
Abstract
Purpose of Review
Sarcomas are rare, heterogeneous group of soft tissue and bone tumors. Precise diagnosis of specific subtypes is challenging using conventional methods. Herein, we review the role of next-generation sequencing (NGS) technology that is used for rapid sequencing of DNA and RNA.
Recent Findings
Recent sarcoma specific studies recommend that molecular genetic testing should be added at diagnosis for appropriate clinical management in addition to diagnosis by expert pathologists. NGS has already been used to identify potentially actionable mutations, copy number alterations, and gene fusions. Rationally, choosing a drug based on an individual patient profile aka: “precision oncology” has been so far limited to few case reports in sarcomas.
Summary
As we improve our ability to deliver personalized medicine using all modalities including conventional therapy, more patients may eventually benefit. As the cost and capacity of NGS outpace Moore’s law, so does the probability of success.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sarcomas are a heterogeneous group of over 100 tumors. Broadly, they arise from cells of mesenchymal origin and are generally tumors from non-epithelial cells. They are classified as either soft tissue sarcomas (STS) or bone sarcomas. The former make up about 12,000 cases per year and the latter about 3000 with 4700 and 1500 deaths, respectively [1]. The scarcity makes sarcomas difficulty to study, a matter complicated by over 70–100 different histologic subtypes. This rarity makes even a diagnosis difficult and often results in divergent pathologic interpretations [2]. Traditionally, sarcomas are managed with a combination of surgery and radiation when localized and occasionally involve neoadjuvant or adjuvant chemotherapy. When the disease recurs or metastasizes, a few chemotherapy options exist and have waning efficacy over subsequent cycles [3]. Therefore, a need exists for more precise diagnosis and new actionable drug targets in this lethal disease. The precise diagnosis of specific sarcoma subtypes requires expert pathologists and has been quite challenging using conventional methods. Given the unprecedented advances in molecular biology, we are at an exciting time where we can interrogate different pathologies at the cellular level.
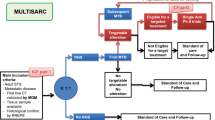
Next-generation sequencing (NGS) is a technology that allows for fast and inexpensive sequencing of both DNA and RNA. This technology led to a rapid decrease in the cost of sequencing an entire genome from $340,000 in 2008 to just $4200 in 2015 and sub-$1000 in 2016 [4]. Herein, we review the current evidence from the literature for using NGS in sarcomas as a research and diagnostic aid as well as a clinical decision tool that can guide therapy determination (Fig. 1).

MindMap outlining the use of NGS in sarcoma research, diagnosis, and treatment
NGS in Sarcoma Research: Review of Literature
After a drastic cost reduction in sequencing, the technology became available to cancer researchers. Specifically with sarcomas, this presented an opportunity to investigate genomes beyond the known translocations. Most investigators have shied away from whole genome sequencing partly because it introduces additional cost and complexity, and partly because of the limited number of potentially therapeutic options. Instead, investigators have chosen targeted next-generation sequencing of a limited number of cancer-associated genes. As NGS became more robust, the number of genes in the targeted sequencing panel has increased from approximately 50-gene to 400+ gene panels. Researchers have turned to NGS in specific sarcoma subtypes to identify recurrent mutations with potential therapeutic implications. Brenca and Maestro reviewed massive parallel sequencing in sarcoma pathobiology [5]. Their comprehensive work is a compendium of over 130 different cytogenetic aberrations. Many of these were discovered using older techniques, but nearly a quarter were identified using NGS. Since the publication of this work in 2015, the preferred method of novel genomic aberration detection has become NGS.
Jour et al. [6] evaluated the utility of NGS in identifying targetable mutations in sarcomas. They used a validated multiplexed panel to fully sequence the coding regions of 194 cancer-related genes. They analyzed 25 soft tissue sarcomas and were able to identify both actionable and non-actionable mutations. The most frequent aberrations in their data set included TP53, PTEN, and CDKN2A. More importantly, they found potentially actionable pathway aberrations in MAP2K4, AURKA, and C-MYC, as well as NOTCH1, PIK3CA, and PDGFR-β. These findings are consistent with our own reports of 102 diverse sarcoma patients who received NGS as part of their clinical care and demonstrate the potential clinical use of many targeted agents in the treatment of sarcomas [7••].
While a shotgun approach may be appropriate for personalized treatment of sarcomas in general, Behjati et al. [8•] focused on a specific subtype. They used whole genome sequencing and an initial set of only three angiosarcomas to identify truncating mutations in PTPRB gene, a tyrosine phosphatase specific to vascular endothelium. The truncation prevents inhibition of angiogenesis and is postulated to be integral to development of angiosarcomas. They expanded this finding to a validation set of 36 angiosarcomas and found that 26% had mutations in the PTPRB gene, thus identifying a potential, although difficult, therapeutic target. Perhaps the most important lesson from their work is that NGS allows a very small number of patient samples, as few as three, to lead to new discoveries about the mechanisms of a rare disease.
Perry et al. [9] took a similar approach with osteosarcomas. They used a comprehensive approach of whole genome sequencing, whole exome sequencing, and RNA sequencing in 59 tumor/normal pairs. They found the genome of osteosarcomas to be complex with high mutation rates and a median of 230 genomic rearrangements. TP53 and RB1 were frequently altered. Importantly, the PI3K/MTOR pathway was altered in 24% of patients and 34% of patient samples had an alteration with potential clinical implications. They further characterized the PI3K/MTOR pathway in a mouse model and found it integral to proliferation of osteosarcoma and a possible therapeutic target. Indeed, a phase II trial was completed using mTOR inhibition in advanced sarcomas [10] and advanced to a phase III trial. Unfortunately, the phase III SUCCEED trial of mTOR inhibitor ridaforolimus did not succeed leading to rejection by the US FDA for approval owing to clinically insignificant results [11]. The patients with sarcoma assigned to ridaforolimus had a median PFS of 17.7 weeks, compared with 14.6 weeks for placebo (P = .0001). The difference in median OS (20.8 months for ridaforolimus vs. 19.6 months for placebo) was non-significant. One major drawback for the study was it did not have a biomarker selection for sarcomas that have aberrant activation of mTOR pathway.
Harnessing the ability of NGS to yield clinically relevant results in a small number of patients, Kohsaka et al. [12] examined embryonal origin rhabdomyosarcomas. This is a disease without the characteristic PAX-FOXO1 fusions. They applied matched normal-tumor whole exome and whole transcriptome NGS to 11 patient samples. In addition to previously identified RAS and FGFR4 genes, they identified two patients with identical c.365T > G point mutations in the MYOD1 gene. An expansion dataset of 93 samples found 8 additional cases with the same mutation. These cases also had frequent co-occurrence of alterations in PI3K-AKT pathway. Review of survival data showed an overall poor prognosis for these patients reflecting a need for more aggressive therapy.
Because of the high yield of data from a small amount of DNA, NGS presents an opportunity to study rare neoplasms. De la Vega et al. [13•] applied an NGS 409 gene panel to a small cohort of 11 soft tissue sarcomas characterized by CIC-DUX4 fusion. While they did not find any recurrent driver mutations, they were able to identify copy number alterations of chromosome 1P corresponding to specific non-sense mutations of ARID1A pointing to a potential epigenetic driver of this disease.
Vlenterie et al. [14] used NGS to evaluate mutations in synovial sarcomas other than the well-characterized t(X;18). They used a 409 cancer gene panel to sequence 26 synovial sarcoma samples. They found that while all of the tumors possessed the canonical translocation, other mutations were not conserved among different patient samples. This led the authors to conclude that the t(X;18) is both the initiating event and driver for synovial sarcoma development and growth. They were able to correlate higher mutation load with adult rather than pediatric tumors and also with a worse overall prognosis. They could not, however, make the same correlation with response to chemotherapy.
Hong et al. [15] attempted to answer the question of drug resistance using NGS. They performed whole exome sequencing on a single patient with dermatofibrosarcoma protuberans (DFSP) that previously responded to imatinib, but eventually had disease progression. The pre- and post-progression biopsies were sequenced with identification of the classic COL1A1-PDGFRβ fusion gene in both samples. Additionally, eight new non-synonymous mutations were found, none in the fusion gene. The authors postulated that activation of NF-kB pathway via mutations in CARD10 gene was potentially responsible for imatinib resistance and creates a novel therapeutic target.
Missiaglia and Sheperd et al. [16] used high-throughput sequencing to derive miRNA expression profiles which help characterize the behavior of rhabdomyosarcomas. They were able to cluster miRNA expression to fusion protein as well as non-fusion protein rhabdomyosarcomas. This led the investigators to postulate that there is an interplay between these fusions and miRNA that contributed to a malignant phenotype.
Challenges With Heterogeneity
One of the challenges to designing a “one-size-fits-all” approach is the tremendous heterogeneity within a single histologic subtype. Zhang et al. [17] illustrated this with a NGS-based 50-gene panel on Ewing sarcoma samples. In just 20 patients, they identified 62 non-synonymous mutations in 26 cancer-related genes. Five of the mutations were previously unidentified in Ewing sarcoma. More than half of the patients had a potentially treatable mutation, but these were not conserved among different patients. This study highlights the need for a truly personalized rather than a disease wide approach using NGS.
NGS for Diagnosis: the Molecular Microscope
Sarcoma heterogeneity and rarity makes establishing a diagnosis difficult. Mathias et al. [18] described a case of a poorly differentiated round cell sarcoma that was originally diagnosed as Ewing sarcoma. Next-generation sequencing was performed and showed a complex genome without characteristic Ewing-associated rearrangements; instead, RB1, PTCH1, and ATRX inactivation pointed to a diagnosis of osteosarcoma. The patient’s chemotherapy was changed to an osteosarcoma regimen, demonstrating the ability of NGS to alter the diagnosis and treatment. Similarly, Doyle et al. [19] describe a case of an atypical carcinoid tumor that, upon performing NGS, was found to have EWSR1-ERG fusion. That patient’s therapy was changed to the Ewing sarcoma regimen.
Italiano et al. [20••] conducted a multicenter observational study of sarcoma tissue. A sarcoma-expert pathologist used tissue to establish a primary diagnosis as well as up to two alternative diagnoses based on histology alone. The pathologist then classified their certainty of diagnosis. The investigators used comparative genomic hybridization, FISH, and RT-PCR to molecularly characterize the tissue. A total of 384 sarcomas were included. The diagnosis was modified in 53 patients based on molecular genetics. Ultimately, this would have led to change in prognosis or initial management in 45 cases. The authors concluded that molecular diagnosis should be mandatory in the diagnosis of sarcomas, even when the diagnosis is made by an expert pathologist. While corrections in diagnosis were made in all levels of certainty, the lower pathologic certainty specimens benefited most from molecular diagnosis.
Identifying Fusion Proteins
Fusion genes are a defining feature of many sarcomas. When pathogenic, they combine a regulatory segment with a protein-coding segment to create a new RNA-expressing protein that has downstream oncogenic effects. The classic Ewing sarcoma EWSR1-FLI1 fusion protein is a transcription factor in the activation of a myriad of downstream genes that are not yet fully mapped out. Transcription factors are notoriously difficult to develop drugs for, and while the EWSR1-FLI1 fusion was first reported by Aurias et al. in 1983 [21], no drug has been able to exploit this clinically.
Fusion proteins can be used for diagnostic purposes. For example, some sarcomas have bimorphic histology such as mesenchymal chondrosarcoma. Biopsy samples may contain only the well-differentiated cartilage component or the small round cell component. This may result in the erroneous diagnosis of osteosarcoma, Ewing sarcoma, or another round cell sarcoma. Wang et al. detected such a chondrosarcoma with recurrent fusion of Hey1-NCOA2 by NGS and demonstrate the utility of NGS in diagnosing fusion protein sarcomas [22].
Guseva et al. [23] describe a particular scenario where fluorescent in situ hybridization (FISH), the traditional method of detecting fusions, fails. They describe a solitary fibrous tumor which has a recently discovered NAB2-STAT6 fusion protein. The two genes are located in close proximity on chromosome 12 and this proximity makes detection by FISH difficult. Instead they used a NGS-based RNA fusion detection kit to identify this fusion in ten patients with a single base pair resolution.
NGS for Prognosis
Often, sarcoma histology can be divided into low-grade and high-grade subtypes. Li et al. [24] used an NGS-based fusion transcript detection assay as a prognostic platform for endometrial stromal sarcomas. They differentiated low- from high-grade tumors based on the presence of JAZF1 fusion genes in the former and YWHAE-NUTM2A/B fusion genes in the latter. All of the low-grade tumors were correctly identified based on this panel and the authors concluded that an NGS-based fusion assay can detect endometrial stromal sarcoma fusions. Similarly, Hoang et al. [25] used NGS in endometrial stromal sarcomas to identify ZC3H7B-BCOR fusion genes with a particularly high-grade and aggressive phenotype. While not used in clinical practice yet, this is an example of NGS applied in prognosis of sarcomas.
Lesluyes et al. [26] attempted to reestablish the complexity index in sarcomas (CINSARC) gene expression signature using NGS (RNA seq expression). CINSARC is a prognostic algorithm based on microarray expression in soft tissue sarcomas. The advantage of NGS is that formalin-fixed, paraffin-embedded tissue can be used instead of frozen tumors. They found that overall there was 77% agreement between the original microarray-based score and their NGS-based technique. They were able to validate their expression signature using TCGA. CINSARC needs to be prospectively validated in a clinical context and the RNA extraction technology needs to be perfected.
Yang et al. [27] performed targeted NGS of 44 cancer-related genes on 54 leiomyosarcoma cases. Leiomyosarcoma is a prototypical complex karyotype sarcoma. They found only two recurrent gene aberrations: TP53 and ATRX. While TP53 mutations correlated with a uterus or retroperitoneal primary and larger size, it did not have prognostic significance. ATRX mutations, on the other hand, correlated with poor differentiation and worse overall survival, thereby identifying a potential prognostic marker for this disease. While prognosis is important to patients, it is not able to change the natural history of a disease.
Recurrent Mutations Identified by NGS
Lee et al. [28] applied an NGS-based comprehensive cancer panel consisting of 151 canonical cancer genes to leiomyosarcomas. They selected 25 cases with either primary or metastatic disease to examine for recurrent potentially targetable alterations. The majority were uterine or pelvic in origin. They found that copy number losses were common especially on chromosomes 10 and 13, and gains were seen on chromosomes 7 and 17. They saw missense mutations in TP53, ATM, ATRX, EGFR, and RB1. The frequent loss of tumor suppressors among a complex genomic landscape is postulated to be a hallmark of leiomyosarcomas and this can be used as a differentiating factor in difficult to characterize cases.
Similarly to Lee, Asano et al. [29] used an NGS panel of 104 cancer-associated genes to evaluate liposarcomas—the most common soft tissue sarcoma in adults. MDM2 and CDK4 gene amplifications are a hallmark of this disease, but single agent MDM2 or CDK4 inhibitors do not have sufficient clinical activity. Genomic profiling was applied to 19 well-differentiated and 37 de-differentiated liposarcomas. Applying their results to the COSMIC database, they found a very low number of potential driver point mutations. These occurred frequently in TP53, KIT, FGFR1, ARID1A, CHEK2, and ROCK1. None of these point mutations was recurrent. Other than MDM2/CDK4 amplifications, the authors identified a high number of receptor tyrosine kinase (RTK) amplifications (32% of samples). These RTK genes had highly increased expression by RT-PCR suggesting that the amplification is relevant. One patient had a previous well-differentiated liposarcoma that recurred as a de-differentiated liposarcoma years later with new amplification of RTK genes, suggesting that this may be a necessary step in acquiring malignant potential. The diversity of these RTK amplifications indicates that a personalized approach may be necessary in treating this neoplasm.
Murali et al. [30] sought to identify novel genetic alterations in a cohort of 34 angiosarcomas. They used NGS-based sequencing consisting of 341 genes involved in tumorigenesis. While TP53 was the most commonly mutated gene (35%), MAPK pathway aberrations were identified in a majority of patients (53%) and all mutations were mutually exclusive. Previously described MYC and KDR amplifications were seen in a third of patients, as well as previously identified PTPRB and PLCG1 gene mutations. MYC amplifications were seen only in radiation-associated angiosarcomas and were mutually exclusive with CDKN2A loss. Their study suggests that the MAPK pathway is a rational therapeutic target in angiosarcomas.
NGS for Precision Oncology Treatment Decisions
Driver Aberrations and Matching Patients to Targeted Therapy
Andersson et al. [31] used an NGS-based panel covering 50 cancer-associated genes to look for driver mutations in 55 sarcoma samples. The majority of these were Ewing, synovial, GIST, or myxoid liposaroma. They were not able to identify recurrent driver mutations in their cohort. However, they were able to identify individual potential driver mutations in nine cases including a BRAFV600E mutation in a Ewing-like sarcoma which is a target for BRAF inhibitors [32].
Our own sequencing of diverse sarcomas [7••] in a similar fashion to Andersson led to finding substantially more potentially actionable mutations. We used a larger NGS panel of 236 or 315 cancer genes to sequence 102 patients with diverse sarcomas. We found that more than half (62%) of all patients had a potentially actionable mutation targetable by a drug either available on the market off label or in a clinical trial for other indications. While such sequencing results are encouraging, from a practical perspective, matching them to a targeted therapy was limited by patient access to these drugs either by paucity of trials in sarcomas for that particular aberration or lack of insurance coverage for off label use of FDA-approved drugs in other indications.
NGS in a patient with chemoresistant sarcoma identified a KIAA1549-BRAF fusion gene with PTEN loss [33•]. The patient was started on a matched therapy with Sorafenib, Temsirolimus, and Bevacizumab targeted the fusion and mTOR pathway likely activated by PTEN loss. She had a 25% reduction in her lesions, along with clinical improvement for 11 cycles.
Doebele et al. [34••] published a similar report of an undefined soft tissue sarcoma. NGS identified an LMNA-NTRK1 fusion protein. This was suspected to be a driver fusion and she was referred for a phase I clinical trial of LOXO-101, an NTRK inhibitor. She had dramatic clinical and radiographic tumor regression after cycle one, and almost complete response after cycle five. This case demonstrates the power of NGS to detect fusions and the ability of fusion proteins to be oncogenic drivers.
Similar to Doebele, Subbiah and Holmes [35] reported a fusion-driven sarcoma that responded to targeted therapy. The case was a malignant gastrointestinal neuroectodermal tumor that recurred in the liver and bones after a primary resection. That patient underwent NGS-based comprehensive genomic profiling of 405 cancer-related genes which resulted in identification of an EWSR1-CREB1 fusion. She was treated with a combination of crizotinib and pazopanib as part of a clinical trial and achieved a partial response that was durable for 2.8 years with minimal toxicity. The authors, in an attempt to identify more patients with this fusion, used a large database of comprehensive genomic profiling to find 11 others with the same profile that could have benefit from similar therapy.
In another demonstration of fusion protein-driven sarcomas, Subbiah et al. [36] reported a case of smooth muscle tumor of uncertain malignant potential (STUMP). The patient had multiple metastatic recurrences after resections and finally underwent NGS-based comprehensive genomic profiling of 315 cancer-associated genes. A DCTN1-ALK fusion protein was identified. Based on ALK expression, this was reclassified as an inflammatory myofibroblastic tumor with myxoid features. The patient was treated on a phase I trial with crizotinib and pazopanib and achieved a partial response based on RECIST. When the authors looked at 139 cases of leiomyosarcoma, they found five additional ALK rearranged tumors.
Jiang et al. [37] applied whole exome sequencing to Ewing sarcoma and desmoplastic small round cell tumors. They identified several novel mutations including PTPRD and GRB10 genes that seem to predispose to IGF1R sensitivity. Two patients treated with an IGF1R inhibitor experienced a complete response and one achieved a partial response when treated on a clinical trial, again identifying a target for personalized medicine [38].
In a recent study, NGS showed response mechanisms of Ewing sarcoma to pazopanib. Responder vs non-responder analysis revealed divergence in the genotype of Ewing sarcoma in a three-gene signature consisting of FGFR3, FGFR4, and FLT4, which are all targets for pazopanib [39].
Failure of NGS to Provide Therapeutic Benefit
We reported two cases of osteosarcoma patients that were treated based on NGS, but without therapeutic benefit [40]. Both patients had chemotherapy-resistant metastatic osteosarcoma. The tumors were sequenced with NGS capture of > 300 genes. Patient no. 1 was shown to have MET amplification, PIK3CA mutation, CCNE1 amplification, and PTPRD mutation. Immunohistochemistry-based analysis showed c-met, COX-2, and SPARC expression as well as mTOR/AKT pathway activation. The patient was treated with sirolumus, crizotinib, nab-Paclitaxel, and celecoxib. Unfortunately, his best response was progressive disease. Patient no. 2 had NF2 loss, PDGFRα, and TP53. He was treated with Temsirolumus, Sorafenib, and Bevacizumab, but had no clinical benefit from this regimen. These two cases illustrate that targeted therapy is more complex given the complicated signaling networks in refractory patients showing that simple drug matching may not yield benefit in most patients. Such negative studies need to be published too as we see a publication bias of only positive cases that may not reflect the real-world patients who do not benefit from targeted therapy.
NGS for Identification of Resistance Mechanisms to Immunotherapy
George et al. [41••] describe a case of previously untreated uterine leiomyosarcoma that had a remarkable response to Pembrolizumab. She had multiple metastases with regression as well as one that had continued growth during this period. The single growing metastasis was surgically resected and the patient continued to have response for over 2 years on Pembrolizumab. PD-L1 staining was < 5% in the sensitive metastases and < 1% in the resistant metastasis. The authors performed RNA seq and whole exome sequencing on both the responsive and resistant tumors to try to explain the difference in response to immunotherapy. Both samples had low overall mutational load and no microsatellite instability defects. The two tumors shared most mutations with the exception of 12 non-synonymous mutations exclusive to the primary and 16 missense mutations exclusive to the resistant tumor. Only one of these 16 mutations had oncogenic potential: PTEN gene, with an additional heterozygous loss of the wild-type copy. The authors postulated that PTEN loss leads to increased expression of immunosuppressive cytokines, but this link has not yet been formally established. George et al. further investigated predicted neoantigens based on WES and found that two of the neoantigens were expressed at lower levels in the resistant sample. With the case report, the investigators describe a potential method for evaluating resistance to immunotherapy using NGS.
Conclusion
Next-generation sequencing is a rapidly evolving technology that will potentially transform the way we study, diagnose, and treat sarcomas. We know little about the pathogenesis of sarcomas beyond the canonical translocations. The ability of NGS to incorporate not only exome sequencing of large numbers of genes but also RNA expression and even miRNA is expanding our understanding of this group of diseases. Already we are discovering new translocations, driver mutations, and predictive signatures. Hopefully, this will lead to better treatments and even cures for patients [42]. What is clear is that NGS is redefining the way we diagnose sarcomas, a disease that is difficult to fully characterize even for an expert sarcoma pathologist. It is in the hands of the pathologists that NGS will make its greatest near-term impact, allowing more precision. We hope that diagnostic disambiguation will lead to better clinical outcomes and undoubtedly there will be patients that benefit. Some of the examples described in this review of clinical responses to targeted therapies are astounding. They demonstrate the clear potential of rationally choosing a drug based on an individual patient profile. These cases need to become the norm rather than the exceptional responder. As we enhance our ability to deliver personalized medicine, more sarcoma patients will undoubtedly benefit. In order to demonstrate the utility of personalized medicine, prospective trials need to be done. The groundwork has already been laid in early pilot trials and as the cost and capacity of NGS outpaces Moore’s law, so does the probability of success.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
A snapshot of sarcoma. National Cancer Institute.
Arbiser ZK, Folpe AL, Weiss SW. Consultative (expert) second opinions in soft tissue pathology analysis of problem-prone diagnostic situations. Am J Clin Pathol. 2001;116(4):473–6.
Subbiah V, Hess KR, Khawaja MR, et al. Evaluation of novel targeted therapies in aggressive biology sarcoma patients after progression from US FDA approved therapies. Sci Rep. 2016;6:35448.
The cost of sequencing a human genome. National Human Genome Research Institute (NHGRI).
Brenca M, Maestro R. Massive parallel sequencing in sarcoma pathobiology: state of the art and perspectives. Expert Rev Anticancer Ther. 2015;15(12):1473–88.
Jour G, Scarborough JD, Jones RL, et al. Molecular profiling of soft tissue sarcomas using next-generation sequencing: a pilot study toward precision therapeutics. Hum Pathol. 2014;45(8):1563–71.
•• Groisberg R, Hong DS, Holla V, et al. Clinical genomic profiling to identify actionable alterations for investigational therapies in patients with diverse sarcomas. Oncotarget. 2017;5(0). Clinical NGS of Sarcomas for novel agents and its utility.
• Behjati S, Tarpey PS, Sheldon H, et al. Recurrent PTPRB and PLCG1 mutations in angiosarcoma. Nat Genet. 2014;46(4):376–9. Identifying a novel gene targets in angiosarcomas
Perry JA, Kiezun A, Tonzi P, et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc Natl Acad Sci U S A. 2014;111(51):E5564–73.
Chawla SP, Staddon AP, Baker LH, et al. Phase II study of the mammalian target of rapamycin inhibitor ridaforolimus in patients with advanced bone and soft tissue sarcomas. J Clin Oncol. 2012;30(1):78–84.
Demetri GD, Chawla SP, Ray-Coquard I, et al. Results of an international randomized phase III trial of the mammalian target of rapamycin inhibitor ridaforolimus versus placebo to control metastatic sarcomas in patients after benefit from prior chemotherapy. J Clin Oncol. 2013;31(19):2485–92.
Kohsaka S, Shukla N, Ameur N, et al. A recurrent neomorphic mutation in MYOD1 defines a clinically aggressive subset of embryonal rhabdomyosarcoma associated with PI3K-AKT pathway mutations. Nat Genet. 2014;46(6):595–600.
• Lazo de la Vega L, Hovelson DH, Cani AK, et al. Targeted next-generation sequencing of CIC-DUX4 soft tissue sarcomas demonstrates low mutational burden and recurrent chromosome 1p loss. Hum Pathol. 2016;58:161–70. Identification of recurrent aberrations in CIC-DUX4 sarcoma
Vlenterie M, Hillebrandt-Roeffen MHS, Flucke UE, et al. Next generation sequencing in synovial sarcoma reveals novel gene mutations. Oncotarget. 2015;6(33):34680–90.
Hong JY, Liu X, Mao M, et al. Genetic aberrations in imatinib-resistant dermatofibrosarcoma protuberans revealed by whole genome sequencing. PLoS One. 2013;8(7):e69752.
Missiaglia E, Shepherd CJ, Aladowicz E, et al. MicroRNA and gene co-expression networks characterize biological and clinical behavior of rhabdomyosarcomas. Cancer Lett. 2017;385:251–60.
Zhang N, Liu H, Yue G, Zhang Y, You J, Wang H. Molecular heterogeneity of Ewing sarcoma as detected by ion torrent sequencing. PLoS One. 2016;11(4):e0153546.
Mathias MD, Chou AJ, Meyers P, et al. Osteosarcoma with apparent Ewing sarcoma gene rearrangement. J Pediatr Hematol Oncol. 2016;38(5):e166–8.
Doyle LA, Wong K-K, Bueno R, et al. Ewing sarcoma mimicking atypical carcinoid tumor: detection of unexpected genomic alterations demonstrates the use of next generation sequencing as a diagnostic tool. Cancer Genetics. 2014;207(7–8):335–9.
•• Italiano A, Di Mauro I, Rapp J, et al. Clinical effect of molecular methods in sarcoma diagnosis (GENSARC): a prospective, multicentre, observational study. Lancet Oncol. 2016. Multicenter study showing that molecular genetic testing should be mandatory for diagnostic accuracy of sarcoma and appropriate clinical management, even when histological diagnosis is made by pathologist experts in this field.
Aurias A, Rimbaut C, Buffe D, Zucker JM, Mazabraud A. Translocation involving chromosome 22 in Ewing’s sarcoma. A cytogenetic study of four fresh tumors. Cancer Genet Cytogenet. 1984;12(1):21–5.
Identification of a novel, recurrent HEY1-NCOA2 fusion in mesenchymal chondrosarcoma based on a genome-wide screen of exon-level expression data (PDF Download Available). ResearchGate.
Guseva NV, Tanas MR, Stence AA, et al. The NAB2–STAT6 gene fusion in solitary fibrous tumor can be reliably detected by anchored multiplexed PCR for targeted next-generation sequencing. Cancer Genetics. 2016;209(7–8):303–12.
Li X, Anand M, Haimes JD, et al. The application of next-generation sequencing-based molecular diagnostics in endometrial stromal sarcoma. Histopathology. 2016;69(4):551–9.
Hoang LN, Aneja A, Conlon N, et al. Novel high-grade endometrial stromal sarcoma: a morphologic mimicker of myxoid leiomyosarcoma. Am J Surg Pathol. 2017;41(1):12–24.
Lesluyes T, Pérot G, Largeau MR, et al. RNA sequencing validation of the Complexity INdex in SARComas prognostic signature. Eur J Cancer. 2016;57:104–11.
Yang C-Y, Liau J-Y, Huang W-J, et al. Targeted next-generation sequencing of cancer genes identified frequent TP53 and ATRX mutations in leiomyosarcoma. Am J Transl Res. 2015;7(10):2072–81.
Lee PJ, Yoo NS, Hagemann IS, et al. Spectrum of mutations in leiomyosarcomas identified by clinical targeted next-generation sequencing. Exp Mol Pathol. 2017;102(1):156–61.
Asano N, Yoshida A, Mitani S, et al. Frequent amplification of receptor tyrosine kinase genes in well-differentiated/dedifferentiated liposarcoma. Oncotarget. 2017;8(8):12941–52.
Murali R, Chandramohan R, Möller I, et al. Targeted massively parallel sequencing of angiosarcomas reveals frequent activation of the mitogen activated protein kinase pathway. Oncotarget. 2015;6(34):36041–52.
Andersson C, Fagman H, Hansson M, Enlund F. Profiling of potential driver mutations in sarcomas by targeted next generation sequencing. Cancer Genetics. 2016;209(4):154–60.
Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015;373(8):726–36.
• Subbiah V, Westin SN, Wang K, et al. Targeted therapy by combined inhibition of the RAF and mTOR kinases in malignant spindle cell neoplasm harboring the KIAA1549-BRAF fusion protein. J Hematol Oncol. 2014;7(1):8. Study showing that BRAF fusion is actionable in spindle cell sarcoma
•• Doebele RC, Davis LE, Vaishnavi A, et al. An oncogenic NTRK fusion in a patient with soft-tissue sarcoma with response to the tropomyosin-related kinase inhibitor LOXO-101. Cancer Discovery. 2015;5(10):1049–57. Study showing that NTRK fusion is actionable in soft tissue sarcoma
Subbiah V, Holmes O, Gowen K, et al. Activity of c-Met/ALK inhibitor crizotinib and multi-kinase VEGF inhibitor pazopanib in metastatic gastrointestinal neuroectodermal tumor harboring EWSR1-CREB1 fusion. Oncology. 2016;91(6):348–53.
Subbiah V, McMahon C, Patel S, et al. STUMP un “stumped”: anti-tumor response to anaplastic lymphoma kinase (ALK) inhibitor based targeted therapy in uterine inflammatory myofibroblastic tumor with myxoid features harboring DCTN1-ALK fusion. J Hematol Oncol. 2015;8:66.
Jiang Y, Subbiah V, Janku F, et al. Novel secondary somatic mutations in Ewing’s sarcoma and desmoplastic small round cell tumors. PLoS One. 2014;9(8):e93676.
Subbiah V, Naing A, Brown RE, et al. Targeted morphoproteomic profiling of Ewing’s sarcoma treated with insulin-like growth factor 1 receptor (IGF1R) inhibitors: response/resistance signatures. PLoS One. 2011;6(4):e18424.
Subbiah V, Meyer C, Zinner R, et al. Phase Ib/II study of safety & efficacy of combination therapy with multikinase VEGF inhibitor pazopanib and MEK inhibitor trametinib in advanced soft tissue sarcoma. Clinical Cancer Research. 2017.
Subbiah V, Wagner MJ, McGuire MF, et al. Personalized comprehensive molecular profiling of high risk osteosarcoma: implications and limitations for precision medicine. Oncotarget. 2015;6(38):40642–54.
•• George S, Miao D, Demetri GD, et al. Loss of PTEN is associated with resistance to anti-PD-1 checkpoint blockade therapy in metastatic uterine leiomyosarcoma. Immunity. 2017;46(2):197–204. Study showing loss of PTEN as a mechanism of resistance to anti-PD1 checkpoint blockade in uterine leiomyosarcoma
Subbiah V, Kurzrock R. Universal genomic testing needed to win the war against cancer: genomics is the diagnosis. JAMA Oncol. 2016;2(6):719–20.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Roman Groisberg declares that he has no conflict of interest.
Jason Roszik declares that he has no conflict of interest.
Anthony Conley declares that he has no conflict of interest.
Shreyaskumar R. Patel has received research funding through grants from Janssen, Eisai, and Morphotek, and has received compensation from Janssen, Eisai, Morphotek, EMD-Serono, CytRx, Bayer, Eli Lilly, Epizyme, and Novartis for service as a consultant.
Vivek Subbiah has received research funding through grants from Roche/Genentech, Novartis, Bayer, GlaxoSmithKline, PharmaMar, FUJIFILM Pharmaceuticals, AbbVie, and NanoCarrier.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Sarcomas
Rights and permissions
About this article

Cite this article
Groisberg, R., Roszik, J., Conley, A. et al. The Role of Next-Generation Sequencing in Sarcomas: Evolution From Light Microscope to Molecular Microscope. Curr Oncol Rep 19, 78 (2017). https://doi.org/10.1007/s11912-017-0641-2
Published:
DOI: https://doi.org/10.1007/s11912-017-0641-2