
Abstract
Humans depend on our commensal bacteria for nutritive, immune-modulating, and metabolic contributions to maintenance of health. However, this commensal community exists in careful balance that, if disrupted, enters dysbiosis; this has been shown to contribute to the pathogenesis of colon, gastric, esophageal, pancreatic, laryngeal, breast, and gallbladder carcinomas. This development is closely tied to host inflammation, which causes and is aggravated by microbial dysbiosis and increases vulnerability to pathogens. Advances in sequencing technology have increased our ability to catalog microbial species associated with various cancer types across the body. However, defining microbial biomarkers as cancer predictors presents multiple challenges, and existing studies identifying cancer-associated bacteria have reported inconsistent outcomes. Combining metabolites and microbiome analyses can help elucidate interactions between gut microbiota, metabolism, and the host. Ultimately, understanding how gut dysbiosis impacts host response and inflammation will be critical to creating an accurate picture of the role of the microbiome in cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The relationship between specific pathogenic bacteria and human carcinogenesis has been the subject of extensive investigation. Historically, most of this research has focused on individual pathogens, such as Helicobacter pylori, and their potential to initiate and perpetuate disease. Previous research focus was on the disease process rather than beneficial gut–microbe interactions. More recently, extensive research supports commensal bacteria playing a role in protection of host health via nutritive, immune-modulating, and metabolic processes [1, 2]. In addition, the more holistic approach of characterizing entire communities of gut bacteria and their interactions is now possible through use of high-throughput DNA sequencing technology. Characterization of the gut microbiome as a whole has furthered our understanding of intestinal microbial ecology to include community-level functions and changes. In healthy individuals, the gut microbiome functions as a symbiont that can offer protection from invading pathogens and prevent tumorigenesis [3•]. However, this commensal community exists in careful balance that, if disrupted, enters dysbiosis and contributes to host disease processes, including cancer [4–7]. Although recent findings still support individual microorganisms influencing carcinogenesis, greater emphasis is on microbial dysbiosis and its larger role in cancer initiation and progression. The focus of this review is on gut microbial community dynamics that shift state from symbiosis to dysbiosis and the subsequent host immune and pathogen response, which drastically alters initiation and progression of multiple types of cancer.
Proposed Mechanisms for Microbiome Involvement in Colorectal Cancer
Multiple studies report different gut microbiome composition in individuals diagnosed with colorectal cancer (CRC) versus healthy individuals [8•, 9, 10, 11•]. In fact, gut microbiota can play a role in either promotion or prevention of CRC, often through modulation of the inflammatory process due to close contact with host colonic mucosa [5]. For example, chemically induced injury and proliferation induced by azoxymethane and dextran sulfate sodium was enhanced in germ-free mice, which lack protective commensals. In addition, tumor development in the germ-free mice resulted in significantly more and larger tumors than in specific-pathogen-free mice [12]. Balance of the gut microbial community, or eubiosis, can be disrupted by an inflammatory environment in the host. For example, host inflammation may influence microbiota composition through generation of specific metabolites such as nitric oxide synthase 2. Nitrate provides a unique energy source for facultative anaerobic bacteria, allowing them to outcompete bacteria that cannot utilize nitrates [13], disrupting balance of the gut microbiome and resulting in dysbiosis. Proinflammatory responses can also compromise barrier and immune function to allow bacterial translocation through intestinal tight junctions and intensify the inflammatory response [14].
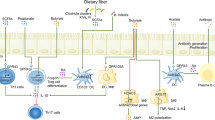
How inflammation interacts with the gut microbiome to influence CRC has been recently synthesized in several hypotheses that summarize our understanding of the interactions to date (Fig. 1). The “alpha-bug¨ hypothesis suggests that a keystone pathogen species, such as enterotoxigenic Bacteroides fragilis (ETBF), remodels the colonic microbiota to promote CRC, possibly via IL-17 and TH17 cell-mediated inflammation. This process could also be initiated by microbial-independent host-mediated inflammation, and may be blocked by beneficial commensal microbiota [15]. Similarly, the bacterial driver–passenger model suggests that “driver” bacteria, such as ETBF, cause or aggravate inflammation and produce genotoxins that lead to cell proliferation and mutations. Subsequently, an adenoma forms and is colonized by “passenger” bacteria such as Fusobacterium spp. that encourage tumor progression [16•]. Following tumor formation, the intestinal barrier is damaged by the continual inflammation and allows bacteria access to tumor tissue. These bacteria and their metabolites stimulate additional inflammatory signals, including IL-17 cytokines, promoting cancer progression [17]. Inflammatory signals may also stimulate macrophages, via induction to an M1 phenotype, to produce chromosome-breaking factors through a bystander effect, damaging DNA and inducing chromosomal instability in neighboring cells [3•]. Likely CRC initiation and progression is engendered by aspects of each of these models.

The progression of the gut microbial community from a state of balance (eubiosis) to imbalance (dysbiosis) is associated with physiologic, metabolic, and cellular responses in the host that modulate cancer risk. BST Bacteroides fragilis toxin, CDT cytolethal distending toxin, CNF cytotoxic necrotizing factor, DCA deoxycholic acid, EREG epiregulin, ERK extracellular-signal-regulated kinase, LCA lithocholic acid, NF-κB nuclear factor κB, NLR NOD-like receptor, NOS2 nitric oxide synthase 2, PGE2 prostaglandin E2, PGE3 prostaglandin E3, RNS reactive nitrogen species, ROS reactive oxygen species, TLR Toll-like receptor
Once bacteria translocate beyond a damaged intestinal epithelium, the host immune system responds with activation of multiple pattern recognition receptors. Pattern recognition receptors important to the CRC process include membrane-bound Toll-like receptors (TLRs) and cytoplasmic NOD-like receptors (NLRs) [6]. Specifically, TLR2 and TLR4 have been shown to be important for tumor formation in murine models, and recent associations between human genetic polymorphisms in TLR2 and TLR4 and CRC risk support a role in humans [18]. Furthermore, activation of nuclear factor (NF)-κB plays a role in CRC tumor initiation by enhancing both cytokines [4, 19] and Wnt signaling, which can convert intestinal epithelial nonstem cells into tumor-initiating cells [20•]. The role of NF-κB in CRC is complex and involves additional signaling pathways which have recently been extensively reviewed [21]. Alternatively, in colitis-associated CRC, TLR signaling in tumor-associated fibroblasts initiates an inflammatory cascade independent of NF-κB via epiregulin. Epiregulin stimulates the extracellular-signal-regulated kinase pathway, which encourages tumor proliferation [22]. Two NLRs are associated with CRC risk: NOD2, which is activated by the bacterial peptidoglycan muramyl dipeptide, and NOD-, leucine-rich-repeat-, and pyrin-domain-containing 6 (NLRP6). With NOD2 deficiency, dysbiosis alone was sufficient for CRC development in mice [23]. However, recent research by Shanahan et al. [24] reveals that NOD2-associated dysbiosis can be overcome by co-housing NOD2 mutants with wild-type mice. Further research is necessary to clarify the role of NOD2 and NLRP6 in gut microbial regulation. However, the role of bacterial translocation across intestinal epithelia in activation of TLR and NLR and in promoting inflammation is strongly supported [5, 6, 25, 26].
Also “driving” the cancer initiative process are pathogens that have been shown to promote tumorigenesis via genotoxic effects, including Escherichia coli, Enterococcus faecalis, and B. fragilis. Pathogenic strains of E. coli generally belong to groups B2 and D and produce genotoxic virulence factors, called cyclomodulins. Cyclomodulins can modulate cellular differentiation, apoptosis, and proliferation [27] and include colibactin, cytotoxic necrotizing factor, and cytolethal distending toxin. Group B2 E. coli that produce cyclomodulins are highly prevalent in colonic mucosa of CRC patients [28]. E. faecalis indirectly increases genotoxin production in the form of DNA-damaging reactive oxygen species and reactive nitrogen species by inducing an M1 phenotype in host macrophages [3•]. ETBF releases fragilysin (also known as B. fragilis toxin ), a toxic virulence factor that induces DNA damage in vivo [29]. All of these organisms have also been shown to play a role in carcinogenesis via induction of inflammatory pathways [7]. In addition, Fusobacterium was recently associated with an upregulation of NF-κB-driven inflammatory genes and was identified as being enriched in colonic tumors [30]. Although specific organisms exert these genotoxic effects, the effects are made possible and intensified through a prior state of dysbiosis.
Microorganisms Associated with Tumor Occurrence and Formation in CRC
A major goal of the Human Microbiome Project has been to define a “core” microbiome that could be useful in identifying deviations from a normal, healthy state. Although the identification of a healthy core intestinal microbiome has remained elusive, numerous comparative studies have begun to reveal the relationship of the microbial community to CRC. The importance of the microbiome in tumor initiation and development has been elegantly demonstrated in murine models. Transfer of the microbiota from tumor-bearing mice induces tumor formation in healthy animals [11•], and mice with a genetic predisposition to develop CRC are spared when treated with antibiotics [31]. Retrospective human cohort studies encompass a range of sample types and populations, addressing questions related to global differences in the microbiome of healthy individuals relative to those with CRC or adenomatous polyps, and differences in the intestinal microclimates between healthy tissue and tumor tissue of an affected individual. Taken together, these studies are beginning to define a CRC-associated microbiome.
Although no bacteria have consistently been associated with CRC across all studies, the Gram-negative oral commensal Fusobacterium nucleatum has been most strongly linked to CRC. Several studies examining the colon tumor microenvironment by comparing tumor tissue with adjacent healthy tissue reported an overabundance of Fusobacterium associated with tumors [8•, 30, 32]. A Chinese study reported a trend for increased Fusobacterium content in tumor tissue relative to matched controls, but failed to achieve significance, which may be a result of the small study size (n = 8), but could also indicate that Fusobacterium association with CRC is not consistent across different ethnicities [33]. Additional studies have confirmed that Fusobacterium spp. are enriched in precancerous adenomas, particularly those displaying high-grade dysplasia [30, 34]. Kostic et al. [30] also reported higher stool levels of Fusobacterium in adenoma and CRC patients than in healthy controls. They also observed that Apc Min/+ mice infected with F. nucleatum had increased tumor multiplicity and selective recruitment of tumor-promoting myeloid cells. Activation of β-catenin signaling, which regulates inflammatory and oncogenic responses via binding of the FadA adhesin produced by F. nucleatum to E-cadherin in host membranes, provides further evidence for the role of Fusobacterium as a driver of CRC initiation and progression [35].
Although it is known that mucosa-adherent bacteria differ significantly from those found in the intestinal lumen, the identification of a CRC-associated stool microbiota is appealing for diagnostic and prognostic purposes. Unfortunately, there appears to be little consensus in the existing published literature of specific bacterial associations, and even more general measures such as bacterial community diversity do not appear to consistently predict CRC. Sobhani et al. [9] reported no differences in bacterial community diversity between case and control stool samples, but did note enrichment in Bacteroides/Prevotella in CRC stool samples, which was corroborated in mucosa samples from tissue biopsies. They also reported depletion of Bifidobacterium longan, Clostridium clostridioforme, and Ruminococcus species. Another study reported higher levels of Akkermansia muciniphila and Citrobacter farmeri in CRC cases, and decreased levels of butyrate-producing species such as Ruminococcus and Roseburia species relative to controls [10]. Akkermansia is a common commensal in the intestines of humans, and its depletion was previously associated with Crohn’s disease and inflammatory bowel disease [36]; however, it was demonstrated to be important in CRC tumor development in a murine model [11•]. In the largest study to date examining stool microbes, a decrease in the microbial diversity of CRC patients was observed, as well as decreased abundance of Clostridium species [37•]. This study also reported higher abundance of Fusobacterium present in stool samples from CRC patients, suggesting possible utility of stool in reflecting mucosa levels of this tumor-associated bacterium. However, the composition of stool microbial communities appears to be a poor predictor of CRC presence on the basis of current knowledge, and more large cohort studies are needed before effective diagnostic or prognostic tests can be developed using bacterial biomarkers in stool samples.
Microbiome Involvement in Gastric and Esophageal Cancers
Stomach and esophageal linings come in close contact with microbiota, and recent evidence supports that the microbiome also influences these cancers. The longest-known and most extensively characterized association between these cancers and a gut microbe is with H. pylori infection. Study of Mongolian gerbils, whose gastric system more closely resembles humans than does that of the widely implemented mouse models, showed that 37 % of animals infected with H. pylori developed adenocarcinomas, whereas no tumor development occurred in uninfected controls [38]. More recent work with this animal model suggests that long-term H. pylori infection disrupts the gut microbial community. H. pylori negative gerbils were observed to have decreased abundance of Bifidobacterium spp. , Clostridium coccoides group, and Clostridium leptum subgroup but a higher abundance of Atopobium cluster [39]. In addition, three lactobacillus species—Lactobacillus reuteri, Lactobacillus johnsonii, and Lactobacillus murinus—inhibit H. pylori growth in vitro, suggesting that some gut microbes may help prevent H. pylori infection [40].
Human studies comparing stomach microbiota in cancer patients and healthy controls indicate that microbes other than H. pylori must be present to facilitate mucosal movement toward gastric cancer development [41•, 42]. In fact, many people infected with H. pylori do not develop gastric cancer [42]. Aviles-Jimenez et al. [41•] noted decreases in the abundance of Porphyromonas, Meisseria, and Streptococcus sinesis and increased abundance of Lactobacillus coleohominis, Pseudomonas, and Lachnospiraceae among gastric cancer patients. The noted increase in abundance of L. coleohominis, a species previously thought to be beneficial, is supported by Dicksved et al. [42], who measured an increase in the abundance of terminal restriction fragments corresponding to lactobacilli in samples from gastric cancer patients. Further investigation of this phenomenon and of H. pylori interactions with the gut microbiome is required to better understand its role in the disease process.
Eradication of H. pylori has been shown to correlate with a decrease in incidence of gastric cancer [42]. Shin et al. [43•] showed a decrease in the methylation of the LOX tumor suppressor gene with eradication of Helicobacter felis, the murine equivalent of H. pylori. A study by Cai et al. [44] indicates that eradication therapy is most effective in restoring parietal cells and reducing dysplasia when the H. felis infection duration is less than 6 months. Infections lasting longer than this, when dysplasia and metaplasia are severer, resulted in only partial reversion of these lesions. Results from human studies of H. pylori eradication for prevention of gastric cancers are conflicting, and studies need to be conducted on larger cohorts with longer follow-ups in order to assess the effectiveness of this strategy for chemoprevention.
Several mechanisms have been proposed by which H. pylori induces development of gastric cancer. Helicobacter pylori infection increases cell proliferation, leading to increased turnover of the gastric mucosa, which could lead to a higher incidence of mutation and less time for DNA repair [38]. Mice lacking secretory phospholipase A2, such as C57BL/6 mice, the showed increased levels of apoptosis after oral infection with H. felis and expansion of aberrant gastric mucosa cell lineages, indicating that secretory phospholipase A2 influences the response of gastric mucosa to H. felis infection [45]. Raf kinase inhibitor protein regulates the cell cycle and apoptosis in the gastric mucosa. In infected mucosa, H. pylori phosphorylates Raf kinase inhibitor protein, removing apoptotic control and inducing proliferation by removing control of the cell cycle [46]. Another tumor suppressor gene, LOX, was shown by Shin et al. [43•] to be methylated in transgenic mice infected with H. felis. The downregulation of these tumor-suppressing proteins allows gastric adenocarcinoma to develop in the presence of H. pylori infection.
Helicobacter pylori infection has also been implicated in the development of esophageal cancer, but its role is unclear [47]. Anderson et al. [48] showed an increase in seropositivity for H. pylori in junctional tumors, those involving the esophagus and gastric cardia. However, in tumors that do not involve the gastric cardia, H. pylori is associated with a lowered risk of tumor development. More is known about the microbiome of reflux esophagitis and Barrett metaplasia, which are precursor states to esophageal cancer. In these conditions, dominance shifts from Gram-positive bacteria to mostly Gram-negative bacteria, suggesting that dysbiosis plays a role in the disease process [49]. It is likely that other microbes are also involved in tumor development in the esophagus. Cancerous esophageal tissue shows a higher prevalence of Treponema denticola, Streptococcus mitis, and Streptococcus anginosus as compared with normal tissue. These pathogens induce inflammation by cytokines, possibly supporting tumor development [50].
Microbiome Involvement in Other Forms of Cancer
The microbiome has also been implicated in the development of pancreatic [51•], laryngeal [52•], and gallbladder [53] carcinoma. Farrell et al. [51•] noted significant shifts in oral microbial composition between healthy and pancreatic cancer groups. Among cancer groups, significant decreases in abundance of Neisseria elongata and S. mitis (a pathogen also implicated in esophageal cancer [50]) were noted. These significant changes in oral microbiota with the development of pancreatic cancer indicate potential for oral N. elongata and S. mitis to serve as biomarkers for pancreatic cancer occurrence.
In gallbladder cancer, Salmonella infection has been shown to be of particular importance [53]. The gallbladder is a known reservoir of Salmonella, leading to increases in secondary bile acid (SBA) concentrations, which is linked to tumor promotion [54]. Sharma et al. [53] showed an association between the typhoid carrier state and gallbladder cancer. In addition, bile culture positivity is associated with increase in gallbladder carcinogenesis, especially positivity for the Vi antigen (a capsular antigen associated with Salmonella). These associations indicate the relevance of the microbiome in the pathogenesis of multiple cancer types.
Viruses are also a component of the gut microbiome and can influence cancer risk. For example, DNA from human papillomavirus (HPV) is detected in almost all cervical cancers [55]. Extensive study indicates that viral antigens E6 and E7 contribute to the malignancy of HPV-induced cervical cancer [56]. However, estrogen is required for the development of cervical cancer from HPV infection. In mice and rats, 83 % of HPV-infected animals develop cervical cancer after estrogen treatment [57]. Estrogen treatment leads to increased transcription of viral antigens E6 and E7, contributing to cervical carcinogenesis [56]. In addition, the presence of estrogen receptor α (ERα) is necessary in the development of cervical cancer from HPV infection [58, 59], as estrogen receptor α knockout mice do not develop cervical cancer when infected with HPV [58]. As intestinal microbes affect circulating estrogen levels [60], these commensal organisms may be involved in the development of cervical cancer from HPV infection; however, further study is needed to support this link.
HPV, in conjunction with H. pylori, has also been implicated in laryngeal cancer [52•, 61]. Gong et al. [52•] associated a total of 15 additional genera with laryngeal carcinoma tissue, with noted increases in the abundance of Fusobacterium, Prevotella, and Gemella. Fusobacterium and Prevotella, in particular, are thought to be associated with the development of biofilms that stimulate an inflammatory response, leading to laryngeal cancer development [62]. Although HPV and H. pylori are both involved in laryngeal cancer development, not much is currently known about how viral and bacterial members of the microbiome interact, an intriguing topic for future research.
Role of Microbial Metabolites in Cancer Development and Progression
Changes in bacterial metabolism can modulate cancer risk and often accompany dysbiosis of the gut microbiome. Specific bacterial metabolites associated with increased CRC risk include prostaglandin E2 [63] and multiple SBAs [10]. Conversely, decreased CRC risk is associated with indole [64], antioxidants [63], and the antiproliferative metabolites butyrate [10] and ursodeoxycholic acid (UDCA) [10]. Indole, a bacterial quorum-sensing molecule produced by catabolism of tryptophan, enhances barrier function of colonic epithelial cells in vitro. In vivo experiments suggest indole is a byproduct of gut microbial metabolism as indole level is significantly lower in germ-free mice than in specific-pathogen-free mice. In vivo experiments also suggest that indole enhances function of both tight junctions and adherens junctions in both germ-free and specific-pathogen-free mice [64]. Butyrate has known antitumorigenic and antiproliferative effects due to its regulation of genes that inhibit cell proliferation and induce apoptosis via histone deacetylase inhibition [65]. UDCA, a microbial metabolite of a primary bile acid, has been shown to prevent colorectal tumor development in animal and preclinical models [66]. UDCA has been administered in clinical trials as a chemopreventive agent, and a systemic review of UDCA’s effect on the incidence or recurrence of CRC is currently under way [67]. However, some evidence also exists to suggest that UDCA may be procarcinogenic at higher doses [68].
SBAs such as deoxycholic acid (DCA) and lithocholic acid (LCA) are produced as products of microbial metabolism of primary bile acids produced by the host. The promotion of CRC by DCA and LCA and other SBAs has recently been extensively reviewed [69]. Recent evidence points to bacteria in Clostridium cluster IX as a possible source of increased abundance of DCA and increased cancer risk in obese mice [70]. The abundance of DCA, in particular, was found to increase rapidly, within 24 h, for an animal-based diet and was linked to overgrowth of inflammation-causing microorganisms associated with inflammatory bowel disease [71]. However, DCA also acts as a ligand of the FXR receptor [72], which has been shown to reduce liver and intestinal tumor growth and metastasis [73]. Similarly, LCA may prevent DNA damage, and therefore tumorigenesis, through stimulation of xenobiotic metabolism and excretion [74]. Although LCA and DCA are predominantly characterized as promoting CRC, future research in this area may reveal a more complex role for these metabolites in the CRC process.
Extensive study indicates a role of intestinal microbes in the metabolism of dietary estrogens. In patients treated with ampicillin, fecal excretion of estrogen metabolites increases, indicating that reabsorption into the bloodstream is reduced with diminished abundance of intestinal microflora [75]. Adding further support to the involvement of intestinal microflora in estrogen metabolism, fecal microbes have been shown to conduct oxidation and reduction reactions on estrogens and can shift intestinal concentrations of estrone and estradiol [60]. Although no definite link has been observed between intestinal microflora estrogen metabolism and cancer development, it is reasonable to anticipate the existence of such a mechanism.
Definitive linkage between estrogen levels and breast cancer development has been shown [76]. In rat models, implanted estrogen leads to cyst formation in mammary tissue [77]. In addition, the presence of antiestrogen antibodies—decreasing estrogen concentrations—delays the onset and growth of mammary tumors in rats and mice [78]. Specifically, 16α hydroxylation of estrogen, a reaction shown to be conducted by the intestinal microflora [60], is associated with an increase in the risk of development of breast cancer [79]. Considering these results and the similarity between the development of CRC and breast cancer, Hill et al. [78] hypothesized a link between breast cancer development and metabolism of estrogen by intestinal microflora.
Recent techniques combine analyses of changing metabolites and microorganisms in an effort to understand interactions between gut microbiota, metabolism, and the host [10, 80, 81]. Further research in this area will deepen the mechanistic understanding of microbial metabolism in the cancer disease process.
Conclusion
The role of microorganisms in cancer initiation and progression can no longer be simply described as a pathogen–disease relationship. Evidence that our microbiome also functions to promote health and prevent disease by encouraging apoptosis and limiting proliferation and inflammation is growing. A microbiome in a state of balance helps to sustain human health, but as this balance is disrupted via inflammatory processes, the community changes and becomes vulnerable to invasion by pathogenic organisms. If these pathogens successfully establish themselves, then a disrupted state of dysbiosis occurs, allowing further inflammation and production of genotoxins and other carcinogenic microbial metabolites. In addition, dysbiosis was recently hypothesized to contribute to the evolution of pathogens, which could potentially raise cancer risk [82].
However, as we begin to understand better the gradient of eubiosis to dysbiosis (Fig. 1), we can develop methods to manipulate the gut microbiome to promote health. As an example, we already know that diet plays a large role in determining the bacterial species of the microbiome, their metabolites, and cancer risk. A recent study looked at rural Africans, who exhibit significantly lower risk of CRC compared with African Americans. Rural Africans were shown to have increased abundance of Prevotella spp. and butyrate as compared with African Americans, who had higher abundance of Bacteroides spp. and SBAs [80]. These differences may be a consequence of rural Africans having higher resistant starch intake and African Americans having higher meat and fat intakes [83]. Dietary choices can affect cancer risk [80, 81, 84], and changing diet to potentially reduce risk is the exciting topic of much current study [85, 86]. Diet is just one example of how to apply our growing knowledge of gut microbiome dynamics toward health promotion and disease prevention. Other potential therapies to modulate the gut microbiome include fecal transplants [87], probiotics [88, 89], exercise [90], and likely many more that we may have failed to mention. Future studies should focus on these therapies and their mechanisms to improve applications in a clinical setting.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Thomas LV, Ockhuizen T. New insights into the impact of the intestinal microbiota on health and disease: a symposium report. Br J Nutr. 2012;107(S1):S1–13.
Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–14.
Yang Y, Wang X, Huycke T, Moore DR, Lightfoot SA, Huycke MM. Colon macrophages polarized by commensal bacteria cause colitis and cancer through the bystander effect. Transl Oncol. 2013;6(5):596. This study demonstrates that specific human commensals can polarize macrophages to the M1 phenotype, which then serve as effectors for bacterially induced bystander effects. The authors propose targeting M1 macrophages as a chemopreventive strategy.
Candela M, Turroni S, Biagi E, Carbonero F, Rampelli S, Fiorentini C, et al. Inflammation and colorectal cancer, when microbiota-host mutualism breaks. World J Gastroenterol. 2014;20(4):908–22.
Jobin C. Colorectal cancer: looking for answers in the microbiota. Cancer Discov. 2013;3(4):384–7.
Schwabe RF, Jobin C. The microbiome and cancer. Nat Rev Cancer. 2013;13(11):800–12.
Sears CL, Garrett WS. Microbes, microbiota, and colon cancer. Cell Host Microbe. 2014;15(3):317–28.
Marchesi JR, Dutilh BE, Hall N, Peters WH, Roelofs R, Boleij A, et al. Towards the human colorectal cancer microbiome. PLoS One. 2011;6(5):e20447. This study profiles the colon tumor microbiome in contrast to non-malignant colon mucosa revealing differing microbial colonization patterns between the two sites.
Sobhani I, Tap J, Roudot-Thoraval F, Roperch JP, Letulle S, Langella P, et al. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS One. 2011;6(1):e16393.
Weir TL, Manter DK, Sheflin AM, Barnett BA, Heuberger AL, Ryan EP. Stool microbiome and metabolome differences between colorectal cancer patients and healthy adults. PLoS One. 2013;8(8):e70803.
Zackular JP, Baxter NT, Iverson KD, Sadler WD, Petrosino JF, Chen GY, et al. The gut microbiome modulates colon tumorigenesis. AmBio. 2013;4(6):e00692–13. This study demonstrates the important role of the microbiome in CRC development using fecal transplants from tumor-bearing mice to conventionalize germ-free animals, which resulted in an increased number of inflammation-induced tumors.
Zhan Y, Chen P-J, Sadler WD, Wang F, Poe S, Núñez G, et al. Gut microbiota protects against gastrointestinal tumorigenesis caused by epithelial injury. Cancer Res. 2013;73(24):7199–210.
Winter SE, Winter MG, Xavier MN, Thiennimitr P, Poon V, Keestra AM, et al. Host-derived nitrate boosts growth of E coli in the inflamed gut. Science. 2013;339(6120):708–11.
Brenchley JM, Douek DC. Microbial translocation across the GI tract. Annu Rev Immunol. 2012;30:149.
Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nat Rev Microbiol. 2012;10(10):717–25.
Tjalsma H, Boleij A, Marchesi JR, Dutilh BE. A bacterial driver–passenger model for colorectal cancer: beyond the usual suspects. Nat Rev Microbiol. 2012;10(8):575–82. This study proposes the “driver and passenger” hypothesis of bacterially induced tumor formation and progression.
Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. 2012;491(7423):254–8.
Pimentel-Nunes P, Teixeira AL, Pereira C, Gomes M, Brandão C, Rodrigues C, et al. Functional polymorphisms of Toll-like receptors 2 and 4 alter the risk for colorectal carcinoma in Europeans. Dig Liver Dis. 2013;45(1):63–9.
Richmond A. NF-κB, chemokine gene transcription and tumour growth. Nat Rev Immunol. 2002;2(9):664–74.
Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Göktuna SI, Ziegler PK, et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell. 2013;152(1):25–38. This article demonstrates the role of inflammation and outlines signaling pathways involved in the bidirectional conversion of tumor-initiating stem cells and nonstem cells.
Zubair A, Frieri M. Role of nuclear factor-ĸB in breast and colorectal cancer. Curr Allergy Asthma Rep. 2013;13(1):44–9.
Neufert C, Becker C, Türeci Ö, Waldner MJ, Backert I, Floh K, et al. Tumor fibroblast–derived epiregulin promotes growth of colitis-associated neoplasms through ERK. J Clin Investig. 2013;123(4):1428.
Couturier-Maillard A, Secher T, Rehman A, Normand S, De Arcangelis A, Haesler R, et al. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Investig. 2013;123(2):700.
Shanahan MT, Carroll IM, Grossniklaus E, White A, von Furstenberg RJ, Barner R, et al. Mouse Paneth cell antimicrobial function is independent of Nod2. Gut. 2014;63(6):903–10.
Nagi RS, Bhat AS, Kumar H. Cancer: a tale of aberrant PRR response. Front Immunol. 2014;5:161.
Pradere J, Dapito D, Schwabe R. The yin and yang of Toll-like receptors in cancer. Oncogene. 2014;33(27):3485–95.
Collins D, Hogan AM, Winter DC. Microbial and viral pathogens in colorectal cancer. Lancet Oncol. 2011;12(5):504–12.
Buc E, Dubois D, Sauvanet P, Raisch J, Delmas J, Darfeuille-Michaud A, et al. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS One. 2013;8(2):e56964.
Goodwin AC, Shields CED, Wu S, Huso DL, Wu X, Murray-Stewart TR, et al. Polyamine catabolism contributes to enterotoxigenic Bacteroides fragilis-induced colon tumorigenesis. Proc Natl Acad Sci U S A. 2011;108(37):15354–9.
Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe. 2013;14(2):207–15.
Bongers G, Pacer ME, Geraldino TH, Chen L, He Z, Hashimoto D, et al. Interplay of host microbiota, genetic perturbations, and inflammation promotes local development of intestinal neoplasms in mice. J Exp Med. 2014;211(3):457–72.
Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012;22(2):299–306.
Geng J, Fan H, Tang X, Zhai H, Zhang Z. Diversified pattern of the human colorectal cancer microbiome. Gut Pathog. 2013. doi:10.1186/1757-4749-5-2.
Flanagan L, Schmid J, Ebert M, Soucek P, Kunicka T, Liska V, Bruha J, Neary P, Dezeeuw N, Tommasino M. Fusobacterium nucleatum associates with stages of colorectal neoplasia development, colorectal cancer and disease outcome. Eur J Clin Microbiol Infect Dis. 2014;33(8):1381-90. doi:10.1007/s10096-014-2081-3.
Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe. 2013;14(2):195–206.
Png CW, Lindén SK, Gilshenan KS, Zoetendal EG, McSweeney CS, Sly LI, et al. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am J Gastroenterol. 2010;105(11):2420–8.
Ahn J, Sinha R, Pei Z, Dominianni C, Wu J, Shi J, et al. Human gut microbiome and risk for colorectal cancer. J Natl Cancer Inst. 2013;105(24):1907–11. This is the largest comparative study of human stool samples from CRC patients and control subjects and was the first to show decreased microbial community diversity in stool.
Watanabe T, Tada M, Nagai H, Sasaki S, Nakao M. Helicobacter pylori infection induces gastric cancer in Mongolian gerbils. Gastroenterology. 1998;115(3):642–8.
Osaki T, Matsuki T, Asahara T, Zaman C, Hanawa T, Yonezawa H, et al. Comparative analysis of gastric bacterial microbiota in Mongolian gerbils after long-term infection with Helicobacter pylori. Microb Pathog. 2012;53(1):12–8.
Zaman C, Osaki T, Hanawa T, Yonezawa H, Kurata S, Kamiya S. Analysis of the microbial ecology between Helicobacter pylori and the gastric microbiota of Mongolian gerbils. J Med Microbiol. 2014;63(Pt 1):129–37.
Aviles-Jimenez F, Vazquez-Jimenez F, Medrano-Guzman R, Mantilla A, Torres J. Stomach microbiota composition varies between patients with non-atrophic gastritis and patients with intestinal type of gastric cancer. Sci Rep. 2014. doi:10.1038/srep04202. As a comprehensive study of microbiota changes in gastric cancer patients, as compared with healthy controls, this study indicates that other microbes are involved in gastric cancer development.
Dicksved J, Lindberg M, Rosenquist M, Enroth H, Jansson JK, Engstrand L. Molecular characterization of the stomach microbiota in patients with gastric cancer and in controls. J Med Microbiol. 2009;58(4):509–16.
Shin CM, Kim N, Lee HS, Park JH, Ahn S, Kang GH, et al. Changes in aberrant DNA methylation after Helicobacter pylori eradication: a long‐term follow‐up study. Int J Cancer. 2013;133(9):2034–42. This study indicates that the eradication of H. pylori, a treatment thought to be effective in reducing the risk of gastric cancer, is not successful in limiting long-term development of gastric cancer. Changes in methylation provide a mechanism for this phenomenon.
Cai X, Carlson J, Stoicov C, Li H, Wang TC, Houghton J. Helicobacter felis eradication restores normal architecture and inhibits gastric cancer progression in C57BL/6 mice. Gastroenterology. 2005;128(7):1937–52.
Wang TC, Goldenring JR, Dangler C, Ito S, Mueller A, Jeon WK, et al. Mice lacking secretory phospholipase A2 show altered apoptosis and differentiation with Helicobacter felis infection. Gastroenterology. 1998;114(4):675–69.
Moen EL, Wen S, Anwar T, Cross-Knorr S, Brilliant K, Birnbaum F, et al. Regulation of RKIP function by Helicobacter pylori in gastric cancer. PLoS One. 2012;7(5):e37819.
Yang L, Chaudhary N, Baghdadi J, Pei Z. Microbiome in reflux disorders and esophageal adenocarcinoma. Cancer J. 2014;20(3):207–10.
Anderson LA, Murphy SJ, Johnston BT, Watson R, Ferguson H, Bamford KB, et al. Relationship between Helicobacter pylori infection and gastric atrophy and the stages of the oesophageal inflammation, metaplasia, adenocarcinoma sequence: results from the FINBAR case–control study. Gut. 2008;57(6):734–9.
Yang L, Lu X, Nossa CW, Francois F, Peek RM, Pei Z. Inflammation and intestinal metaplasia of the distal esophagus are associated with alterations in the microbiome. Gastroenterology. 2009;137(2):588–97.
Narikiyo M, Tanabe C, Yamada Y, Igaki H, Tachimori Y, Kato H, et al. Frequent and preferential infection of Treponema denticola, Streptococcus mitis, and Streptococcus anginosus in esophageal cancers. Cancer Sci. 2004;95(7):569–74.
Farrell JJ, Zhang L, Zhou H, Chia D, Elashoff D, Akin D, et al. Variations of oral microbiota are associated with pancreatic diseases including pancreatic cancer. Gut. 2012;61(4):582–8. This study indicates that changes of the oral microbiota may be useful biomarkers in the detection of pancreatic cancer. It provides an illustration of microbial changes between cancer patients and healthy controls, indicating species that are relevant to the development of pancreatic cancer.
Gong H-L, Shi Y, Zhou L, Wu C-P, Cao P-Y, Tao L, et al. The composition of microbiome in larynx and the throat biodiversity between laryngeal squamous cell carcinoma patients and control population. PLoS One. 2013;8(6):e66476. This study provides a comprehensive depiction of microbial changes between cancer patients and healthy controls, implicating H. pylori and a number of other species in the development of laryngeal carcinoma.
Sharma V, Chauhan VS, Nath G, Kumar A, Shukla VK. Role of bile bacteria in gallbladder carcinoma. Hepatogastroenterology. 2007;54(78):1622.
Shukla V, Tiwari S, Roy S. Biliary bile acids in cholelithiasis and carcinoma of the gall bladder. Eur J Cancer Prev. 1993;2(2):155–60.
Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189(1):12–9.
Mitrani-Rosenbaum S, Tsvieli R, Tur-Kaspa R. Oestrogen stimulates differential transcription of human papillomavirus type 16 in SiHa cervical carcinoma cells. J Gen Virol. 1989;70(8):2227–32.
Riley RR, Duensing S, Brake T, Münger K, Lambert PF, Arbeit JM. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res. 2003;63(16):4862–71.
Chung S-H, Wiedmeyer K, Shai A, Korach KS, Lambert PF. Requirement for estrogen receptor α in a mouse model for human papillomavirus–associated cervical cancer. Cancer Res. 2008;68(23):9928–34.
Elson DA, Riley RR, Lacey A, Thordarson G, Talamantes FJ, Arbeit JM. Sensitivity of the cervical transformation zone to estrogen-induced squamous carcinogenesis. Cancer Res. 2000;60(5):1267–75.
Lombardi P, Goldin B, Boutin E, Gorbach SL. Metabolism of androgens and estrogens by human fecal microorganisms. J Steroid Biochem. 1978;9(8):795–801.
D'Souza G, Kreimer AR, Viscidi R, Pawlita M, Fakhry C, Koch WM, et al. Case–control study of human papillomavirus and oropharyngeal cancer. N Engl J Med. 2007;356(19):1944–56.
Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–7.
Nugent JL, McCoy AN, Addamo CJ, Jia W, Sandler RS, Keku TO. Altered tissue metabolites correlate with microbial dysbiosis in colorectal adenomas. J Proteome Res. 2014;13(4):1921–9.
Forsythe P, Bienenstock J. Immunomodulation by commensal and probiotic bacteria. Immunol Investig. 2010;39(4–5):429–48.
Chang PV, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci USA. 2014. doi:10.1073/pnas.1322269111.
Akare S, Jean‐Louis S, Chen W, Wood DJ, Powell AA, Martinez JD. Ursodeoxycholic acid modulates histone acetylation and induces differentiation and senescence. Int J Cancer. 2006;119(12):2958–69.
Miao XP, Ouyang Q, Li HY, Zhao ZQ, Pan Y, Wang ZW. Ursodeoxycholic acid for the prevention of colorectal adenomas and carcinomas. Cochrane Database Syst Rev. 2008;4:CD007377. doi:10.1002/14651858.CD007377.
Eaton JE, Silveira MG, Pardi DS, Sinakos E, Kowdley KV, Luketic VA, et al. High-dose ursodeoxycholic acid is associated with the development of colorectal neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Am J Gastroenterol. 2011;106(9):1638–45.
Ajouz H, Mukherji D, Shamseddine A. Secondary bile acids: an underrecognized cause of colon cancer. World J Surg Oncol. 2014;12(1):164.
Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499(7456):97–101.
David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505(7484):559–63.
Wang H, Chen J, Hollister K, Sowers LC, Forman BM. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell. 1999;3(5):543–53.
Deuschle U, Schüler J, Schulz A, Schlüter T, Kinzel O, Abel U, et al. FXR controls the tumor suppressor NDRG2 and FXR agonists reduce liver tumor growth and metastasis in an orthotopic mouse xenograft model. PLoS One. 2012;7(10):e43044.
Inagaki T, Moschetta A, Lee Y-K, Peng L, Zhao G, Downes M, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A. 2006;103(10):3920–5.
Adlercreutz H, Martin F, Pulkkinen M, Dencker H, Rimer U, Sjoberg N-O, et al. Intestinal metabolism of estrogens 1. J Clin Endocrinol Metab. 1976;43(3):497–505.
Woolcott CG, Shvetsov YB, Stanczyk FZ, Wilkens LR, White KK, Caberto C, et al. Plasma sex hormone concentrations and breast cancer risk in an ethnically diverse population of postmenopausal women: the Multiethnic Cohort Study. Endocr Relat Cancer. 2010;17(1):125–34.
Mackenzie I. The production of mammary cancer in rats using oestrogens. Br J Cancer. 1955;9(2):284.
Hill M, Goddard P, Williams R. Gut bacteria and aetiology of cancer of the breast. Lancet. 1971;298(7722):472–3.
Muti P, Bradlow HL, Micheli A, Krogh V, Freudenheim JL, Schünemann HJ, et al. Estrogen metabolism and risk of breast cancer: a prospective study of the 2: 16α-hydroxyestrone ratio in premenopausal and postmenopausal women. Epidemiology. 2000;11(6):635–40.
Ou J, Carbonero F, Zoetendal EG, DeLany JP, Wang M, Newton K, et al. Diet, microbiota, and microbial metabolites in colon cancer risk in rural Africans and African Americans. Am J Clin Nutr. 2013;98(1):111–20.
Xie G, Zhang S, Zheng X, Jia W. Metabolomics approaches for characterizing metabolic interactions between host and its commensal microbes. Electrophoresis. 2013;34(19):2787–98.
Stecher B, Maier L, Hardt W-D. 'Blooming' in the gut: how dysbiosis might contribute to pathogen evolution. Nat Rev Microbiol. 2013;11(4):277–84.
O'Keefe SJ, Chung D, Mahmoud N, Sepulveda AR, Manafe M, Arch J, et al. Why do African Americans get more colon cancer than Native Africans? J Nutr. 2007;137(1):175S–82S.
Keszei AP, Goldbohm RA, Schouten LJ, Jakszyn P, van den Brandt PA. Dietary N-nitroso compounds, endogenous nitrosation, and the risk of esophageal and gastric cancer subtypes in the Netherlands Cohort Study. Am J Clin Nutr. 2013;97(1):135–46.
Giacosa A, Barale R, Bavaresco L, Gatenby P, Gerbi V, Janssens J, et al. Cancer prevention in Europe: the Mediterranean diet as a protective choice. Eur J Cancer Prev. 2013;22(1):90–5.
Vipperla K, Ou J, Wahl E, Ruder E, O'Keefe S. A 14-day in-house dietary modification of a ‘Western’ diet to an ‘African’ diet changes the microbiota, its metabolome, and biomarkers of colon cancer risk (825.5). FASEB J. 2014;28(1 Suppl):825.
Song Y, Garg S, Girotra M, Maddox C, von Rosenvinge EC, Dutta A, et al. Microbiota dynamics in patients treated with fecal microbiota transplantation for recurrent Clostridium difficile infection. PLoS One. 2013;8(11):e81330.
Marzotto M, Maffeis C, Paternoster T, Ferrario R, Rizzotti L, Pellegrino M, et al. Lactobacillus paracasei A survives gastrointestinal passage and affects the fecal microbiota of healthy infants. Res Microbiol. 2006;157(9):857–66.
Matsumoto M, Aranami A, Ishige A, Watanabe K, Benno Y. LKM512 yogurt consumption improves the intestinal environment and induces the T‐helper type 1 cytokine in adult patients with intractable atopic dermatitis. Clin Exp Allergy. 2007;37(3):358–70.
Clarke SF, Murphy EF, O'Sullivan O, Lucey AJ, Humphreys M, Hogan A, et al. Exercise and associated dietary extremes impact on gut microbial diversity. Gut. 2014. doi:10.1136/gutjnl-2013-306541.
Compliance with Ethics Guidelines
Conflict of Interest
Amy M. Sheflin, Alyssa K. Whitney, and Tiffany L. Weir declare that they have no conflict of interest
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Integrative Care
Rights and permissions
About this article

Cite this article
Sheflin, A.M., Whitney, A.K. & Weir, T.L. Cancer-Promoting Effects of Microbial Dysbiosis. Curr Oncol Rep 16, 406 (2014). https://doi.org/10.1007/s11912-014-0406-0
Published:
DOI: https://doi.org/10.1007/s11912-014-0406-0