The FDA alert on serotonin syndrome with use of triptans combined with selective serotonin reuptake inhibitors or selective serotonin-norepinephrine reuptake inhibitors: American Headache Society position paper. Headache. 2010;50:1089–99.
Ansari H, Ziad S. Drug-Drug Interactions in Headache Medicine. Headache. 2016;56:1241–8.
Ferrari MD, Färkkilä M, Reuter U, Pilgrim A, Davis C, Krauss M, et al. Acute treatment of migraine with the selective 5-HT1F receptor agonist lasmiditan – A randomised proof-of-concept trial. Cephalalgia. 2010;30:1170–8.
Vila-Pueyo M. Targeted 5-HT1F Therapies for Migraine. Neurotherapeutics. 2018;15:291–303.
Pearlman EM, Wilbraham D, Dennehy EB, Berg PH, Tsai M, Doty EG, et al. Effects of lasmiditan on simulated driving performance: Results of two randomized, blinded, crossover studies with placebo and active controls. Hum Psychopharmacol. 2020;35: e2732.
Drugs for migraine. The Medical Letter. The Medical Letter: Inc; 2020. p. 61.
Krege JH, Rizzoli PB, Liffick E, Doty EG, Dowsett SA, Wang J, et al. Safety findings from Phase 3 lasmiditan studies for acute treatment of migraine: Results from SAMURAI and SPARTAN. Cephalalgia. 2019;39:957–66.
Finch A, Pillans P. P-glycoprotein and its role in drug-drug interactions. Aust Prescr. 2014;37:137–9.
Szkutnik-Fiedler D. Pharmacokinetics, Pharmacodynamics and Drug-Drug Interactions of New Anti-Migraine Drugs-Lasmiditan, Gepants, and Calcitonin-Gene-Related Peptide (CGRP) Receptor Monoclonal Antibodies. Pharmaceutics. 2020;12. https://doi.org/10.3390/pharmaceutics12121180.
de Vries T, Villalón CM, MaassenVanDenBrink A. Pharmacological treatment of migraine: CGRP and 5-HT beyond the triptans. Pharmacol Ther. 2020;211: 107528.
Lasmiditan. 11 Oct 2019 [cited 31 May 2021]. Available: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211280s000lbl.pdf.
Tsai M, Case M, Ardayfio P, Hochstetler H, Wilbraham D. Effects of Lasmiditan on Cardiovascular Parameters and Pharmacokinetics in Healthy Subjects Receiving Oral Doses of Propranolol. Clin Pharmacol Drug Dev. 2020;9:629–38.
van Hoogstraten WS, MaassenVanDenBrink A. The need for new acutely acting antimigraine drugs: moving safely outside acute medication overuse. J Headache Pain. 2019;20:54.
Rau JC, Navratilova E, Oyarzo J, Johnson KW, Aurora SK, Schwedt TJ, et al. Evaluation of LY573144 (lasmiditan) in a preclinical model of medication overuse headache. Cephalalgia. 2020;40:903–12.
Schiff PL. Ergot and its alkaloids. Am J Pharm Educ. 2006;70:98.
Tfelt-Hansen P, Saxena PR, Dahlöf C, Pascual J, Láinez M, Henry P, et al. Ergotamine in the acute treatment of migraine: a review and European consensus. Brain. 2000;123(Pt 1):9–18.
Silberstein SD, Shrewsbury SB, Hoekman J. Dihydroergotamine (DHE) - Then and Now: A Narrative Review. Headache. 2020;60:40–57.
Dahlöf C, Maassen Van Den Brink A. Dihydroergotamine, ergotamine, methysergide and sumatriptan - basic science in relation to migraine treatment. Headache. 2012;52: 707–714.
D.H.E. 45 (dihydroergotamine mesylate). 31 Jul 2002 [cited 31 May 2021]. Available: https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/005929s044lbl.pdf.
Tfelt-Hansen P, Saxena PR. Ergot alkaloids in the acute treatment of migraines. 3rd ed. In: Jes Olesen, Peter J. Goadsby, Nabih M. Ramadan, Peer Tfelt-Hansen, K. Michael A. Welch, editor. The Headaches. 3rd ed. Lippincott Williams & Wilkinz 2006;459–467.
Srisuma S, Lavonas EJ, Wananukul W. Ergotism and factitious hypotension associated with interaction of ergotamine with CYP3A4 inhibitors. Clin Toxicol. 2014;52:674–7.
Cytochrome P450 3A (including 3A4) inhibitors and inducers. 2021 [cited 29 May 2021]. Available: https://www.uptodate.com/contents/image?imageKey=CARD%2F76992&topicKey=ONC%2F4621&source=see_link.
Silberstein SD, Young WB. Safety and efficacy of ergotamine tartrate and dihydroergo t amine in the treatment of migraine and status migrainosus. special article Neurology. 1995;45:577–584.
Saper JR, Silberstein S. Pharmacology of dihydroergotamine and evidence for efficacy and safety in migraine. Headache. 2006;46(Suppl 4):S171–81.
Impel NeuroPharma Announces U.S. FDA Approval of TRUDHESATM (Dihydroergotamine Mesylate) Nasal Spray for the Acute Treatment of Migraine. 3 Sep 2021 [cited 13 Dec 2021]. Available: https://impelnp.com/2021/09/03/impel-neuropharma-announces-u-s-fda-approval-of-trudhesa-dihydroergotamine-mesylate-nasal-spray-for-the-acute-treatment-of-migraine/.
Aurora S, Jeleva M, Hocevar-Trnka J, Hoekman J, Shrewsbury S. A long-term, open-label study of safety and tolerability of precision olfactory delivery (POD®) of DHE in acute migraine (STOP 301): Clinical results. [cited 24 May 2021]. Available: https://impelnp.com/wp-content/uploads/2020/10/PAINWeek_2020_STOP-301-Clinical-Results-Poster_Final_8-25-20_FINAL.pdf.
Clinical Trials. [cited 14 Dec 2021]. Available: https://www.satsumarx.com/our-research/clinical-trials/.
Hauser JM, Azzam JS, Kasi A. Antiemetic Medications. StatPearls. Treasure Island (FL): StatPearls Publishing. 2020.
Marmura MJ. Use of dopamine antagonists in treatment of migraine. Curr Treat Options Neurol. 2012;14:27–35.
Friedman BW. Managing Migraine. Ann Emerg Med. 2017;69:202–7.
Wijemanne S, Jankovic J, Evans RW. Movement Disorders From the Use of Metoclopramide and Other Antiemetics in the Treatment of Migraine. Headache. 2016;56:153–61.
Savitt D, Jankovic J. Tardive syndromes. J Neurol Sci. 2018;389:35–42.
Rao AS, Camilleri M. Review article: metoclopramide and tardive dyskinesia. Aliment Pharmacol Ther. 2010;31:11–9.
Al-Saffar A, Lennernäs H, Hellström PM. Gastroparesis, metoclopramide, and tardive dyskinesia: Risk revisited. Neurogastroenterol Motil. 2019;31: e13617.
McGeeney BE. Dopamine antagonists and migraine. Drug Dev Res. 2007;68:341–5.
Isbister GK, Dawson A, Whyte IM. Citalopram overdose, serotonin toxicity, or neuroleptic malignant syndrome? Canadian journal of psychiatry. Revue canadienne de psychiatrie. 2001;657–659.
Gillman PK. The serotonin syndrome and its treatment. J Psychopharmacol. 1999;13:100–9.
Freedman SB, Uleryk E, Rumantir M, Finkelstein Y. Ondansetron and the risk of cardiac arrhythmias: a systematic review and postmarketing analysis. Ann Emerg Med. 2014;64:19-25.e6.
Lee DY, Trinh T, Roy SK. Torsades de Pointes after Ondansetron Infusion in 2 Patients. Tex Heart Inst J. 2017;44:366–9.
QTDrugs Lists (registration required) :: Crediblemeds. [cited 13 Dec 2021]. Available: https://crediblemeds.org/druglist.
Hargreaves R, Olesen J. Calcitonin Gene-Related Peptide Modulators - The History and Renaissance of a New Migraine Drug Class. Headache. 2019;59:951–70.
Edvinsson L, Warfvinge K. Recognizing the role of CGRP and CGRP receptors in migraine and its treatment. Cephalalgia. 2019;39:366–73.
Russell FA, King R, Smillie S-J, Kodji X, Brain SD. Calcitonin gene-related peptide: physiology and pathophysiology. Physiol Rev. 2014;94:1099–142.
Biohaven Pharmaceutical Holding Company Ltd. Biohaven to Advance Vazegepant into Phase 3 for the Acute Treatment of Migraine Following Successful End of Phase 2 Meeting with FDA. 23 Mar 2020 [cited 23 May 2021]. Available: https://www.prnewswire.com/news-releases/biohaven-to-advance-vazegepant-into-phase-3-for-the-acute-treatment-of-migraine-following-successful-end-of-phase-2-meeting-with-fda-301027933.html.
FDA approves QULIPTATM (atogepant), the first and only oral CGRP receptor antagonist specifically developed for the preventive treatment of migraine. [cited 14 Dec 2021]. Available: https://news.abbvie.com/news/press-releases/fda-approves-qulipta-atogepant-first-and-only-oral-cgrp-receptor-antagonist-specifically-developed-for-preventive-treatment-migraine.htm.
FDA Approves Biohaven’s NURTEC® ODT (rimegepant) for Prevention: Now the First and Only Migraine Medication for both Acute and Preventive Treatment. [cited 29 May 2021]. Available: https://www.biohavenpharma.com/investors/news-events/press-releases/05-27-2021.
Ankrom W, Bondiskey P, Li C-C, Palcza J, Liu W, Dockendorf MF, et al. Ubrogepant Is Not Associated With Clinically Meaningful Elevations of Alanine Aminotransferase in Healthy Adult Males. Clin Transl Sci. 2020;13:462–72.
Goadsby PJ, Tepper SJ, Watkins PB, Ayele G, Miceli R, Butler M, et al. Safety and tolerability of ubrogepant following intermittent, high-frequency dosing: Randomized, placebo-controlled trial in healthy adults. Cephalalgia. 2019;39:1753–61.
Ivans A, Stringfellow J, Coric V, Croop R. Results of a Phase 1, Open-label, Single-dose, Parallel-group Study of Rimegepant 75 mg in Subjects with Hepatic Impairment (2126). Neurology. 2020;94. Available: https://n.neurology.org/content/94/15_Supplement/2126.
Ailani J, Lipton RB, Hutchinson S, Knievel K, Lu K, Butler M, et al. Long-Term Safety Evaluation of Ubrogepant for the Acute Treatment of Migraine: Phase 3, Randomized, 52-Week Extension Trial. Headache. 2020;60:141–52.
Croop R, Lipton RB, Kudrow D, Stock DA, Kamen L, Conway CM, et al. Oral rimegepant for preventive treatment of migraine: a phase 2/3, randomised, double-blind, placebo-controlled trial. Lancet. 2021;397:51–60.
Hutchinson S, Silberstein SD, Blumenfeld AM, Lipton RB, Lu K, Yu SY, et al. Safety and efficacy of ubrogepant in participants with major cardiovascular risk factors in two single-attack phase 3 randomized trials: ACHIEVE I and II. Cephalalgia. 2021; 3331024211000311.
Hutchinson S, Schim J, Lipton R, Croop R, Jensen C, Thiry A, Stock E, Conway C, Lovegren M, Coric V, Hanna M. Oral rimegepant 75 mg is safe and well tolerated in adults with migraine and cardiovascular risk factors: results of a multicenter, long-term, open-label safety study. Presented at: American Academy of Neurology 2021 Virtual Annual Meeting; April 17–21, 2021. Available: https://index.mirasmart.com/AAN2021/PDFfiles/AAN2021-001995.html.
Lipton RB, Croop R, Stock EG, Stock DA, Morris BA, Frost M, et al. Rimegepant, an Oral Calcitonin Gene-Related Peptide Receptor Antagonist, for Migraine. N Engl J Med. 2019;381:142–9.
Mulder IA, Li M, de Vries T, Qin T, Yanagisawa T, Sugimoto K, et al. Anti-migraine Calcitonin Gene-Related Peptide Receptor Antagonists Worsen Cerebral Ischemic Outcome in Mice. Ann Neurol. 2020;88:771–84.
Ubrelvy (ubrogepant) package insert. Madison, NJ: Allergan USA, Inc. Dec 2019 [cited 31 May 2020]. Available: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211765s000lbl.pdf.
Nurtec (rimegepant) package insert. New Haven, CT: Biohaven Pharmaceuticals, Inc. Feb 2020 [cited 31 May 2021]. Available: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212728s000lbl.pdf.
Qulipta (atogepant) package insert. North Chicago, Ill.: AbbVie Inc. Available: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/215206Orig1s000lbl.pdf.
Mao Q, Unadkat JD. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport–an update. AAPS J. 2015;17:65–82.
Tamaki H, Satoh H, Hori S, Ohtani H, Sawada Y. Inhibitory effects of herbal extracts on breast cancer resistance protein (BCRP) and structure-inhibitory potency relationship of isoflavonoids. Drug Metab Pharmacokinet. 2010;25:170–9.
Inhibitors and inducers of P-glycoprotein (P-gp) drug efflux pump (P-gp multidrug resistance transporter). In: UpToDate® [Internet]. 2021 [cited 31 May 2021]. Available: https://www.uptodate.com/contents/image/print?imageKey=EM%2F73326&topicKey=RHEUM%2F1666&source=see_link.
Kalliokoski A, Niemi M. Impact of OATP transporters on pharmacokinetics. Br J Pharmacol. 2009;158:693–705.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Abigail Chua declares receiving payments as speaker for Biohaven, Amgen, and Eli Lilly.
Sandhya Mehla and Yulia Orlova declare no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Episodic Migraine
Rights and permissions
About this article

Cite this article
Orlova, Y.Y., Mehla, S. & Chua, A.L. Drug Safety in Episodic Migraine Management in Adults Part 1: Acute Treatments. Curr Pain Headache Rep 26, 481–492 (2022). https://doi.org/10.1007/s11916-022-01057-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11916-022-01057-3