
Abstract
Purpose of Review
The development of adiposity in the bone marrow, known as marrow adipose tissue (MAT), is often associated with musculoskeletal frailty. Glucocorticoids, which are a key component of the biological response to stress, affect both bone and MAT. These molecules signal through receptors such as the glucocorticoid receptor (GR), but the role of the GR in regulation of MAT is not yet clear from previous studies. The purpose of this review is to establish and determine the role of GR-mediated signaling in marrow adiposity by comparing and contrasting what is known against other energy-storing tissues like adipose tissue, liver, and muscle, to provide better insight into the regulation of MAT during times of metabolic stress (e.g., dietary challenges, aging).
Recent Findings
GR-mediated glucocorticoid signaling is critical for proper storage and utilization of lipids in cells such as adipocytes and hepatocytes and proteolysis in muscle, impacting whole-body composition, energy utilization, and homeostasis through a complex network of tissue cross talk between these systems. Loss of GR signaling in bone promotes increased MAT and decreased bone mass.
Summary
GR-mediated signaling in the liver, adipose tissue, and muscle is critical for whole-body energy and metabolic homeostasis, and both similarities and differences in GR-mediated GC signaling in MAT as compared with these tissues are readily apparent. It is clear that GC-induced pathways work together through these tissues to affect systemic biology, and understanding the role of bone in these patterns of tissue cross talk may lead to a better understanding of MAT-bone biology that improves treatment strategies for frailty-associated diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The skeleton facilitates biomechanical mobility and protection for the body, with a dynamic nature regulated by the coordinated turnover activity of osteoblasts and osteoclasts. This constant restructuring of the skeleton regulates the shape, size, and density of bone, and also facilitates bone’s role as a storehouse of minerals that helps maintain homeostatic calcium and phosphate levels in the blood. In contrast to these well understood roles for the mineralized matrix of bone, the functional versatility of the bone marrow tissue within the medullary cavity is often underappreciated. Bone marrow, in addition to serving as the niche for hematopoiesis, also serves a partially metabolic function with fat deposition starting at birth and gradually accumulating throughout life [1]. This accumulation of marrow adipocytes, or marrow adipose tissue (MAT), is inversely related to hematopoietic and osteoblastic cell abundance, decreasing the volume of bone forming cells [2,3,4]. Although MAT shows a positive correlation with bone mass at early ages, it is negatively associated with metrics of bone mass like bone mineral density (BMD) later in life and is often associated with the development of age-related osteoporosis [5, 6].
Systemic factors that control bone remodeling activity include endogenous hormones and regulatory factors like melatonin, growth hormone (GH), parathyroid hormone (PTH), adiponectin, sclerostin (SOST), sex hormones, and glucocorticoids [6,7,8]. The effects of these factors on bone mass have been studied extensively. For example, low androgen and estrogen levels both contribute to bone loss in men, and estrogen deficiency in females after menopause clearly increases the likelihood of osteoporosis [1, 9]. Similarly, gonadectomy and growth hormone deficiency both negatively affect bone mass and size, whereas melatonin can be beneficial for skeletal integrity [10,11,12]. These same factors can also affect bone marrow adiposity. As one example, osteocyte-secreted SOST, which inhibits bone formation, drives adipogenesis in bone marrow [13]. By regulating adipocyte differentiation and lipid storage in bone marrow, hormonal factors can have both local/paracrine and systemic/endocrine effects on bone formation. This review focuses on our current understanding of marrow fat, with particular attention to the role of glucocorticoid signaling in the regulation of marrow adiposity and osteogenesis.
Marrow Adipose Tissue
Adipocytes are an energy storehouse, but can also be a prime contributor to metabolic and cardiovascular diseases. Visceral deposition of white adipose tissue (WAT), is associated with several diseases [14]. In contrast, brown adipose tissue (BAT) primarily functions in thermogenesis and can be considered beneficial [15]. In light of these characterizations, the question of whether marrow adipose tissue (MAT) is “good” or “bad” is somewhat perplexing, because MAT encompasses features of both WAT and BAT depending upon site, functional demand, age, and disease [3, 16,17,18]. The concept of constitutive and regulated bone marrow adipose tissue (cMAT and rMAT, respectively), as first described by Scheller et al., highlights some of these differences [19]. The cMAT that appears early in life, found in the distal tibia, tail vertebrae, and paws of rodent models, gives marrow a characteristic yellow appearance and shares biological similarity with white adipose tissue (WAT) [20]. In contrast, rMAT develops in the proximal tibia and femur in response to nutritional and environmental changes, and is also influenced by genetic and endocrine factors. This rMAT is related to pathophysiology of skeletal frailty starting even in early life (reviewed in [21]). Physiologic variation of rMAT is also strongly evident in cold exposure and caloric restriction, the latter of which may be partly under the influence of sympathetic tone as MAT abundance during caloric restriction can be reduced via treatment with propranolol [19, 22•, 23]. Interestingly, rMAT shares characteristics of both WAT and BAT, while its role in disease states is not yet fully understood. Therefore, careful classification of MAT, including comparing the quality and quantity of these fat types as different states of marrow adiposity, is important to describe its relationship to stages and progression of skeletal frailty.
Despite the negative association between MAT and bone mass, it is important to recognize that a small amount of fat in the marrow may be beneficial. Some studies show that fatty acids (FA) such as palmitate and l-carnitine are an important fuel source for osteoblasts [24,25,26]; studies in mice showed that defects in FA uptake and catabolism by osteoblasts impaired postnatal bone acquisition, increased body fat, and reduced global energy expenditure [27, 28]. Bone, and its marrow microenvironment, may be a major source of fat metabolism for the body, as well as a site of fat synthesis [29, 30], meaning that bone is well positioned to be a “combustion engine” for fat within marrow. However, long-chain saturated FA, particularly palmitate, also induce autophagy and apoptosis in osteoblasts [31]. Lipotoxicity from these FA causes insulin resistance and defects in insulin receptor signaling, further aggravating energy homeostasis and promoting more marrow fat depositions [32]. Work recently presented at the American Society for Bone and Mineral Research annual meeting demonstrated that genetic deletion of PPAR-gamma under the control of the mesenchymal lineage Prx1 promoter eliminated cMAT and rMAT in the bone, and led to an increase in cortical and trabecular bone mass, bone formation, and bone strength [33]. This parallels an earlier report where deletion of PPAR-gamma with Col1 − 3.6 kb Cre increased trabecular bone mass and metrics of osteoblastic bone formation in 6-month-old mice [34], as well as a recent report indicating that treatment with an inhibitor of FA synthase (cerulenin) prevented fatty infiltration of the bone marrow and bone loss induced by ovariectomy [35], and a study showing that treatment of skeletally mature (9-month-old) male mice with the PPAR-gamma antagonist bisphenol-A-diglycidyl ether (BADGE) increased bone volume, improved bone quality, and increased metrics of osteoblastogeneiss while decreasing marrow adiposity [36], all of which suggest a deleterious role for MAT in skeletal homeostasis. However, it is important to note that work presented at the same meeting demonstrated that neither genetic deletion of PPAR-gamma with Dermo1-Cre nor treatment of aged mice with the PPAR-gamma antagonist GW9662 protected against aging-related trabecular bone loss [37, 38]. Thus, the role of PPAR-gamma-expressing marrow adipocytes, particularly in the pathogenesis of aging-related bone loss, remains somewhat unresolved.
Glucocorticoids
Glucocorticoids (GC) are a component of stress responses and circadian rhythm, playing a critical role in the regulation of plasma glucose during times of stress by promoting metabolic homeostasis in multiple cell types, for example increasing gluconeogenesis in the liver or decreasing uptake and utilization of glucose in WAT and skeletal tissues [39, 40]. GC also affect endocrine and immune function through genomic and non-genomic pathways, resulting primarily in modulation of anti-inflammatory systems. Exogenous GC have been used for decades in this capacity to treat a variety of diseases, although they can engender a multitude of tissue-specific side effects including growth retardation, osteoporosis, and muscle atrophy in musculoskeletal system when used at high dosages or for an extended period of time.
In the context of the skeleton, while much attention has been paid to the negative effects of exogenous, pharmacologic GC effects in bone (e.g., glucocorticoid-induced osteoporosis, or GIO), it is important to note that endogenous GC are both necessary and important for proper bone cell function [41]. However, dysregulated glucocorticoid signaling may also contribute to age-related bone loss through effects on osteoblastic senescence and osteoprogenitor lineage selection (i.e., affecting the ratio of osteoblastic versus adipogenic differentiation of progenitor cells) even in young subjects [42,43,44,45]. Local activation of endogenous GC is enhanced during aging; in humans, this contributes to increased cortisol levels and alterations in circadian cortisol variation, both of which may be associated with bone frailty [46,47,48].
GC-induced signaling can be mediated through glucocorticoid receptor (GR, also known as the gene Nr3c1) in target tissues, affecting the transcription of multiple genes, either by the GR interacting with other transcription factors directly or by occupying the glucocorticoid response element (GRE) in the promoter regions of target genes as a GR–GR dimer complex or GR-transcription factor complex. Regulation of GC activity can be initiated in the brain, resulting in modulation of both circulating levels of GC and their biological activity. In addition, local regulation of GC activity and downstream signaling is possible through the activity of enzymes like 11β-hydroxysteroid dehydrogenase types 1 and 2 (Hsd11b1 and Hsd11b2, respectively), which catalyze the local conversion between inactive GC (cortisone in humans and 11-dehydrocorticosterone or 11-DHC in rodents) and active GC (cortisol in humans and corticosterone in rodents) (Fig. 1). Tight regulation of each of these factors is necessary to maintain proper homeostasis. Importantly, alterations in either GC level or activity, as occurs with aging, dietary changes, and other biological challenges, can have direct downstream effects on the skeletal system through bone cells (reviewed in [41, 49]).

“Classical” GR signaling: Conversion between inactive and active forms of glucocorticoids (GC) is catalyzed by hydroxysteroid dehydrogenase enzymes Hsd11b1 and Hsd11b2. The active GC binds the GR, which then translocates to nucleus to bind to DNA, inducing or affecting various transcriptional activities. 11-DHC: 11-Dehydrocorticosterone. Hsd11b1: 11β-Hydroxysteroid dehydrogenase type 1, Hsd11b2: 11β-Hydroxysteroid dehydrogenase type 2
The role of endogenous GC in bone marrow, and specifically in MAT, is less clear. Along with understanding the function of GC in bone marrow adipocytes, the role of GC-based signaling in osteoblastic lipid storage and utilization, and the possible contribution of these mechanisms to fat storage in bone marrow, must be carefully considered, as previous research suggests that osteoblast-lineage cells store lipids under the influence of GC signaling, and that this process is enhanced by aging [30]. It is possible that insights into the role of endogenous GCs and their activity in the bone marrow niche may be better appreciated through understanding the role of endogenous GC signaling in other tissues capable of lipid infiltration, utilization, and storage, such as traditional adipose depots (WAT and BAT), liver, and muscle [30, 50]. In the next section, we will summarize known effects of GR-mediated signaling in these systems, particularly with regard to the metabolic responses to biological stressors such as dietary modification (caloric restriction and high fat diet) and aging. These effects will be contrasted against what is known with regard to endogenous GC and in the regulation of bone and bone marrow adiposity.
Tissue-Specific Knockouts of the GR in Fat, Liver, and Skeletal Muscle
Adipocytes
Several recent studies have investigated the role of GR-based signaling in adipocytes through the creation of adipocyte-specific GR-knockout models. Mouse embryonic fibroblasts isolated from GR-knockout mice (created with GR-floxed mice and actin-Cre) formed fat pads, but tissues and adipocytes formed were smaller as compared with cells isolated from wildtype mice [51]. This indicates that while GR signaling is not required for adipogenesis, it plays an important role in processes of adipocyte differentiation and growth. With regard to cellular metabolism, postnatal deletion of the GR in adiponectin-expressing adipocytes (via a tamoxifen-inducible adiponectin-Cre) promoted stress-induced lipid storage in fat tissue and decreased lipolysis during fasting (as measured by expression of adipose triglyceride lipase) [52]. Similarly, loss of GR in adiponectin-expressing adipocytes (with a constitutive adiponectin-Cre) prevented WAT from undergoing lipolysis due to defects in adrenergic signal transduction [53•]. This altered homeostasis on a whole-body scale, where adipocyte-specific GR knockouts were protected against systemic fat deposition in models of aging and high-fat diet (HFD) administration, but adversely responded to caloric restriction as seen by a disproportionate loss of lean mass because muscle tissues were broken down when fat could not be metabolized as an energy source during fasting. Accordingly, GR-based signaling in adipocytes was deemed essential for proper substrate mobilization in response to metabolic challenges [53•]. These seemingly disparate systemic effects between HFD and caloric restriction in adipocyte-specific GR-knockout mice highlight the important, interconnected metabolic biology of organ systems such as adipose tissue and liver, where adipocyte-specific GR-knockout mice were protected against HFD-induced hepatic steatosis due to decreased circulating FA load from impaired adipocyte lipolysis [53•]. Likewise, adipocyte-specific GR-knockout mice were protected against hepatic steatosis induced by corticosterone treatment, likely by promoting increased storage and sequestration of lipids in adipose tissues [52]. However, some debate exists, as one study found that deletion of the GR in adiponectin-expressing adipocytes was not protective against increases in weight, fat mass, adipocyte size, or hepatic lipid content in mice subjected to a high-fat–high-sucrose diet (HFSD) [54], and another study found that adipocyte-specific GR-knockout mice responded similarly to a HFD in terms of weight gain and fat pad histology as compared with wildtype mice [55]. Still, taken together, these studies suggest that GR expression and downstream signaling in adipocytes is an essential component of the body’s response to metabolic stressors. The role of GR-mediated signaling in adipose tissues under non-stressed conditions, however, is still a matter of debate. Despite reported differences in the observed responses to stressful challenges, it is interesting to note that several independent studies reported that major phenotypic differences between adipocyte-specific GR-knockout mice and wildtype mice were not seen under baseline (non-stressed) conditions. Indeed, ~ 6-month-old male adiponectin-Cre-driven GR-knockout mice showed no differences in body weight, fat mass, lean mass, or percent fat as compared with wildtype littermates on a normal chow diet [53•, 54,55,56].
Hepatocytes
In the liver, glucocorticoid signaling is implicated in various stages of nonalcoholic fatty liver deposition [57]; however, loss of GR in hepatocytes surprisingly induced liver dysfunction in mice including the development of fatty liver under normal and HFD feeding conditions [58]. There is some disagreement about this phenomenon, however, as another study of hepatocytic GR deficiency found no differences in liver triglyceride content under non-stressed conditions [56]. As seen in adipocyte-specific knockouts of the GR, changes in GR-mediated signaling in the liver may also cause compensatory changes elsewhere in the body. For example, a hepatocyte-specific knockout of the GR (created with albumin-Cre) triggered increased gluconeogenesis activity in the kidney, although no major changes in body weight, lean mass, or fat mass were seen [56]. However, upon treatment with the GR agonist dexamethasone, insulin sensitivity was improved and overall fat mass was decreased in these liver-specific GR-knockout mice [56]. This suggests that GR-mediated signaling in the liver, like in adipose tissue, is important for whole body energy and metabolic homeostasis.
Myocytes
Fatty infiltration of skeletal muscle is associated with musculoskeletal frailty [59,60,61], similar to how increasing fat storage in bone marrow is associated with poor skeletal outcomes. This process is correlated to the inflammatory profile of muscle tissue in obesity and ageing [62, 63]. At the opposite end of the spectrum from obesity, increased intramyocellular lipid storage is also seen in cancer cachexia despite an overall reduction in total body fat mass with increased lipolysis, driven in part by inflammatory mediators like TNF-alpha [64, 65]. Intramyocellular protein and lipid metabolism is regulated by glucocorticoids [66, 67], although the specific role of the GR in myosteatosis related to intramyocellular lipid accumulation has not been addressed in previous studies. However, it is interesting to note that intermuscular fat, attributed to the presence of adipocytes within the muscle tissue, is less sensitive to GC than other depots like subcutaneous and visceral WAT, and expresses lower levels of the GR than these other depots [68]. GR-mediated signaling does play a major role in muscle structure and biology. Muscle hypertrophy was observed in muscle-specific knockout mouse models of the GR created using skeletal muscle actin (Acta1)-Cre and fast myosin light chain (Mlcf)-Cre, but did not occur when the knockout was created with muscle creatinine kinase (Mck)-Cre [69,70,71,72]. Interestingly, this effect on muscle mass may occur across species, as a recent report of a GR-deficient zebrafish model (created with CRISPR-Cas9 technology) also presented with increased muscle mass [73]. As with liver- and fat-specific GR knockouts, systemic changes elsewhere in the body were induced by GR knockout in the muscle. For example, knockout of the GR with skeletal muscle actin (Acta1)-Cre produced smaller adipose tissues due to drastic shift of energy utilization in liver and adipose tissue [69], and GR-deficient zebrafish demonstrated increases in whole-body protein and lipid content [73].
Bone and Bone Marrow
Mice with deletion of the GR under the control of a macrophage-specific promoter (deleting the GR in osteoclast-lineage cells) demonstrated few differences as compared with wildtype mice in the absence of treatment with exogenous GC [74]. One of the first studies reporting the effects of an osteoblast-lineage-specific GR knockout (created using Runx2-Cre) demonstrated that GR-deficient mice presented with reduced trabecular bone mass [75]. Surprisingly, these bone-targeted GR knockouts showed no changes in histological indices of osteoblast nor osteoclast abundance that could explain the low trabecular bone mass, although primary osteoblasts from these mice were impaired in their ability to proliferate and produce a mineralized matrix [75]. These results provided support for the idea that GR-mediated signaling is important for promoting osteoblast expansion under the influence of endogenous GC, but the response of MAT to loss of the GR in this study was not reported [75]. A recent report demonstrated that the GR is critical for proper fracture healing, but again did not address the role of MAT in the skeletal response to loss of the GR [76]. To address this gap in knowledge, we recently developed a mouse model with conditional knockout of the GR in osteoprogenitors using Osx1-Cre, subjecting these mice to either ad libitum or caloric restriction feeding to determine the effects of GR deficiency in the regulation of bone and MAT [77••]. These GR-deficient mice presented with reduced cortical and trabecular bone mass under both feeding conditions, resulting from decreased bone formation activity. The abundance of MAT was drastically increased in GR-deficient mice under both ad libitum and caloric restriction conditions, and surprisingly, treatment with the β-adrenergic receptor antagonist propranolol was unable to rescue CR-induced marrow fat in either WT or GR-deficient mice [23, 77••]. The mechanism for increased MAT in these GR-deficient mice is not yet known, but may reflect processes such as impaired MAT lipolysis (as in adipocyte-specific knockouts of the GR) [52, 53•], altered mesenchymal progenitor cell differentiation patterns, or altered cross talk between the bone and other body systems. Notably, unlike the adipocyte- and liver-targeted GR-knockout models described above, the effects of caloric restriction on body mass and fat mass were not exacerbated nor altered in osteoprogenitor-specific GR-knockout mice, but this does not rule out the possibility for cross talk between the skeleton and other body systems that is mediated by GR-based signaling in bone [77••]. It will be of interest to determine how these bone-specific GR-knockout mice respond to the stress of aging and other metabolic challenges. From a clinical perspective, a recent study found that higher adrenal secretion of endogenous GC, while still in the range of what is considered normal and physiological, was associated with higher bone marrow fat content in children [43].
Summary and Future Directions
In understanding the role of GC and the GR in MAT biology, it must also be acknowledged that GC can bind other receptors that facilitate signaling outside of the “classical” GR action. It has been reported that GC bind the mineralocorticoid receptor (MR) at up to 10-fold higher affinity than the GR [78], and that the MR may mediate some of the harmful actions of exogenous GC in bone [79]. Likewise, the “GR” actually presents in two possible confirmations: GR-alpha, and GR-beta, with GR-alpha mediating the classically known effects of the GR as described above and GR-beta demonstrating an inhibitory role [80, 81]. The MR, GR-alpha, and GR-beta are all expressed in the skeletal system [82], although the specific roles of each of these receptors in MAT biology are largely unknown.
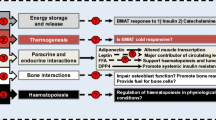
Reflecting upon the role of the GR- and GC-induced signaling in the body systems described above, similarities and differences among tissues are readily apparent (Fig. 2). Several independent studies support the assertion that endogenous GC-induced and GR-mediated signaling is important for lipolysis in fat [52, 53•, 55] and proteolysis in muscle [70,71,72,73], suggesting commensurate induction of energy metabolism responses between different tissues. However, during fasting, GR-mediated signaling induces lipolysis in WAT, whereas MAT expands during caloric restriction, although this may occur independently from GR action [53•, 77••]. Regardless, it is clear that these GC-induced pathways work together to affect systemic biology, such as how the muscle-liver-fat signaling axis is well established for lipolytic action in adipose tissue through GR [69]. In future studies, it will be critical to determine the molecular mechanisms by which GR-mediated endogenous GC signaling affects the biology of bone and MAT. This may, in turn, lead to a better understanding of the skeleton’s role in the systemic response to endogenous glucocorticoids, and how these processes affect whole body homeostasis during metabolic challenges such as altered diet, stress, and aging.

Reported effects of tissue specific GR knockout (KO), including potential pathways for tissue cross talk
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Hawkes CP, Mostoufi-Moab S. Fat-bone interaction within the bone marrow milieu: impact on hematopoiesis and systemic energy metabolism. Bone. 2019;119:57–64.
Sulston RJ, Cawthorn WP. Bone marrow adipose tissue as an endocrine organ: close to the bone? Horm Mol Biol Clin Invest. 2016;28:21–38.
Veldhuis-Vlug AG, Rosen CJ. Clinical implications of bone marrow adiposity. J Intern Med. 2018;283:121–39.
Li J, Li J, Zuo B, Zhang L, Dai L, Zhang X. Osteoblast versus adipocyte: bone marrow microenvironment-guided epigenetic control. Case Rep Orthop Res. 2018;1:2–18.
Newton LA, Hanks JL, Davis M, Casazza K. The relationships among total body fat, bone mineral content and bone marrow adipose tissue in early-pubertal girls. Bonekey Rep. 2013;2:315.
Rharass T, and Lucas S (2018) Mechanisms in endocrinology: bone marrow adiposity and bone, a bad romance? Eur J Endocrinol
Li T, Jiang S, Lu C, Yang W, Yang Z, Hu W, et al. Melatonin: another avenue for treating osteoporosis? J Pineal Res. 2019;66:e12548.
Yang S, Liu H, Liu Y, Liu L, Zhang W, and En L (2019) Effect of adiponectin secreted from adipose-derived stem cells on bone-fat balance and bone defect healing. J Tissue Eng Regen Med
Manolagas SC, O’Brien CA, Almeida M. The role of estrogen and androgen receptors in bone health and disease. Nat Rev Endocrinol. 2013;9:699–712.
Zhang XZ, Kalu DN, Erbas B, Hopper JL, Seeman E. The effects of gonadectomy on bone size, mass, and volumetric density in growing rats are gender-, site-, and growth hormone-specific. J Bone Miner Res Off J Am Soc Bone Miner Res. 1999;14:802–9.
Bao T, Zeng L, Yang K, Li Y, Ren F, Zhang Y, et al. Can melatonin improve the osteopenia of perimenopausal and postmenopausal women? A meta-analysis. Int J Endocrinol. 2019;2019:5151678.
Ikegame M, Hattori A, Tabata MJ, Kitamura KI, Tabuchi Y, Furusawa Y, et al. Melatonin is a potential drug for the prevention of bone loss during space flight. J Pineal Res. 2019;67:e12594.
Fairfield H, Falank C, Harris E, Demambro V, McDonald M, Pettitt JA, et al. The skeletal cell-derived molecule sclerostin drives bone marrow adipogenesis. J Cell Physiol. 2018;233:1156–67.
Tchernof A, Despres JP. Pathophysiology of human visceral obesity: an update. Physiol Rev. 2013;93:359–404.
Bartelt A, Bruns OT, Reimer R, Hohenberg H, Ittrich H, Peldschus K, et al. Brown adipose tissue activity controls triglyceride clearance. Nat Med. 2011;17:200–5.
Devlin MJ, Rosen CJ. The bone-fat interface: basic and clinical implications of marrow adiposity. Lancet Diabetes Endocrinol. 2015;3:141–7.
Hardouin P, Rharass T, Lucas S. Bone marrow adipose tissue: to be or not to be a typical adipose tissue? Front Endocrinol (Lausanne). 2016;7:85.
Ghali O, Al Rassy N, Hardouin P, Chauveau C. Increased bone marrow adiposity in a context of energy deficit: the tip of the iceberg? Front Endocrinol (Lausanne). 2016;7:125.
Scheller EL, Doucette CR, Learman BS, Cawthorn WP, Khandaker S, Schell B, et al. Region-specific variation in the properties of skeletal adipocytes reveals regulated and constitutive marrow adipose tissues. Nat Commun. 2015;6:7808.
Li Y, Meng Y, Yu X. The unique metabolic characteristics of bone marrow adipose tissue. Front Endocrinol (Lausanne). 2019;10:69.
Scheller EL, Cawthorn WP, Burr AA, Horowitz MC, MacDougald OA. Marrow adipose tissue: trimming the fat. Trends Endocrinol Metab. 2016;27:392–403.
• Cawthorn WP, Scheller EL, Parlee SD, Pham HA, Learman BS, Redshaw CM, et al. Expansion of bone marrow adipose tissue during caloric restriction is associated with increased circulating glucocorticoids and not with hypoleptinemia. Endocrinology. 2016;157:508–21 This study provided some of the first evidence that GC may drive MAT expansion during caloric restriction.
Baek K, Bloomfield S. A. (2012) Blocking beta-adrenergic signaling attenuates reductions in circulating leptin, cancellous bone mass, and marrow adiposity seen with dietary energy restriction. J Appl Physiol. 1985;113:1792–801.
Hooshmand S, Balakrishnan A, Clark RM, Owen KQ, Koo SI, Arjmandi BH. Dietary l-carnitine supplementation improves bone mineral density by suppressing bone turnover in aged ovariectomized rats. Phytomedicine. 2008;15:595–601.
Adamek G, Felix R, Guenther HL, Fleisch H. Fatty acid oxidation in bone tissue and bone cells in culture. Characterization and hormonal influences. Biochem J. 1987;248:129–37.
Dirckx N, Moorer MC, Clemens TL, Riddle RC. The role of osteoblasts in energy homeostasis. Nat Rev Endocrinol. 2019;15:651–65.
Frey JL, Li Z, Ellis JM, Zhang Q, Farber CR, Aja S, et al. Wnt-Lrp5 signaling regulates fatty acid metabolism in the osteoblast. Mol Cell Biol. 2015;35:1979–91.
Kim SP, Li Z, Zoch ML, Frey JL, Bowman CE, Kushwaha P, et al. Fatty acid oxidation by the osteoblast is required for normal bone acquisition in a sex- and diet-dependent manner. JCI Insight. 2017;2:92704.
Rendina-Ruedy E, Guntur AR, Rosen CJ. Intracellular lipid droplets support osteoblast function. Adipocyte. 2017;6:250–8.
McGee-Lawrence ME, Carpio LR, Schulze RJ, Pierce JL, McNiven MA, Farr JN, et al. Hdac3 deficiency increases marrow adiposity and induces lipid storage and glucocorticoid metabolism in osteochondroprogenitor cells. J Bone Miner Res Off J Am Soc Bone Miner Res. 2016;31:116–28.
Gunaratnam K, Vidal C, Gimble JM, Duque G. Mechanisms of palmitate-induced lipotoxicity in human osteoblasts. Endocrinology. 2014;155:108–16.
Wei J, Ferron M, Clarke CJ, Hannun YA, Jiang H, Blaner WS, et al. Bone-specific insulin resistance disrupts whole-body glucose homeostasis via decreased osteocalcin activation. J Clin Invest. 2014;124:1–13.
Tommasini S, Nelson T, Faulkner-Filosa C, Rodeheffer MS, Rosen C, Lindskog D, and Horowitz M (2018) Selective deletion of marrow adipocytes leads to increased mesenchymal precursors, a shift in lineage allocation, and increased bone mass with improved bone biomechanics. In ASBMR Annual Meeting, American Society for Bone and Mineral Research, Montreal, Quebec, Canada
Cao J, Ou G, Yang N, Ding K, Kream BE, Hamrick MW, et al. Impact of targeted PPARgamma disruption on bone remodeling. Mol Cell Endocrinol. 2015;410:27–34.
Bermeo S, Al Saedi A, Vidal C, Khalil M, Pang M, Troen BR, et al. Treatment with an inhibitor of fatty acid synthase attenuates bone loss in ovariectomized mice. Bone. 2019;122:114–22.
Duque G, Li W, Vidal C, Bermeo S, Rivas D, Henderson J. Pharmacological inhibition of PPARgamma increases osteoblastogenesis and bone mass in male C57BL/6 mice. J Bone Miner Res Off J Am Soc Bone Miner Res. 2013;28:639–48.
Rosario R, Baban B, Hamrick M, Isales C, Shi X. PPARγ in inflammation and aging. Denver: In ASBMR Annual Meeting, American Society for Bone and Mineral Research; 2017.
Rosario R, Ajith A, Ding K, Elsayed R, Su Y, Horuzsko A, et al. Complex role for PPARγ in bone, inflammation and immune function in aging animals. In: In ASBMR Annual Meeting. Montreal: American Society for Bone and Mineral Research; 2018.
Kuo T, McQueen A, Chen TC, Wang JC. Regulation of glucose homeostasis by glucocorticoids. Adv Exp Med Biol. 2015;872:99–126.
Lee RA, Harris CA, Wang JC. Glucocorticoid receptor and adipocyte biology. Nucl Receptor Res. 2018;5:101373.
Zhou H, Cooper MS, Seibel MJ. Endogenous glucocorticoids and bone. Bone Res. 2013;1:107–19.
Vande Berg BC, Malghem J, Lecouvet FE, Devogelaer JP, Maldague B, Houssiau FA. Fat conversion of femoral marrow in glucocorticoid-treated patients: a cross-sectional and longitudinal study with magnetic resonance imaging. Arthritis Rheum. 1999;42:1405–11.
Esche J, Shi L, Hartmann MF, Schonau E, Wudy SA, Remer T. Glucocorticoids and body fat inversely associate with bone marrow density of the distal radius in healthy youths. J Clin Endocrinol Metab. 2019;104:2250–6.
Canalis E, Mazziotti G, Giustina A, Bilezikian JP. Glucocorticoid-induced osteoporosis: pathophysiology and therapy. Osteoporos Int. 2007;18:1319–28.
Hartmann K, Koenen M, Schauer S, Wittig-Blaich S, Ahmad M, Baschant U, et al. Molecular actions of glucocorticoids in cartilage and bone during health, disease, and steroid therapy. Physiol Rev. 2016;96:409–47.
Zhao ZY, Lu FH, Xie Y, Fu YR, Bogdan A, Touitou Y. Cortisol secretion in the elderly. Influence of age, sex and cardiovascular disease in a Chinese population. Steroids. 2003;68:551–5.
Johar H, Emeny RT, Bidlingmaier M, Reincke M, Thorand B, Peters A, et al. Blunted diurnal cortisol pattern is associated with frailty: a cross-sectional study of 745 participants aged 65 to 90 years. J Clin Endocrinol Metab. 2014;99:E464–8.
Reynolds RM, Dennison EM, Walker BR, Syddall HE, Wood PJ, Andrew R, et al. Cortisol secretion and rate of bone loss in a population-based cohort of elderly men and women. Calcif Tissue Int. 2005;77:134–8.
Henneicke H, Gasparini SJ, Brennan-Speranza TC, Zhou H, Seibel MJ. Glucocorticoids and bone: local effects and systemic implications. Trends Endocrinol Metab. 2014;25:197–211.
Hamrick MW, McGee-Lawrence ME, Frechette DM. Fatty infiltration of skeletal muscle: mechanisms and comparisons with bone marrow adiposity. Front Endocrinol (Lausanne). 2016;7:69.
Bauerle KT, Hutson I, Scheller EL, Harris CA. Glucocorticoid receptor signaling is not required for in vivo adipogenesis. Endocrinology. 2018;159:2050–61.
Dalle H, Garcia M, Antoine B, Boehm V, Do TTH, Buyse M, et al. Adipocyte glucocorticoid receptor deficiency promotes adipose tissue expandability and improves the metabolic profile under corticosterone exposure. Diabetes. 2019;68:305–17.
• Mueller KM, Hartmann K, Kaltenecker D, Vettorazzi S, Bauer M, Mauser L, et al. Adipocyte glucocorticoid receptor deficiency attenuates aging- and HFD-induced obesity and impairs the feeding-fasting transition. Diabetes. 2017;66:272–86 This study demonstrates a key role for GR-mediated GC signaling in adipocyte lipolysis.
Desarzens S, Faresse N. Adipocyte glucocorticoid receptor has a minor contribution in adipose tissue growth. J Endocrinol. 2016;230:1–11.
Shen Y, Roh HC, Kumari M, Rosen ED. Adipocyte glucocorticoid receptor is important in lipolysis and insulin resistance due to exogenous steroids, but not insulin resistance caused by high fat feeding. Mol Metab. 2017;6:1150–60.
Bose SK, Hutson I, Harris CA. Hepatic glucocorticoid receptor plays a greater role than adipose gr in metabolic syndrome despite renal compensation. Endocrinology. 2016;157:4943–60.
Woods CP, Hazlehurst JM, Tomlinson JW. Glucocorticoids and non-alcoholic fatty liver disease. J Steroid Biochem Mol Biol. 2015;154:94–103.
John K, Marino JS, Sanchez ER, Hinds TD Jr. The glucocorticoid receptor: cause of or cure for obesity? Am J Physiol Endocrinol Metab. 2016;310:E249–57.
Tuttle LJ, Sinacore DR, Mueller MJ. Intermuscular adipose tissue is muscle specific and associated with poor functional performance. J Aging Res. 2012;2012:172957.
Choi SJ, Files DC, Zhang T, Wang ZM, Messi ML, Gregory H, et al. Intramyocellular lipid and impaired myofiber contraction in normal weight and obese older adults. J Gerontol A Biol Sci Med Sci. 2016;71:557–64.
Reinders I, Murphy RA, Brouwer IA, Visser M, Launer L, Siggeirsdottir K, et al. Muscle quality and myosteatosis: novel associations with mortality risk: the Age, Gene/Environment Susceptibility (AGES)-Reykjavik study. Am J Epidemiol. 2016;183:53–60.
Khan IM, Perrard XY, Brunner G, Lui H, Sparks LM, Smith SR, et al. Intermuscular and perimuscular fat expansion in obesity correlates with skeletal muscle T cell and macrophage infiltration and insulin resistance. Int J Obes. 2015;39:1607–18.
Addison O, Drummond MJ, LaStayo PC, Dibble LE, Wende AR, McClain DA, et al. Intramuscular fat and inflammation differ in older adults: the impact of frailty and inactivity. J Nutr Health Aging. 2014;18:532–8.
Seelaender MC, Batista ML. Adipose tissue inflammation and cancer cachexia: the role of steroid hormones. Horm Mol Biol Clin Invest. 2014;17:5–12.
Stephens NA, Skipworth RJ, Macdonald AJ, Greig CA, Ross JA, Fearon KC. Intramyocellular lipid droplets increase with progression of cachexia in cancer patients. J Cachexia Sarcopenia Muscle. 2011;2:111–7.
Morgan SA, Gathercole LL, Simonet C, Hassan-Smith ZK, Bujalska I, Guest P, et al. Regulation of lipid metabolism by glucocorticoids and 11beta-HSD1 in skeletal muscle. Endocrinology. 2013;154:2374–84.
Morgan SA, Hassan-Smith ZK, Doig CL, Sherlock M, Stewart PM, Lavery GG. Glucocorticoids and 11beta-HSD1 are major regulators of intramyocellular protein metabolism. J Endocrinol. 2016;229:277–86.
Chu W, Wei W, Han H, Gao Y, Liu K, Tian Y, et al. Muscle-specific downregulation of GR levels inhibits adipogenesis in porcine intramuscular adipocyte tissue. Sci Rep. 2017;7:510.
Shimizu N, Maruyama T, Yoshikawa N, Matsumiya R, Ma Y, Ito N, et al. A muscle-liver-fat signalling axis is essential for central control of adaptive adipose remodelling. Nat Commun. 2015;6:6693.
de Theije CC, Schols A, Lamers WH, Ceelen JJM, van Gorp RH, Hermans JJR, et al. Glucocorticoid receptor signaling impairs protein turnover regulation in hypoxia-induced muscle atrophy in male mice. Endocrinology. 2018;159:519–34.
Watson ML, Baehr LM, Reichardt HM, Tuckermann JP, Bodine SC, Furlow JD. A cell-autonomous role for the glucocorticoid receptor in skeletal muscle atrophy induced by systemic glucocorticoid exposure. Am J Physiol Endocrinol Metab. 2012;302:E1210–20.
Hu Z, Wang H, Lee IH, Du J, Mitch WE. Endogenous glucocorticoids and impaired insulin signaling are both required to stimulate muscle wasting under pathophysiological conditions in mice. J Clin Invest. 2009;119:3059–69.
Faught E, Vijayan MM. Loss of the glucocorticoid receptor in zebrafish improves muscle glucose availability and increases growth. Am J Physiol Endocrinol Metab. 2019;316:E1093–104.
Kim HJ, Zhao H, Kitaura H, Bhattacharyya S, Brewer JA, Muglia LJ, et al. Glucocorticoids suppress bone formation via the osteoclast. J Clin Invest. 2006;116:2152–60.
Rauch A, Seitz S, Baschant U, Schilling AF, Illing A, Stride B, et al. Glucocorticoids suppress bone formation by attenuating osteoblast differentiation via the monomeric glucocorticoid receptor. Cell Metab. 2010;11:517–31.
Rapp AE, Hachemi Y, Kemmler J, Koenen M, Tuckermann J, Ignatius A. Induced global deletion of glucocorticoid receptor impairs fracture healing. FASEB J: official publication of the Federation of American Societies for Experimental Biology. 2018;32:2235–45.
•• Pierce JL, Ding KH, Xu J, Sharma AK, Yu K, Del Mazo Arbona N, Rodriguez-Santos Z, Bernard P, Bollag WB, Johnson MH, Hamrick MW, Begun DL, Shi XM, Isales CM, and McGee-Lawrence ME (2019) The glucocorticoid receptor in osteoprogenitors regulates bone mass and marrow fat. J Endocrinol. This study was the first to show that loss of the GR in bone promoted increased MAT deposition.
Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, et al. Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science. 1987;237:268–75.
Fumoto T, Ishii KA, Ito M, Berger S, Schutz G, Ikeda K. Mineralocorticoid receptor function in bone metabolism and its role in glucocorticoid-induced osteopenia. Biochem Biophys Res Commun. 2014;447:407–12.
Hinds TD Jr, Ramakrishnan S, Cash HA, Stechschulte LA, Heinrich G, Najjar SM, et al. Discovery of glucocorticoid receptor-beta in mice with a role in metabolism. Mol Endocrinol. 2010;24:1715–27.
Oakley RH, Cidlowski JA. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol. 2013;132:1033–44.
Beavan S, Horner A, Bord S, Ireland D, Compston J. Colocalization of glucocorticoid and mineralocorticoid receptors in human bone. J Bone Miner Res Off J Am Soc Bone Miner Res. 2001;16:1496–504.
Funding
The authors are supported by funding provided by the National Institute on Aging (NIA AG036675) and the American Diabetes Association (1-16-JDF-062).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Anuj K. Sharma, Xingming Shi, Carlos M. Isales, and Meghan E. McGee-Lawrence declare no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Bone Marrow and Adipose Tissue
Rights and permissions
About this article

Cite this article
Sharma, A.K., Shi, X., Isales, C.M. et al. Endogenous Glucocorticoid Signaling in the Regulation of Bone and Marrow Adiposity: Lessons from Metabolism and Cross Talk in Other Tissues. Curr Osteoporos Rep 17, 438–445 (2019). https://doi.org/10.1007/s11914-019-00554-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-019-00554-6