
Abstract
During embryonic and fetal development much of the skeleton initiates as a cartilaginous scaffold, which is progressively resorbed and replaced by bone. Endochondral bone formation continues until the growth plates fuse during puberty. At all life stages adequate delivery of mineral is required for the skeleton to achieve and maintain appropriate mineral content and strength. During fetal development the placenta actively transports calcium, phosphorus, and magnesium. Postnatally passive and then active absorption from the intestines becomes the main supply of minerals to the skeleton. Animal and human data indicate that fetal bone development requires parathyroid hormone (PTH) and PTH-related protein but not vitamin D/calcitriol, calcitonin, or (possibly) sex steroids. During the postnatal period, when intestinal calcium absorption becomes an active process, skeletal development begins to depend upon vitamin D/calcitriol but this requirement can be bypassed by increasing the calcium content of the diet or by administering intermittent calcium infusions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Skeletal patterning begins in the embryo but it is in the fetal and neonatal periods that bone formation and mineralization accelerate. The placenta actively transports calcium, magnesium, and phosphorus to appropriately mineralize the skeleton before birth. With loss of the placental pump at birth, the neonate becomes dependent upon intestinal absorption of minerals. Initially, the intestines absorb calcium passively but with increased postnatal age this becomes active and vitamin D-dependent. The calciotropic hormones play different roles during the fetal and neonatal periods in regulating skeletal development and mineralization.
Overview of Fetal and Neonatal Mineral Metabolism
Within the developing human, serum calcium, ionized calcium, magnesium, and phosphorus are raised above maternal values [1••]. Studies in fetal mice suggest that these high mineral concentrations are needed for normal skeletal accretion of minerals [2, 3•, 4] but not for fetal survival to term [2, 5, 6].
The fetal circulation in humans and other mammals is characterized by low levels of parathyroid hormone, 1,25-dihydroxyvitamin D (calcitriol), and fibroblast growth factor-23, and high levels of parathyroid hormone–related protein (PTHrP) and calcitonin [1••], [7]. 25-hydroxyvitamin D [25(OH)D] readily crosses the placenta and results in cord blood 25(OH)D levels that are within 75% to 100% of the maternal value at term [8, 9, 10•]. The low calcitriol levels are due to suppression of the renal 1α-hydroxylase (CYP27b1) by high serum calcium and phosphorus, and low parathyroid hormone. But if fetal hyperparathyroidism is present then fetal calcitriol levels markedly increase [11].
Animal studies have shown that absence of parathyroids, parathyroid hormone, or PTHrP each cause fetal hypocalcemia and hyperphosphatemia [1••, 2, 3•, 5, 6], whereas absence of calcitonin [4], vitamin D [12–14], calcitriol [15], or the vitamin D receptor [16] do not disturb fetal blood calcium or phosphorus.
Minerals enter the fetus predominantly via the placenta, whereas kidneys and intestines enable calcium to be voided into the amniotic fluid and then swallowed and reabsorbed. PTHrP regulates placental calcium and possibly magnesium transfer, although parathyroid hormone (PTH) may also play a role [3•, 5, 17–19]. During the interval of active calcium transfer the placenta markedly upregulates expression of genes involved in calcium transport, and the fetal skeleton accretes mineral rapidly [16, 20–22]. Human skeletons accrete 80% of required calcium in the third trimester [23], whereas rats attain 95% of skeletal calcium during the last 5 days of gestation [24].
At birth the total and ionized calcium levels fall due to loss of the placental calcium infusion and calciotropic hormones, and a breathing-induced rise in blood pH [25]. Phosphorus increases over the same interval and then declines. PTH rises to near adult normal values by 24 to 48 h after birth and this precedes an increase in calcitriol [1••].
The neonate is dependent upon the intestines for supply of minerals. Calcium absorption in newborns is largely passive and nonsaturable [26, 27]. The high lactose content of milk increases paracellular diffusion of calcium in the distal small bowel and net bioavailability of dietary calcium in humans [28, 29] and rodents [30]. As the neonate matures, passive absorption of calcium declines, enterocytes upregulate expression of the vitamin D receptor and associated calcium-transporting genes and proteins, and calcium absorption becomes active and vitamin D-dependent [26, 31, 32]. This developmentally programmed maturation of the neonatal intestines explains why preterm babies do not respond to calcitriol but are dependent upon passive absorption of mineral until they are more mature.
Overview of Endochondral Bone Development
Early patterning of the embryonic skeleton is dependent upon a multitude of signaling pathways, which include Hox genes, Wnts, Hedgehogs, bone morphogenetic proteins, fibroblast growth factors, Notch/Delta, and other factors [33•]. Mesenchymal cells are laid down in spatiotemporal patterns where bones of the axial and appendicular skeletons will form. These osteochondral progenitors become osteoblasts at the site of intramembranous bone formation (vault of the skull and a few other bones), whereas in the bulk of the developing skeleton they become chondroblasts that initiate endochondral bone formation.
By 8 weeks of gestation in humans a complete cartilaginous scaffold with digits and joints is present. The scaffold of each long bone lengthens at both ends, with the oldest cells nearer the center undergoing differentiation, hypertrophy, and then apoptosis. Chondroclasts and osteoclasts resorb the apoptosed chondrocytes and surrounding matrix, vascular invasion occurs, and osteoblasts lay down the primary spongiosa (osteoid) that will become mineralized. These primary ossification centers form in the vertebrae and long bones between the 8th to 12th weeks, but it is not until the third trimester that the bulk of skeletal mineralization occurs. Resorption and remodeling of the primary spongiosa to create secondary spongiosa occurs in utero and resorption will abnormally increase when the fetus is stressed by maternal hypocalcemia. At the 34th week of gestation, secondary ossification centers form in the femurs—creating true growth plates—but otherwise most epiphyses are cartilaginous until after birth [34]. The growth plates become fused during puberty, after which linear growth is no longer possible.
Role of PTHrP
PTHrP is produced by the perichondrium and proliferating chondrocytes, whereas the PTH/PTHrP receptor (PTH1R) is expressed further down the growth plate within pre-hypertrophic chondrocytes [35, 36, 37•]. The critical role that PTHrP plays in regulating endochondral bone formation was first made evident by Pthrp-null fetuses, which have a chondrodysplasia characterized by dwarfed long bones and deformed growth plates [38]. Further study has determined that PTHrP acts locally to delay terminal differentiation and hypertrophy of chondrocytes. Without PTHrP, hypertrophy begins early and bone formation starts before the cartilaginous template has reached its intended length; consequently, the long bones are shortened [37•, 39, 40]. Conversely, overexpression of PTHrP or expression of an activating mutation of the PTH1R within fetal chondrocytes results in delayed chondrocyte hypertrophy, and skeletons that are largely cartilaginous at birth [41, 42]. PTHrP and Indian hedgehog interact in a negative feedback loop that determines the length of the columns of proliferative chondrocytes, and PTHrP also interacts with the Wnt signaling pathway to determine the lifespan and fate of chondrocytes [37•, 40].
Preosteoblasts and osteoblasts express PTHrP and the PTH1R, and thus one might expect the Pthrp-null skeleton to show decreased expression in osteoblast-specific genes and reduced bone formation and mineralization. However, Pthrp-null growth plates have normal expression of collagen α1(I) and collagenase-3, and upregulated expression of osteocalcin and osteopontin [38, 43]. Acceleration of skeletal maturation in the Pthrp-null results in bones becoming mineralized in utero that normally do not mineralize until after birth, and the normally cartilaginous parts of the ribs and sternum also become bone [38]. Pthrp nulls have threefold higher levels of PTH than normal [2] and this hyperparathyroidism likely rescues any deficit in osteoblast function that loss of PTHrP might otherwise cause. Further studies in mice lacking PTH or the PTH1R support this notion (see below).
Targeted deletion of PTHrP within preosteoblasts and osteoblasts leads to reduced bone formation and low bone mass at 6 weeks of age, confirming that osteoblast-derived PTHrP regulates bone formation postnatally [44]. However, the fetal skeletons of these mice have not been examined and thus it remains unknown whether osteoblast-derived PTHrP is important for fetal bone formation.
Pthrp-null fetuses lack circulating PTHrP, are hypocalcemic (ionized calcium reduced to the maternal level), and have a lower rate of placental calcium transfer [2, 5]. Despite these changes and shorter long bones, the Pthrp-null skeleton has a normal ash weight and content of calcium, magnesium, and phosphorus [2, 5]. Why is skeletal mineral content not reduced? The Pthrp-null skeleton accretes more mineral than normal due to its accelerated and abnormal calcification of skeletal structures, and thus its mineral content cannot be meaningfully compared with the normal skeleton.
Overall, the evidence is that PTHrP participates in fetal skeletal development by upregulating placental calcium transfer, maintaining a high serum calcium, determining the fate of chondrocytes within the scaffold, and possibly regulating preosteoblasts and osteoblasts.
Role of PTH
There is mixed evidence for parathyroid hormone’s role in regulating endochondral bone formation and mineralization. Aparathyroid Hoxa3-null fetuses in a Black Swiss background have low skeletal mineral content but otherwise show no abnormality of endochondral bone development or expression of osteoblast-specific genes including collagen α1(I), collagenase-3, osteocalcin, and osteopontin [2, 6]. They are markedly hypocalcemic with an ionized calcium reduced well below the maternal level, whereas the serum phosphorus is increased above the fetal norm [6]. Placental calcium transport and circulating PTHrP levels are normal [2, 6]. It is evident that, despite its normally low circulating levels in the fetus, PTH plays an important role in regulating serum minerals and skeletal mineral accretion.
Double mutants lacking Hoxa3 and Pthrp have an undermineralized form of the Pthrp-null chondrodysplasia and are globally smaller than either single mutant [2]. These findings confirm that the chondrodysplasia seen in Pthrp nulls is largely due to absence of PTHrP and not the compensatory secondary hyperparathyroidism, whereas undermineralization of the skeleton results from loss of parathyroids and/or PTH [2].
In contrast to the aparathyroid Hoxa3 phenotype, Pth-null fetuses in a C57BL/6 background have slightly shortened tibial metaphyseal lengths, shorter metacarpals and metatarsals, smaller vertebrae, reduced trabecular bone volumes, and fewer osteoclasts and osteoblasts [45]. Modest changes are also seen in expression of genes involved in chondrocyte maturation and apoptosis, mineralization, and vascular invasion of the growth plate [45]. Overall, the structural changes are largely in bone parameters whereas the cartilaginous indices are little different from wild-type (wt) siblings. Pth/Pthrp double mutants are smaller and display an undermineralized form of Pthrp-null chondrodysplasia (similar to the Hoxa3/Pthrp double mutants) [45]. The data from this study indicate that PTH regulates bone formation in utero but not chondrocyte development.
A third study backcrossed Pth nulls and Gcm2 nulls (which have little or no circulating PTH) from the original inbred C57BL/6 strains into outbred Black Swiss to compare to Hoxa3-null fetuses [3•]. In both Pth and Gcm2 nulls the ionized calcium is reduced to the maternal level, equal to the Pthrp null but not to the extreme seen in the Hoxa3 null [2, 3•, 5]. Skeletons of both mutants are undermineralized but to a lesser extent than in Hoxa3 null [2, 3•]. There is no shortening of the long bones or alteration in trabecular bone volumes or skeletal morphology [3•]. However, studies of gene expression within the Pth-null and Gcm2-null growth plates were not done. It is possible that alterations in serum calcium (which is approximately 0.25 mmol/L higher in Black Swiss [5]), or genetic differences in background strain, may explain why some alterations in bone parameters are seen in Pth-null fetuses within one but not both background strains.
Placental calcium transfer is normal in Pth-null fetuses (the same as in Hoxa3 nulls) but upregulated significantly in Gcm2 nulls [3•]. Circulating PTHrP is normal in both mutants [3•]. Further study found that the PTH gene is expressed in wt placenta and upregulated in Gcm2-null placentas, whereas Pth-null placentas have reduced expression of genes involved in the transport of calcium and other cations [3•]. PTH may therefore regulate placental calcium and cation transport independent of PTHrP.
Overall, the evidence indicates that PTH maintains the fetal blood calcium at a level sufficient to facilitate mineral accretion by the developing skeleton. Within developing endochondral bones PTH does not control chondrocytes, whereas its effects on osteoblast development vary with the genetic background.
Role of PTHrP and PTH in Combination
Loss of the PTH1R blocks action of amino-terminal PTH and PTHrP, leading to a phenotype that combines aspects of Pthrp-null and Pth-null fetuses. These Pthr1-null fetuses are globally smaller than wt and display the Pthrp-null phenotype of accelerated endochondral ossification and dysplasia combined with the Hoxa3- or Pth-null phenotype of significant undermineralization of the skeleton [39]. The human equivalent of absence of the PTH1R, Blomstrand chondrodysplasia, shows similar features [46, 47].
There are notable differences between Pthrp- and Pthr1-null skeletons. Pthrp-null fetuses have normal expression of collagenase-3, upregulation of osteocalcin and osteopontin, and increased mineralization [43]. In contrast, Pthr1 fetuses have reduced expression of collagenase-3, osteocalcin, and osteopontin, and reduced mineralization [43]. This suggests that PTH upregulates expression of these osteoblast-specific genes in the Pthrp null, whereas blocking PTH action downregulates these genes in the Pthr1 nulls.
Pthr1-null fetuses have the lowest ionized calcium level, equivalent to Hoxa3/Pthrp double mutants [2, 5]. PTH and PTHrP evidently have additive roles in regulating the fetal blood calcium and skeletal mineralization because loss of both ligands or their common receptor leads to the greatest decline in blood calcium and skeletal mineral content [2]. However, it remains unclear why the ionized calcium is lower in aparathyroid Hoxa3-null fetuses than it is in Pth or Gcm2-null fetuses within the same genetic background. Placental calcium transfer is upregulated 50% in Pthr1-null fetuses, presumably reflecting the 11-fold increase in systemic PTHrP and its actions on a mid-molecular receptor [2, 5]. This increase is relative to placental blood flow as inferred from the diffusion of 51Cr-EDTA across the placenta; the absolute amount of 45Ca transferred to the Pthr1-null fetuses is reduced compared with their wt littermates, in keeping with their much smaller size and lower skeletal mineral content [48•].
Role of Estradiol
Sex steroids (estradiol and testosterone) circulate at low levels during fetal development [49] and the role that each might play in fetal calcium homeostasis and skeletal development is unclear. Mice lacking estrogen receptor α or β, or the aromatase, have normal skeletal lengths at birth and do not develop altered skeletal metabolism until later [50–52]. However, no studies have examined whether absence of sex steroids alters fetal calcium homeostasis or endochondral bone development and mineralization.
Role of Calcitonin
Calcitonin appears unimportant for fetal and neonatal skeletal development because in its absence the mutant fetuses have normal placental calcium transfer, ionized calcium, calciotropic hormone levels, endochondral bone development, gene expression, and mineral content [4]. However, serum magnesium and skeletal mineral content are both lower than normal, suggesting that absence of calcitonin disturbs magnesium homeostasis through unknown mechanisms [4].
Role of Vitamin D/Calcitriol
The role of vitamin D/calcitriol in regulating fetal and neonatal skeletal development has come under more intense interest in recent years and will be discussed in greater detail.
Animal Data
Severe vitamin D deficiency, ablation of vitamin D receptor, and absence of 1α-hydroxylase each result in normal skeletal lengths, morphology, ash weight, mineral content (by atomic absorption spectroscopy), and radiologic bone mineral content at term; moreover, the blood calcium, phosphorus, and PTH are normal. Animals examined include offspring of severely vitamin D-deficient rats [12–14], 1α-hydroxylase-null pigs [15], and both Vdr heterozygous and Vdr-null mice [16]. 1α-hydroxylase-null mice appear normal at birth but extensive fetal studies have not been reported [53, 54]. The normal, heterozygous, and Vdr-null fetuses born of Vdr heterozygous mothers are indistinguishable from each other, whereas heterozygous and null fetuses born of Vdr-null mothers are smaller and weigh less, but have normal mineral content after adjusting for smaller size [16].
The placenta evidently provides calcium to the fetus without requiring calcitriol, as suggested by studies in vitamin D-deficient rats [55] and Vdr-null fetuses [16]. Calbindin-D 9 K and Ca2+-ATPase both show normal placental expression in severely vitamin D-deficient and Vdr-null placentas [16, 55–57]. Placental calcium transfer and placental expression of PTHrP and the calcium channel TRPV6 are upregulated in placentas of Vdr-null mice [16].
It is in the weeks after birth—particularly after weaning—that hypocalcemia, hypophosphatemia, secondary hyperparathyroidism, and rickets begin to develop [12, 13, 15, 53, 54, 58, 59]. This parallels the developmental change of intestinal calcium absorption from a nonsaturable, passive process in the newborn to an active, saturable, calcitriol-dependent process [60, 61]. Additional animal data have shown that calcitriol’s role can be completely bypassed by using high-dose oral calcium or calcium infusions, resulting in a normal skeleton despite absence of vitamin D, vitamin D receptor, or 1α-hydroxylase [62–64].
Collectively these findings indicate that fetal calcium homeostasis, skeletal development, and mineralization do not require vitamin D, calcitriol, or its receptor. The animal studies predict that human babies born of vitamin D-replete and vitamin D-deficient mothers should be indistinguishable in blood calcium, phosphorus, skeletal morphology, and mineral content at birth. Calcitriol controls active intestinal calcium absorption in the older neonate but its role can be completely bypassed by increasing the calcium content of the diet.
Human Data: Observational Studies and Case Reports
A systematic study of newborns who died of obstetrical accidents found skeletal ash weight and mineral content (by atomic absorption spectroscopy) to be the same between neonates born of normal mothers versus those with extreme vitamin D deficiency and osteomalacia; moreover, there were no radiologic signs of rickets and centers of ossification were normal [65]. A few case reports indicate that skeletal changes suggestive of rickets (usually craniotabes) can be detected at birth [66–68], but other investigators have concluded that craniotabes is a nonspecific finding that should not be used to indicate the presence of rickets [69]. In multiple reports that describe craniotabes or rickets being present “at birth” the diagnosis was actually made within the first or second week [68, 70–74]. In one such case radiographic findings were absent at day 2 after birth but had developed by day 16 [74]. In many cases in which skeletal abnormalities were found soon after birth, the mothers had significant malnutrition, malabsorption (eg, celiac disease, pancreatic insufficiency), or very low intakes of both calcium and vitamin D [67, 68, 73]. Consequently, the skeletal changes may not have been due to vitamin D deficiency alone in cases of early neonatal rickets.
The reported global experience is that vitamin D-deficient rickets usually does not develop (or become recognized) until weeks to months after birth with a peak incidence between 6 and 18 months, even in regions where severe vitamin D deficiency during pregnancy is endemic and clinicians are especially vigilant for it [75–78]. The maturation of intestinal calcium absorption to a vitamin D-dependent process likely explains why vitamin D-deficient rickets does not usually develop until later.
Children with 1α-hydroxylase deficiency (vitamin D-dependent rickets type I; VDDR-I) or those lacking the vitamin D receptor (vitamin D-dependent rickets type II or hereditary vitamin D-resistant rickets; VDDR-II) are normal at birth [79–82]. In both conditions hypocalcemia, hypophosphatemia, and rickets eventually develop. VDDR-I presents late in the first year, whereas VDDR-II presents in the second year or later [79–82]. The deficiency in intestinal calcium absorption in VDDR-II patients can be bypassed by repeated calcium infusions or high oral dose calcium, thereby correcting the biochemical abnormalities and preventing or healing rickets [81, 83, 84]. Thus, the genetic disorders in humans confirm the animal data that hypocalcemia and rickets are not present at birth and can be prevented with calcium alone.
Congdon et al. [69] found that forearm bone mineral content, measured within 5 days of birth, did not differ by use of vitamin D supplementation during pregnancy. More recently, Weiler et al. [85•] measured bone mineral content of the lumbar spine, femur, and whole body within 15 days after birth in 50 healthy term infants. There was no difference in bone mineral content at any site of vitamin D-sufficient versus -insufficient whites, whereas the mineral content of the lumbar spine (but not whole body or femur) was lower in Asian and First Nations babies compared with whites. The authors concluded that genetic differences affected bone mineral content whereas vitamin D insufficiency did not [85•].
Human Data: Intervention Studies
Several randomized clinical trials of vitamin D supplementation during pregnancy found no effect on cord blood calcium or phosphorus, anthropometric measurements, or radiologic evidence of rickets [86–91]. This includes a study of 126 babies in which controls were severely vitamin D deficient (10 nmol/L 25(OH)D on cord blood), whereas babies from vitamin D-treated mothers had 25(OH)D levels of 138 nmol/L [86].
Hollis et al. [92••] recently completed two large randomized trials of vitamin D supplementation beginning at 12 to 16 weeks of pregnancy. In the first study, 494 women received 400, 2,000, or 4,000 IU of vitamin D3 daily, whereas in the second study, 257 women received 2,000 or 4,000 IU of vitamin D3 per day. Results of the first study have been recently published and show that use of 400 to 4,000 IU of vitamin D had no effect on gestational age at delivery, birth weight, mode of delivery, or need for level II or III neonatal care [92••]. Moreover, at a presentation to the Centers for Disease Control and Prevention in Atlanta, Wagner [93••] revealed that neither trial showed an effect on cord blood calcium, preterm birth, preterm labor, preeclampsia, or infection. The first study’s primary objective included measuring bone mineral density of the newborns but this was apparently not carried out. The limited published results clearly indicate the amount of maternal vitamin D intake required to achieve various target levels of 25(OH)D in mother and cord blood, but do not show any clinical benefit of higher intakes for mother or baby or provide any evidence as to which target level of 25(OH)D is required for optimal skeletal health of the fetus or neonate.
Beyond preventing rickets, at best a transient effect on bone mass has been seen through the use of vitamin D supplements during infancy. In one randomized study healthy term infants achieved 25(OH)D levels of 95 nmol/L on vitamin D versus 50 nmol/L on placebo. Bone mineral content of the radius and ulna was 23% higher than in the placebo group by 12 weeks of age but this difference vanished by 6 and 12 months [94, 95]. Another randomized study found breastfed infants achieved 25(OH)D levels of 92.4 nmol/L on vitamin D versus 47 nmol/L on placebo, but this did not affect bone mineral content at 3 or 6 months of age [96].
Human Data: Associational Studies
Several studies have examined associations between single measurements of serum 25(OH)D during pregnancy in the mother and various skeletal outcomes in the fetus, neonate, or child. In none of these was an association found with birth weight, skeletal lengths, and bone mineral density [10•, 97, 98•, 99•]. Morley et al. [97] measured 25(OH)D at 28 to 32 weeks in 374 mothers and reported in a subanalysis that a 25(OH)D level below 28 nmol/L was associated with a slightly shorter knee-heel length. However, the difference was not statistically significant after correcting for gestational age [97]. Mahon et al. [98•] measured maternal 25(OH)D levels at 36 weeks in 424 women and found that 25(OH)D levels below 50 nmol/L were associated with greater distal metaphyseal cross-sectional area and a novel parameter called the “femoral splaying index.” The greater cross-sectional area was inferred by the authors to represent early rachitic change. In contrast, Viljakainen et al. [10•] found that mothers above a median 25(OH)D level of 42.6 nmol/L had babies with a slightly greater tibial cross-sectional area. These authors interpreted the increased cross-sectional area to indicate stronger bone. The two studies exemplify how subjective the interpretation has been, with greater cross-sectional area of the metaphysis considered an adverse effect in one study and a benefit in another.
A well-publicized study by Javaid et al. [99•] found no associations between maternal serum 25(OH)D and birth weight, length, placental weight, abdominal circumference, head circumference, or cord blood calcium. At the 9-month follow-up there was still no association of maternal 25(OH)D with skeletal and anthropometric parameters in the infants. However, a maternal serum 25(OH)D level below 27.5 nmol/L during pregnancy was associated with a modestly lower bone mineral content in offspring at 9 years of age compared with offspring of mothers whose 25(OH)D levels were 50 nmol/L or higher. These findings have been used to promote the theory that vitamin D exposure during fetal development programs childhood peak bone mass [100].
There are significant caveats about these associational studies. Unstated are which radiologic parameters were prespecified and how many were examined before a single statistically significant finding was reported in each study. Why the tibia in one study and the femur in another, and not both sites? The possibility of chance findings cannot be excluded. Notably associational studies are hypothesis-generating and do not prove causality. Moreover, these associational studies are confounded by factors that contribute to low 25(OH)D levels, including maternal obesity, lower socioeconomic status, poorer nutrition, lack of exercise, prenatal care or vitamin supplementation, etc. Therefore, is lower 25(OH)D simply a marker of a less healthy pregnant woman? In the Javaid et al. [99•] study, much time elapsed between birth (when no effect was seen) and 9 years of age (when lower bone mineral content was found). Did low 25(OH)D in utero program lower bone mineral content at 9 years of age? Or does the lower maternal 25(OH)D status in late pregnancy indicate that lower socioeconomic status, poorer nutrition, and other factors in the mother remain unchanged and will be shared with the child?
Overall, the current evidence (reviewed in more detail in [101••]) indicates that the human fetus may suffer no skeletal problems as a consequence of vitamin D deficiency and insufficiency, but after birth hypocalcemia and progressive rickets will develop in those with severe vitamin D deficiency. It remains unclear whether 25(OH)D levels in the fetus or pregnant mother have a direct influence on neonatal or childhood bone mass. Large randomized controlled trials of vitamin D supplementation during pregnancy are needed to control the confounding and answer these questions. The American Academy of Pediatrics and the Institute of Medicine both concluded that a target 25(OH)D blood level of 50 nmol/L is appropriate for pediatric ages and to date there is no conclusive evidence that higher target 25(OH)D blood levels confer any additional benefit [102••].
Conclusions
At all life stages adequate delivery of mineral is required for the skeleton to achieve and maintain appropriate mineral content and strength (Fig. 1). The placenta supplies mineral to the fetus while passive and then active absorption from the intestines supplies the neonate. Fetal and postnatal bone development require PTH and PTH-related protein. When intestinal calcium absorption becomes an active process, skeletal development begins to depend upon vitamin D/calcitriol but this requirement can be bypassed by increasing the calcium content of the diet or by administering intermittent calcium infusions. Key messages of interest to clinicians are summarized in Table 1. Much remains unknown about the roles of the calciotropic hormones during fetal development, particularly potential skeletal and extraskeletal roles of vitamin D, and thus suggested priorities for future research are outlined in Table 2.

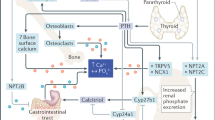
Relative roles of PTH, PTHrP, and calcitriol during fetal and neonatal life. The placenta is the main source of mineral during fetal life. PTH and PTHrP are expressed within the placenta but may also act on it from systemic sources to stimulate calcium transfer. The intestines are a trivial source of mineral in the fetus but are the main source for the neonate. Intestinal calcium absorption is initially passive but later becomes active, saturable, and calcitriol-dependent. Within the endochondral skeleton, PTHrP is produced by proliferating chondrocytes and perichondrial cells (arrowheads) and delays terminal differentiation of pre-hypertrophic chondrocytes. PTHrP is also produced within pre-osteoblasts and osteoblasts and stimulates bone formation (semicircular arrows). During fetal life PTHrP and PTH both maintain high blood calcium and phosphorus levels to facilitate mineralization. Calcitriol is not required to regulate blood calcium, endochondral bone formation, or skeletal mineralization in the fetus. PTH—parathyroid hormone; PTHrP—parathyroid hormone–related protein; PTH1R— parathyroid hormone receptor type 1 AKA PTH/PTHrP receptor
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
•• Kovacs CS: Fetal Mineral Homeostasis. In: Pediatric Bone: Biology and Diseases, 2nd Edition. Edited by Glorieux FH, Pettifor JM, Jüppner H. San Diego: Elsevier / Academic Press; 2011:247–275. In-depth, comprehensive review of the regulation of fetal mineral homeostasis using available animal and human data.
Kovacs CS, Chafe LL, Fudge NJ, et al. PTH regulates fetal blood calcium and skeletal mineralization independently of PTHrP. Endocrinology. 2001;142:4983–93.
• Simmonds CS, Karsenty G, Karaplis AC, Kovacs CS: Parathyroid Hormone Regulates Fetal-Placental Mineral Homeostasis. J Bone Miner Res 2010, 25:594–605. Studies in Pth-null and Gcm2-null fetuses clarify the role of PTH in regulating fetal mineral homeostasis and placental mineral transfer. PTH is expressed in the placenta.
McDonald KR, Fudge NJ, Woodrow JP, et al. Ablation of calcitonin/calcitonin gene related peptide-α impairs fetal magnesium but not calcium homeostasis. Am J Physiol Endocrinol Metab. 2004;287:E218–26.
Kovacs CS, Lanske B, Hunzelman JL, et al. Parathyroid hormone-related peptide (PTHrP) regulates fetal-placental calcium transport through a receptor distinct from the PTH/PTHrP receptor. Proc Natl Acad Sci U S A. 1996;93:15233–8.
Kovacs CS, Manley NR, Moseley JM, et al. Fetal parathyroids are not required to maintain placental calcium transport. J Clin Invest. 2001;107:1007–15.
Takaiwa M, Aya K, Miyai T, et al. Fibroblast growth factor 23 concentrations in healthy term infants during the early postpartum period. Bone. 2010;47:256–62.
Haddad Jr JG, Boisseau V, Avioli LV. Placental transfer of vitamin D3 and 25-hydroxycholecalciferol in the rat. J Lab Clin Med. 1971;77:908–15.
Seki K, Furuya K, Makimura N, et al. Cord blood levels of calcium-regulating hormones and osteocalcin in premature infants. J Perinat Med. 1994;22:189–94.
• Viljakainen HT, Saarnio E, Hytinantti T, et al.: Maternal vitamin D status determines bone variables in the newborn. J Clin Endocrinol Metab 2010, 95:1749–1757. This is associational study that found increased metaphyseal area with higher 25(OH)D levels and concluded that this means stronger bone; this is in contrast with the study by Mahon et al. [98•].
Kovacs CS, Ho-Pao CL, Hunzelman JL, et al. Regulation of murine fetal-placental calcium metabolism by the calcium-sensing receptor. J Clin Invest. 1998;101:2812–20.
Halloran BP, De Luca HF. Effect of vitamin D deficiency on skeletal development during early growth in the rat. Arch Biochem Biophys. 1981;209:7–14.
Miller SC, Halloran BP, DeLuca HF, Jee WS. Studies on the role of vitamin D in early skeletal development, mineralization, and growth in rats. Calcif Tissue Int. 1983;35:455–60.
Brommage R, DeLuca HF. Placental transport of calcium and phosphorus is not regulated by vitamin D. Am J Physiol. 1984;246:F526–9.
Lachenmaier-Currle U, Harmeyer J. Placental transport of calcium and phosphorus in pigs. J Perinat Med. 1989;17:127–36.
Kovacs CS, Woodland ML, Fudge NJ, Friel JK. The vitamin D receptor is not required for fetal mineral homeostasis or for the regulation of placental calcium transfer. Am J Physiol Endocrinol Metab. 2005;289:E133–44.
Care AD, Abbas SK, Pickard DW, et al. Stimulation of ovine placental transport of calcium and magnesium by mid-molecule fragments of human parathyroid hormone-related protein. Exp Physiol. 1990;75:605–8.
Husain SM, Birdsey TJ, Glazier JD, et al. Effect of diabetes mellitus on maternofetal flux of calcium and magnesium and calbindin9K mRNA expression in rat placenta. Pediatr Res. 1994;35:376–81.
Barri M, Abbas SK, Pickard DW, et al. Fetal magnesium homeostasis in the sheep. Exp Physiol. 1990;75:681–8.
Glazier JD, Atkinson DE, Thornburg KL, et al. Gestational changes in Ca2+ transport across rat placenta and mRNA for calbindin9K and Ca2+-ATPase. Am J Physiol. 1992;263:R930–5.
Bruns ME, Fausto A, Avioli LV. Placental calcium binding protein in rats. Apparent identity with vitamin D-dependent calcium binding protein from rat intestine. J Biol Chem. 1978;253:3186–90.
Delorme AC, Danan JL, Ripoche MA, Mathieu H. Biochemical characterization of mouse vitamin D-dependent calcium-binding protein. Evidence for its presence in embryonic life. Biochem J. 1982;205:49–57.
Trotter M, Hixon BB. Sequential changes in weight, density, and percentage ash weight of human skeletons from an early fetal period through old age. Anat Rec. 1974;179:1–18.
Comar CL. Radiocalcium studies in pregnancy. Ann N Y Acad Sci. 1956;64:281–98.
Loughead JL, Mimouni F, Tsang RC. Serum ionized calcium concentrations in normal neonates. Am J Dis Child. 1988;142:516–8.
Giles MM, Fenton MH, Shaw B, et al. Sequential calcium and phosphorus balance studies in preterm infants. J Pediatr. 1987;110:591–8.
Barltrop D, Oppe TE. Calcium and fat absorption by low birthweight infants from a calcium-supplemented milk formula. Arch Dis Child. 1973;48:580–2.
Kobayashi A, Kawai S, Obe Y, Nagashima Y. Effects of dietary lactose and lactase preparation on the intestinal absorption of calcium and magnesium in normal infants. Am J Clin Nutr. 1975;28:681–3.
Kocian J, Skala I, Bakos K. Calcium absorption from milk and lactose-free milk in healthy subjects and patients with lactose intolerance. Digestion. 1973;9:317–24.
Buchowski MS, Miller DD. Lactose, calcium source and age affect calcium bioavailability in rats. J Nutr. 1991;121:1746–54.
Shaw JC. Evidence for defective skeletal mineralization in low-birthweight infants: the absorption of calcium and fat. Pediatrics. 1976;57:16–25.
Senterre J, Salle B. Calcium and phosphorus economy of the preterm infant and its interaction with vitamin D and its metabolites. Acta Paediatr Scand Suppl. 1982;296:85–92.
• Yang Y: Skeletal morphogenesis during embryonic development. Crit Rev Eukaryot Gene Expr 2009, 19:197–218. Detailed review of the molecular control of skeletal morphogenesis including endochondral bone formation.
Moore KL, Persaud TVN. The Developing Human. 6th ed. Philadelphia, PA: W. B. Saunders; 1998.
Lee K, Deeds JD, Bond AT, et al. In situ localization of PTH/PTHrP receptor mRNA in the bone of fetal and young rats. Bone. 1993;14:341–5.
Lee K, Deeds JD, Segre GV. Expression of parathyroid hormone-related peptide and its receptor messenger ribonucleic acids during fetal development of rats. Endocrinology. 1995;136:453–63.
• Karsenty G, Kronenberg HM, Settembre C: Genetic control of bone formation. Annu Rev Cell Dev Biol 2009, 25:629–648. Detailed review of the regulation of endochondral bone formation.
Karaplis AC, Luz A, Glowacki J, et al. Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone-related peptide gene. Genes Dev. 1994;8:277–89.
Lanske B, Karaplis AC, Lee K, et al. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science. 1996;273:663–6.
Vortkamp A, Lee K, Lanske B, et al. Indian hedgehog and parathyroid hormone-related protein regulate the rate of cartilage differentiation. Science. 1996;273:613–22.
Weir EC, Philbrick WM, Amling M, et al. Targeted overexpression of parathyroid hormone-related peptide in chondrocytes causes chondrodysplasia and delayed endochondral bone formation. Proc Natl Acad Sci U S A. 1996;93:10240–5.
Schipani E, Lanske B, Hunzelman J, et al. Targeted expression of constitutively active PTH/PTHrP receptors delays endochondral bone formation and rescues PTHrP-less mice. Proc Natl Acad Sci U S A. 1997;94:13689–94.
Lanske B, Divieti P, Kovacs CS, et al. The parathyroid hormone/parathyroid hormone-related peptide receptor mediates actions of both ligands in murine bone. Endocrinology. 1998;139:5192–204.
Miao D, He B, Jiang Y, et al. Osteoblast-derived PTHrP is a potent endogenous bone anabolic agent that modifies the therapeutic efficacy of administered PTH 1–34. J Clin Invest. 2005;115:2402–11.
Miao D, He B, Karaplis AC, Goltzman D. Parathyroid hormone is essential for normal fetal bone formation. J Clin Invest. 2002;109:1173–82.
Karaplis AC, He B, Nguyen MT, et al. Inactivating mutation in the human parathyroid hormone receptor type 1 gene in Blomstrand chondrodysplasia. Endocrinology. 1998;139:5255–8.
Oostra RJ, van der Harten JJ, Rijnders WP, et al. Blomstrand osteochondrodysplasia: three novel cases and histological evidence for heterogeneity. Virchows Arch. 2000;436:28–35.
• Simmonds CS, Kovacs CS: Role of parathyroid hormone (PTH) and PTH-related protein (PTHrP) in regulating mineral homeostasis during fetal development. Crit Rev Eukaryot Gene Expr 2010, 20:235–273. Detailed review of the relative roles of PTH and PTHrP in regulating placental calcium transfer, blood mineral concentrations, and skeletal development and mineralization during fetal life.
Fisher DA. Fetal and neonatal endocrinology. In: DeGroot LJ, Jameson JL, editors. Endocrinology. 5th ed. Philadelphia, PA: Elsevier Saunders; 2006. p. 3369–86.
Mueller SO, Korach KS. Estrogen receptors and endocrine diseases: lessons from estrogen receptor knockout mice. Curr Opin Pharmacol. 2001;1:613–9.
Vidal O, Lindberg MK, Hollberg K, et al. Estrogen receptor specificity in the regulation of skeletal growth and maturation in male mice. Proc Natl Acad Sci U S A. 2000;97:5474–9.
Windahl SH, Andersson G, Gustafsson JA. Elucidation of estrogen receptor function in bone with the use of mouse models. Trends Endocrinol Metab. 2002;13:195–200.
Panda DK, Miao D, Tremblay ML, et al. Targeted ablation of the 25-hydroxyvitamin D 1alpha -hydroxylase enzyme: evidence for skeletal, reproductive, and immune dysfunction. Proc Natl Acad Sci U S A. 2001;98:7498–503.
Dardenne O, Prud’homme J, Arabian A, et al. Targeted inactivation of the 25-hydroxyvitamin D(3)-1(alpha)-hydroxylase gene (CYP27B1) creates an animal model of pseudovitamin D-deficiency rickets. Endocrinology. 2001;142:3135–41.
Glazier JD, Mawer EB, Sibley CP. Calbindin-D9K gene expression in rat chorioallantoic placenta is not regulated by 1,25-dihydroxyvitamin D3. Pediatr Res. 1995;37:720–5.
Marche P, Delorme A, Cuisinier-Gleizes P. Intestinal and placental calcium-binding proteins in vitamin D-deprived or -supplemented rats. Life Sci. 1978;23:2555–61.
Verhaeghe J, Thomasset M, Brehier A, et al. 1,25(OH)2D3 and Ca-binding protein in fetal rats: relationship to the maternal vitamin D status. Am J Physiol. 1988;254:E505–12.
Li YC, Amling M, Pirro AE, et al. Normalization of mineral ion homeostasis by dietary means prevents hyperparathyroidism, rickets, and osteomalacia, but not alopecia in vitamin D receptor-ablated mice. Endocrinology. 1998;139:4391–6.
Li YC, Pirro AE, Amling M, et al. Targeted ablation of the vitamin D receptor: an animal model of vitamin D dependent rickets type II with alopecia. Proc Natl Acad Sci U S A. 1997;94:9831–5.
Halloran BP, DeLuca HF. Calcium transport in small intestine during early development: role of vitamin D. Am J Physiol. 1980;239:G473–9.
Ghishan FK, Parker P, Nichols S, Hoyumpa A. Kinetics of intestinal calcium transport during maturation in rats. Pediatr Res. 1984;18:235–9.
Amling M, Priemel M, Holzmann T, et al. Rescue of the skeletal phenotype of vitamin D receptor-ablated mice in the setting of normal mineral ion homeostasis: formal histomorphometric and biomechanical analyses. Endocrinology. 1999;140:4982–7.
Van Cromphaut SJ, Dewerchin M, Hoenderop JG, et al. Duodenal calcium absorption in vitamin D receptor-knockout mice: functional and molecular aspects. Proc Natl Acad Sci U S A. 2001;98:13324–9.
Dardenne O, Prudhomme J, Hacking SA, et al. Rescue of the pseudo-vitamin D deficiency rickets phenotype of CYP27B1-deficient mice by treatment with 1,25-dihydroxyvitamin D3: biochemical, histomorphometric, and biomechanical analyses. J Bone Miner Res. 2003;18:637–43.
Maxwell JP, Miles LM. Osteomalacia in China. J Obstet Gynaecol Br Empire. 1925;32:433–73.
Mohapatra A, Sankaranarayanan K, Kadam SS, et al. Congenital rickets. J Trop Pediatr. 2003;49:126–7.
Innes AM, Seshia MM, Prasad C, et al. Congenital rickets caused by maternal vitamin D deficiency. Paediatr Child Health. 2002;7:455–8.
Begum R, Coutinho ML, Dormandy TL, Yudkin S. Maternal malabsorption presenting as congenital rickets. Lancet. 1968;1:1048–52.
Congdon P, Horsman A, Kirby PA, et al. Mineral content of the forearms of babies born to Asian and white mothers. Br Med J (Clin Res Ed). 1983;286:1233–5.
Park W, Paust H, Kaufmann HJ, Offermann G. Osteomalacia of the mother–rickets of the newborn. Eur J Pediatr. 1987;146:292–3.
Ford JA, Davidson DC, McIntosh WB, et al. Neonatal rickets in Asian immigrant population. Br Med J. 1973;3:211–2.
Moncrieff M, Fadahunsi TO. Congenital rickets due to maternal vitamin D deficiency. Arch Dis Child. 1974;49:810–1.
Teotia M, Teotia SP, Nath M. Metabolic studies in congenital vitamin D deficiency rickets. Indian J Pediatr. 1995;62:55–61.
Sann L, David L, Thomas A, et al. Congenital hyperparathyroidism and vitamin D deficiency secondary to maternal hypoparathyroidism. Acta Paediatr Scand. 1976;65:381–5.
Campbell DE, Fleischman AR. Rickets of prematurity: controversies in causation and prevention. Clin Perinatol. 1988;15:879–90.
Pereira GR, Zucker AH. Nutritional deficiencies in the neonate. Clin Perinatol. 1986;13:175–89.
Specker BL. Do North American women need supplemental vitamin D during pregnancy or lactation? Am J Clin Nutr. 1994;59:484S–90.
Beck-Nielsen SS, Brock-Jacobsen B, Gram J, et al. Incidence and prevalence of nutritional and hereditary rickets in southern Denmark. Eur J Endocrinol. 2009;160:491–7.
Bouillon R, Verstuyf A, Mathieu C, et al. Vitamin D resistance. Best Pract Res Clin Endocrinol Metab. 2006;20:627–45.
Takeda E, Yamamoto H, Taketani Y, Miyamoto K. Vitamin D-dependent rickets type I and type II. Acta Paediatr Jpn. 1997;39:508–13.
Kitanaka S, Takeyama K, Murayama A, et al. Inactivating mutations in the 25-hydroxyvitamin D3 1alpha-hydroxylase gene in patients with pseudovitamin D-deficiency rickets. N Engl J Med. 1998;338:653–61.
Silver J, Landau H, Bab I, et al. Vitamin D-dependent rickets types I and II. Diagnosis and response to therapy. Isr J Med Sci. 1985;21:53–6.
Balsan S, Garabedian M, Larchet M, et al. Long-term nocturnal calcium infusions can cure rickets and promote normal mineralization in hereditary resistance to 1,25-dihydroxyvitamin D. J Clin Invest. 1986;77:1661–7.
Hochberg Z, Tiosano D, Even L. Calcium therapy for calcitriol-resistant rickets. J Pediatr. 1992;121:803–8.
• Weiler HA, Fitzpatrick-Wong SC, Schellenberg JM: Bone mass in First Nations, Asian and white newborn infants. Growth Dev Aging 2008, 71:35–43. Observational study that found that ethnic background explains bone mineral content in the newborn but not 25(OH)D levels.
Brooke OG, Brown IR, Bone CD, et al. Vitamin D supplements in pregnant Asian women: effects on calcium status and fetal growth. Br Med J. 1980;280:751–4.
Mallet E, Gugi B, Brunelle P, et al. Vitamin D supplementation in pregnancy: a controlled trial of two methods. Obstet Gynecol. 1986;68:300–4.
Delvin EE, Salle BL, Glorieux FH, et al. Vitamin D supplementation during pregnancy: effect on neonatal calcium homeostasis. J Pediatr. 1986;109:328–34.
Marya RK, Rathee S, Dua V, Sangwan K. Effect of vitamin D supplementation during pregnancy on foetal growth. Indian J Med Res. 1988;88:488–92.
Marya RK, Rathee S, Lata V, Mudgil S. Effects of vitamin D supplementation in pregnancy. Gynecol Obstet Invest. 1981;12:155–61.
Yu CK, Sykes L, Sethi M, et al. Vitamin D deficiency and supplementation during pregnancy. Clin Endocrinol (Oxf). 2009;70:685–90.
•• Hollis BW, Johnson D, Hulsey TC, et al.: Vitamin D supplementation during pregnancy: Double blind, randomized clinical trial of safety and effectiveness. J Bone Miner Res 2011, 26:in press. One of two large clinical trials of vitamin D supplementation that nicely demonstrates the amount of maternal intake of vitamin D required to achieve several target blood levels of 25(OH)D in mother and in cord blood. However, no clinical benefit to mother or offspring was demonstrated.
•• Wagner CL: Vitamin D Supplementation during Pregnancy: Impact on Maternal Outcomes. Presented at the Centers for Disease Control and Prevention Conference on Vitamin D Physiology in Pregnancy: Implications for Preterm Birth and Preeclampsia. Atlanta, Georgia; April 26–27, 2011. Additional data were presented from two large clinical trials that demonstrated the amount of maternal vitamin D supplementation required to achieve various target levels of maternal and cord blood 25(OH)D. However, in the intention-to-treat analyses no effect of vitamin D supplementation was seen on cord blood calcium, skeletal, and anthropometric parameters in the newborns, or adverse pregnancy outcomes such as preterm birth, preeclampsia, infections, etc.
Greer FR, Searcy JE, Levin RS, et al. Bone mineral content and serum 25-hydroxyvitamin D concentration in breast-fed infants with and without supplemental vitamin D. J Pediatr. 1981;98:696–701.
Greer FR, Searcy JE, Levin RS, et al. Bone mineral content and serum 25-hydroxyvitamin D concentrations in breast-fed infants with and without supplemental vitamin D: one-year follow-up. J Pediatr. 1982;100:919–22.
Greer FR, Marshall S. Bone mineral content, serum vitamin D metabolite concentrations, and ultraviolet B light exposure in infants fed human milk with and without vitamin D2 supplements. J Pediatr. 1989;114:204–12.
Morley R, Carlin JB, Pasco JA, Wark JD. Maternal 25-hydroxyvitamin D and parathyroid hormone concentrations and offspring birth size. J Clin Endocrinol Metab. 2006;91:906–12.
• Mahon P, Harvey N, Crozier S, et al.: Low maternal vitamin D status and fetal bone development: cohort study. J Bone Miner Res 2010, 25:14–19. Associational study that found increased metaphyseal area with lower 25(OH)D levels and concluded this indicated early rickets; this is in contrast with the study by Viljakainen et al. [10•], which showed the opposite finding and concluded that increased metaphyseal area means stronger bone.
• Javaid MK, Crozier SR, Harvey NC, et al.: Maternal vitamin D status during pregnancy and childhood bone mass at age 9 years: a longitudinal study. Lancet 2006, 367:36–43. Associational study that found reduced skeletal mineral content at 9 years of age (but not at birth or 9 months of age) in children born of mothers in the lower tertile of 25(OH)D levels during pregnancy. Results used to support the theory that 25(OH)D levels during fetal life program childhood bone mass, but the data are confounded by maternal factors that lead to lower 25(OH)D levels in the first place.
Cooper C, Westlake S, Harvey N, et al. Review: developmental origins of osteoporotic fracture. Osteoporos Int. 2006;17:337–47.
•• Kovacs CS: Fetus, Neonate and Infant. In: Vitamin D: Third Edition. Edited by Feldman D, Pike WJ, Adams JS. New York: Academic Press; 2011: 625–646. Detailed and comprehensive review of available animal and human data pertaining the roles of vitamin D, 25(OH)D, and calcitriol in regulating mineral metabolism and skeletal development in the fetus, neonate, and infant.
•• IOM: Dietary reference intakes for calcium and vitamin D. Washington, DC: The National Academies Press 2011. Comprehensive review of all available evidence that concluded that target 25(OH)D blood levels of 50 nmol/L are appropriate for all pediatric ages to ensure optimal skeletal development and health.
Disclosure
Conflicts of interest: C.S. Kovacs: has received grant support from the Canadian Institutes of Health Research, and also the Dairy Farmers of Canada/National Sciences and Engineering Research Council of Canada.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kovacs, C.S. Bone Development in the Fetus and Neonate: Role of the Calciotropic Hormones. Curr Osteoporos Rep 9, 274–283 (2011). https://doi.org/10.1007/s11914-011-0073-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-011-0073-0
Keywords
- Fetus
- Neonate
- Calcium
- Phosphorus
- Endochondral bone development
- Skeletal development
- Mesenchymal bone development
- Parathyroid hormone
- Parathyroid hormone-related protein
- PTH/PTHrP receptor
- Estradiol
- Estrogen receptor
- Calcitonin
- Calcitonin receptor
- Vitamin D
- 25-hydroxyvitamin D
- Calcitriol
- Knockout mice
- Sheep
- Humans
- Randomized clinical trials
- Observational studies
- Associational studies
- RANKL
- Osteoprotegerin
- Indian hedge hog
- Placental calcium transfer
- Bone formation
- Bone resorption