
Abstract
The mammalian/mechanistic target of rapamycin (mTOR) pathway is a key signaling pathway that has been implicated in genetic epilepsy syndromes, neurodegenerative diseases, and conditions associated with autism spectrum disorder and cognitive impairment. The mTOR pathway has become an exciting treatment target for these various disorders, with mTOR inhibitors such as rapamycin being studied for their potential therapeutic applications. In particular, tuberous sclerosis complex (TSC) is a genetic disorder resulting from overactivation of the mTOR pathway, and pharmacologic therapy with mTOR inhibitors has emerged as a viable treatment option for the systemic manifestations of the disease. In this review, we discuss the approved indications for mTOR inhibitors in TSC, the potential future applications of mTOR inhibitors in TSC and other neurological conditions, and the safety considerations applicable to mTOR therapy in the pediatric population.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In 1721, the Dutch explorer Jacob Roggeveen guided an expedition from the Netherlands to the unknown regions of the Pacific Ocean and on Easter Sunday in the year 1722, he and his crew disembarked on a remote landmass they called Easter Island [1]. Easter Island had been inhabited by the Rapa Nui people for many centuries prior to Roggeveen’s arrival and remains one of the most isolated populated islands in the world. In 1972, a bacterium isolated from an Easter Island soil sample was fortuitously found to have antifungal properties, specifically against the yeast Candida albicans. The compound was named rapamycin, for its origin (rapa-) and its antimicrobial (-mycin) properties [2, 3]. Studies following its discovery showed rapamycin to also have potent immunosuppressive and antitumor effects [4, 5]. In the early 1990s, molecular genetic studies in yeast identified the targets of rapamycin as TOR1 and TOR2 [6], which was followed several years later by the discovery of the mammalian homolog of the TOR proteins, otherwise known as the mammalian/mechanistic target of rapamycin (mTOR) [7, 8]. Due to its broad range of antiproliferative activity, including antimicrobial, antitumor, and immunosuppressive properties, rapamycin has been a compound of intense interest. Rapamycin first came into clinical use in 1999, with the Federal Drug Administration (FDA) approval of the mTOR inhibitor sirolimus (rapamycin) as an immunosuppressant to prevent allograft rejection in organ transplantation. In the 25 years since the discovery of mTOR as the target of rapamycin, aberrant mTOR activity has been linked to a wide spectrum of human diseases, including various cancers, metabolic diseases such as obesity and diabetes, and neurological conditions such as epilepsy, autism spectrum disorder, and cognitive disability [9, 10]. The mTOR pathway has been identified as a crucial cellular signaling pathway, with the pharmacological manipulation of mTOR signaling being studied as potential treatments for various disorders. In this review, we discuss the use of mTOR inhibitors in neurology, with a specific focus on the FDA-approved indications in tuberous sclerosis complex (TSC), but also touching on potential applications that are currently under study.
Background
Tuberous Sclerosis Complex
TSC is a rare, autosomal dominant disease with variable penetrance that affects an estimated 1 in 6000 individuals [11]. The constellation of clinical symptoms, specifically intellectual disability, epilepsy, facial rash, and various brain and systemic tumors, was first described by Bourneville in 1880 [12]. The hallmark of the disease is benign tumors in organs including but not limited to the brain (cortical tubers, subependymal giant cell astrocytomas), kidney (angiomyolipomas), heart (rhabdomyomas), and eyes (retinal hamartomas). Although the tumors are typically considered benign, symptoms develop due to mass effect or the displacement of normal tissue. Skin lesions such as hypopigmented macules and facial angiofibromas are also encountered [13]. The tumors are age-dependent, for example, with cortical tubers and cardiac rhabdomyomas typically present at birth and showing either stability or regression, but subependymal giant cell astrocytomas and renal angiomyolipomas growing over time [14]. Neuropsychiatric symptoms are common and include epilepsy, intellectual disability, autism spectrum disorder, anxiety, and mood disorders [15].
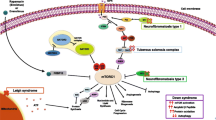
Over a century after Bourneville’s initial clinical description of TSC came the discovery of two causative genes, TSC1 encoding hamartin and TSC2 encoding tuberin, both identified via linkage analysis in multigenerational families [16, 17]. The TSC1-TSC2 protein complex was subsequently shown to act as a tumor suppressor with inhibitory effects on the mTOR cascade (Fig. 1) [18]. Since the elucidation of the underlying genetic and biochemical basis of TSC, rapamycin and mTOR have been intricately linked with TSC. Although TSC has served as the model disease cause by aberrant mTOR signaling, the mTOR pathway has been implicated in various conditions beyond TSC, as described below.

Simplified schematic depicting the mTOR pathway. The mTOR pathway involves two complexes, mTORC1 and mTORC2 (not pictured). mTORC1 integrates signals including growth factors, energy status, oxygen, and amino acids, which act via upstream kinases and control many downstream functions including cell growth, proliferation, survival, ribosome biogenesis, and mRNA translation. Specific genes in the mTOR pathway have been implicated in malformations of cortical development (“mTORopathies”). Elevated levels of phosphorylated S6K1 and eIF4E have been detected in tissues (brain and peripheral lymphocytes) of patients with fragile X syndrome. 4E-BP1 eukaryotic translation factor 4E (eIF4E)-binding protein 1, AKT-AMPK 5′-adenosine monophosphate-activated protein kinase, DEPDC5 disheveled, Egl-10 and Pleckstrin domain-containing protein 5, eIF4E eukaryotic translation factor 4E, LKB1 liver kinase B, mTORC1 mammalian target of rapamycin complex 1, PDK1 phosphoinositide-dependent kinase 1, PI3K phosphoinositide 3-kinase, PMSE polyhydramnios, megalencephaly, symptomatic epilepsy, PTEN phosphatase and tensin homolog on chromosome 10, Rheb Rhas homolog expressed in brain, S6 ribosomal protein S6, S6K1 ribosomal protein S6 kinase, STRADα STE20-related kinase adapter alpha, TSC1 tuberous sclerosis complex 1 protein, TSC2 tuberous sclerosis complex 2 protein
mTOR Pathway
mTOR is a serine-threonine protein kinase that is a member of the phosphoinositide 3-kinase-related kinase family and is the catalytic subunit of two distinct complexes called mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) (Fig. 1) [9, 10, 19]. The TSC1-TSC2 protein complex negatively regulates mTORC1 by converting Rhas homolog expressed in brain (Rheb) from its active to inactive state [20]. Downstream substrates of mTORC1 include S6 kinase (S6K) and eukaryotic translation factor 4E (eIF4E)-binding protein 1 (4EBP1), with mTOR activating S6K and inhibiting 4EBP1, ultimately promoting translation and cell growth [9]. mTORC2 has an important role in cell survival and the maintenance of the actin cytoskeleton, but its role in the mTOR pathway had been less well described. mTORC2 is generally insensitive to rapamycin inhibition, although mTORC2 may show sensitivity after prolonged therapy [21]. The mTORC1 complex integrates upstream signals including nutrient levels, growth factors, energy, and stress, which together act to influence mTORC1 activity and consequently translation, cell growth, and cell survival [9]. These upstream regulators act via kinases including phosphoinositide 3-kinase (PI3K), Akt (also known as PKB, protein kinase B), liver kinase B (LKB1), and AMP kinase (AMPK). Given the central role of the mTOR pathway in promoting cell growth and proliferation, mTOR inhibitors have emerged as a rational, effective treatment for tumors in TSC.
mTOR Inhibitors in the Current Treatment of TSC
Subependymal Giant Cell Astrocytomas
Over a decade after the approval of sirolimus as an immunosuppressant for use in organ transplantation, the mTOR inhibitor everolimus was approved by the FDA in 2010 for the treatment of subependymal giant cell astrocytomas (SEGAs) associated with TSC (Table 1). SEGAs are typically slow-growing glioneuronal tumors arising near the foramen of Monro [22]. SEGAs are reported in up to 20 % of individuals with TSC and are associated with an increased risk of morbidity and mortality secondary to obstructive hydrocephalus, mass effect, and infiltration of surrounding tissues [23, 24]. In the past, surgery was the primary treatment option, given the lack of responsiveness of SEGAs to chemotherapy and radiation [23]. In the era prior to mTOR inhibitors, the debate centered upon when, not if, to operate [25]. Initial case reports suggested that mTOR inhibition could lead to shrinkage of SEGAs associated with TSC [26, 27]. In a prospective, open-label study of everolimus in 28 patients with evidence of serial tumor growth, everolimus led to a clinically meaningful reduction in tumor volume with greater than 30 and 50 % reductions in SEGA volume occurring in up to 80 and 56 % of patients, with no patients developing new lesions or needing to undergo surgical resection or other therapies during the study period [28]. A double-blind, placebo-controlled, phase 3 trial followed (EXIST-1), in which 35 % of patients in the treated group demonstrated a greater than 50 % reduction in tumor volume, compared to no patients in the placebo group demonstrating a reduction in tumor volume [29••]. In the extension phase of EXIST-1, a greater than 50 % reduction in tumor volume was achieved in 49 % of patients, with the median time to SEGA response being 3.58 months and the median duration of exposure being 29.3 months [30]. The current recommendations include mTOR inhibitors for growing but asymptomatic SEGAs [31], although guidelines have not been established regarding the optimal timing or duration of treatment.
Renal Angiomyolipomas and Lymphangioleiomyomatosis
Similar to SEGAs, the treatment of renal angiomyolipomas (AMLs) has shifted from surgical to medical therapy with the introduction of mTOR inhibitors into clinical practice. AMLs develop in over 80 % of patients with TSC, with the lesions often being detected during childhood but causing morbidity and mortality later in life [32, 33]. Although these are typically slow-growing tumors, they carry a risk of renal failure and spontaneous hemorrhage, making them one of the leading causes of death and disability in TSC [34]. In the past, nephrectomy, nephron-sparing surgery, and interventional radiological techniques such as embolization were the therapies most commonly utilized in the treatment of AMLs [35]. In addition to AMLs, lymphangioleiomyomatosis (LAM) is a significant source of morbidity and mortality in TSC patients, often occurring concurrently with AMLs. Although men with TSC can show radiographic evidence of LAM, clinically symptomatic LAM affects women almost exclusively and is characterized by the proliferation of abnormal smooth-muscle cells and cystic changes within the lung parenchyma [36]. LAM is typically diagnosed in young adult women, with approximately 30 % of women with TSC being affected [37]. Presenting symptoms include dyspnea or pneumothorax [14, 36].
In one open-label study, 25 adult patients with TSC, LAM, or both were treated with sirolimus for 1 year and monitored for a change in AML volume or lung function as assessed by pulmonary function testing [38]. The findings were positive, in that AML volume regressed during therapy (53.2 % of baseline value at 12 months), although AML volume tended to increase after therapy was stopped (85.9 % of baseline value at 24 months). An improvement in pulmonary function testing, specifically spirometric measurements and gas trapping, persisted after treatment. This was followed by a double-blind, placebo-controlled, phase 3 trial (EXIST-2) of everolimus, which included 113 adult patients with definite TSC and five patients with sporadic LAM who had at least one AML larger than 3 cm [39••]. The primary endpoint (at least a 50 % reduction in total AML volume) was achieved in 42 % of the individuals treated with everolimus compared to none of the individuals treated with placebo. The median time to response was 2.9 months. Based on these findings, the FDA approved everolimus for the treatment of AMLs in TSC. Everolimus is currently the first-line treatment for asymptomatic but growing AMLs greater than 3 cm, with embolization reserved for acute hemorrhage or as the second-line therapy, and nephrectomy avoided if at all possible [31].
Facial Angiofibromas
Facial angiofibromas occur in over 70 % of patients with TSC and are one of the most highly visible manifestations of the disease. These lesions can be detected at any age but tend to progress during puberty and adolescence [14]. Although the lesions are typically non-painful, they may spontaneously bleed or become a source of emotional distress [40]. Historically, treatment options have included dermabrasion, surgical excision, and laser therapy [41, 42]. One early case report described a patient with TSC who had pronounced regression of her facial angiofibromas while receiving oral rapamycin following renal transplantation [43]. This was followed by the first of a handful of case reports describing the efficacy of a topical rapamycin formulation, hypothesized to avoid the systemic side effects of oral rapamycin [40]. A double-blind, placebo-controlled trial including 28 patients was published in 2012, with 73 % in the treatment arm reporting an improvement with treatment, compared to 38 % in the placebo arm. No adverse events related to the study product were reported, and no detectable systemic absorption of the rapamycin was measured during the study period [44]. Despite definitive clinical evidence of efficacy, mTOR inhibitors are not currently FDA approved for facial angiofibromas.
mTOR Inhibitors—Future Directions
Epilepsy in TSC
Epilepsy is one of the most common neurological symptoms in TSC, affecting an estimated 85 % of patients, two thirds of whom are refractory to currently available medical therapies [15]. Poor cognitive outcomes have been correlated with a history of seizure, earlier age at seizure onset, and refractory epilepsy [15]. The processes underlying the development of seizures and epilepsy in TSC are multifaceted. Cortical tubers are thought to be central to the development of epilepsy [45], with targeted tuber removal leading to seizure freedom in over half of selected individuals [46]. Peri-tuberal cortex has shown hyperexcitability on intracranial electroencephalogram monitoring [47], demonstrates immunohistologic and molecular abnormalities, and has shown dysregulated mTOR activity [48]. Although the pathophysiology of epilepsy in TSC is complex and implicates processes beyond the cortical tubers for which the disease is named, these additional cellular and molecular mechanisms may be amenable to correction by mTOR inhibitors and may represent a novel approach to epilepsy treatment in TSC. Pre-clinical studies have shown that manipulation of the mTOR pathway with rapamycin not only reduces the frequency of seizures in symptomatic animals, but pre-symptomatic treatment can also prevent the development of seizures and histopathologic abnormalities that are typically seen [49]. The pre-clinical data is promising and has provided the groundwork for human trials.
As a secondary outcome in the initial open-label study of everolimus in the treatment of SEGAs, everolimus therapy was associated with a clinically relevant reduction in the overall frequency of clinical and subclinical seizures [28]. In a prospective, open-label, phase 1/2 clinical trial, 20 patients (median age 8 years, range 2–21 years) underwent treatment with everolimus for 12 weeks [50••]. A clinically meaningful reduction in seizure frequency was seen, with the median seizure frequency decreasing by 73 %, with four subjects (20 %) being free of clinical seizures and seven subjects (35 %) having a greater than 90 % reduction in seizure frequency. Additionally, parent-reported improvements in behavior and quality of life were observed. Most recently, a placebo-controlled, randomized, double-blind phase 3 clinical trial of adjunctive everolimus in TSC patients with treatment-resistant epilepsy showed positive results, with the response rate (greater than 50 % reduction in seizures) being significantly higher in the treatment groups (28.2 % in the low-exposure group and 40.0 % in the high-exposure group) as compared to the placebo group (15.1 %) [51••]. These results are promising, with everolimus representing a truly targeted therapy addressing the underlying molecular pathology of TSC, which may soon come into standard clinical use for the treatment for epilepsy in TSC.
Neurocognition in TSC
The clinical manifestations of TSC include neuropsychiatric conditions including autism spectrum disorder, attention-deficit disorder, and mood and anxiety disorders. Additionally, a wide range of intellectual abilities is seen, ranging from severe intellectual disability to normal intelligence. Autism spectrum disorder is diagnosed in an estimated 20–60 % of individuals with TSC, and autism spectrum disorder, epilepsy, and intellectual disability have been shown to be closely linked [52–54]. TSC is one of the most common single-gene disorders associated with autism spectrum disorder [55]. In a recent study, the social communication profile of toddlers with TSC and autism spectrum syndrome demonstrated complete convergence when compared to toddlers with non-syndromic autism spectrum disorder, using measures including the Autism Diagnostic Observation Schedule (ADOS) and the Mullen Scales of Early Learning [56]. Autism spectrum disorder is a behaviorally defined neurodevelopmental disorder, and although the specific cause has yet to be established, a genetic contribution has clearly been implicated. TSC has long been seen as a useful model for the study of autism spectrum disorder, and as the underlying molecular and biochemical mechanisms of TSC are elucidated, these findings may also help to unravel the complex mechanisms underlying autism spectrum disorder.
Pre-clinical studies have suggested that at least some of the neurocognitive impairments in TSC are related to dysregulated mTOR signaling rather than irreversible structural defects, and that treatment with rapamycin can attenuate some of the neurocognitive symptoms. In adult Tsc2 +/− mice deficits in learning and memory (in the absence of neuropathology and seizures), brief treatment with rapamycin reversed learning and memory impairments [57•]. Additionally, Tsc1 +/− and Tsc2 +/− mice have been shown to exhibit behavioral abnormalities and impairments in social interaction, which can also be recovered by treatment with rapamycin [58].
Although neurocognition had been identified as a secondary outcome measure during the open-label study of everolimus in the treatment of SEGAs, substantially below-average baseline intellectual abilities and comorbid behavioral disorders limited the cognitive assessments [28]. Results from an ongoing placebo-controlled, double-blind trial of everolimus in children with TSC between the ages of 6 and 21 years, with the primary endpoint looking at improvements on neurocognitive tests, are pending (NCT01289912).
mTOR Signaling in Other Neurological Syndromes
The extent of human disease involving the mTOR pathway extends far beyond TSC. Recent studies have demonstrated that aberrant mTOR signaling is associated with disorders of cortical development, which may lead to symptomatic epilepsy and intellectual disability [59–61]. Germline or somatic mutations have been found in various genes along the mTOR pathway, with specific examples including STE20-related kinase adapter alpha (STRADα) (polyhydramnios, megalencephaly, and symptomatic epilepsy) [60], AKT3, PI3K, or MTOR (hemimegalencephaly) [59, 62], and disheveled, Egl-10 and Pleckstrin domain-containing protein 5 (DEPDC5) or phosphatase and tensin homolog on chromosome 10 (PTEN) (focal cortical dysplasia type IIb) [63–65]. Collectively, these cortical brain malformations have been referred to as the “mTORopathies,” and they have solidified the crucial role of the mTOR pathway in the normal brain development and generated excitement about the potential for novel therapeutic targets.
Fragile X syndrome (FXS) is the most common inherited form of intellectual disability [66]. Autism spectrum disorder is identified in up to two thirds of males with FXS [67] and seizures are seen in roughly one quarter of children with FXS [68]. The fragile X mental retardation protein (FMRP) is an RNA-binding protein that negatively regulates translation and protein synthesis [69, 71, 70•]. In the hippocampi of juvenile Fmr1 knock-out mice, it has been shown that mTOR phosphorylation and activity are elevated [72]. The dysregulated mTOR signaling seen in Fmr1 KO mice is of great interest, as similar cognitive and social interaction deficits are observed in conditions (e.g., TSC) along the mTOR pathway. Additionally, in brain tissue and peripheral blood lymphocytes from patients with FXS, increased phosphorylation of the downstream mTOR effectors ribosomal protein S6 kinase (S6K1) and eIF4E has been shown [73]. The mTOR pathway may serve as a novel therapeutic target for neurobehavioral disorders, including FXS and other conditions along the mTOR cascade.
Although Alzheimer disease (AD) and Huntington disease (HD) are not diseases of childhood, interestingly, the mTOR pathway has also been implicated in these conditions. AD is a progressive neurodegenerative disorder characterized pathologically by the accumulation of beta-amyloid containing plaques, neurofibrillary tangles, and the loss of cortical neurons [74]. mTOR signaling has been shown to be dysregulated in postmortem samples obtained from the brains of AD patients, with levels of mTOR and downstream targets being correlated with levels of tau and phosphorylated tau [75]. HD is a dominantly inherited neurodegenerative disorder caused by a polyglutamine expansion, which leads to abnormal neuronal inclusions containing the proteins of huntingtin [76]. mTOR has been shown to be sequestered in such inclusions in cell models, transgenic mice, and human brains, with rapamycin being shown in a fly model of HD to protect against neurodegeneration by inducing autophagy and attenuating huntingtin accumulation [77, 78]. Although its role in these conditions is not yet fully understood, the mTOR pathway may potentially represent a novel therapeutic target in the treatment of these neurodegenerative disorders.
Limitations and Challenges
Although the major short-term side effects of oral mTOR inhibitors have been described [29••, 30, 39••], the long-term effects on growth and development in pediatric patients are not fully known. In the extension phase of EXIST-1, no patients terminated treatment (at a median exposure of 29 months) due to adverse events and no differences in height, height velocity, weight, and weight velocity were identified in patients younger than 18 years [30]. Amenorrhea was reported in 5/28 (18 %) of women and girls aged 10–55 years, a finding that has been reported in other studies [39••] and raises the question of the effect of everolimus on growth and sexual maturation.
Questions also remain about the optimal duration of treatment, and whether there is a critical treatment window during which mTOR inhibitors may have a potentially maximal effect. Pre-clinical studies suggest that intermittent dosing might have a role in the treatment of epilepsy in TSC, as rapamycin may have a sustained effect despite drug holidays [79].
Rapamycin as a cancer therapy has had limited success, in large part, because of feedback loops such as the S6K1-mediated feedback loop regulating PI3K signaling, the inhibition of which leads to an upregulation of PI3K signaling and ultimately pro-survival and proliferative signals through Akt and other kinases [19]. Additionally, rapamycin does not fully inhibit all the functions of mTORC1, which may lead to stimulation of other portions of the mTOR cascade (e.g., 4E-BP1) and ultimately dysregulated growth. Lastly, the mTOR pathway plays a crucial role in normal cellular growth and survival, and the effects of its inhibition on the developing brain have yet to be fully understood.
Conclusions
Since the discovery of rapamycin over 40 years, rapamycin derivatives and the mTOR pathway have become fields of great interest. A number of conditions have been traced to the mTOR pathway, with TSC being one of the most extensively studied. The introduction of rapamycin derivatives into clinical practice has led to a shift in the treatment of TSC, with oral and topical rapamycin derivatives replacing more invasive interventions for tumors. Ongoing clinical trials are currently testing whether mTOR inhibitors may also be efficacious for other symptoms of TSC that are not necessarily directly related to tumor growth, such as epilepsy and cognitive deficits.
The mTOR pathway has also been implicated in a number of neurological conditions including structural brain disorders, inherited neurocognitive disorders such as fragile X syndrome, autism spectrum disorder, and progressive neurodegenerative diseases such as Alzheimer disease and Huntington disease. Manipulation of the mTOR pathway may have much broader applications beyond TSC, with the mTOR pathway holding enormous potential for novel, targeted therapies. However, this excitement must be balanced by an understanding of the possible unanticipated effects of mTOR pathway manipulation, particularly in the developing brain.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Rogeveen, J. The journal of Jacob Roggeveen, ed. A. Sharp. 1970: Oxford University Press.
Vezina C, Kudelski A, Sehgal SN. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J Antibiot (Tokyo). 1975;28(10):721–6.
Sehgal SN, Baker H, Vezina C. Rapamycin (AY-22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J Antibiot (Tokyo). 1975;28(10):727–32.
Martel RR, Klicius J, Galet S. Inhibition of the immune response by rapamycin, a new antifungal antibiotic. Can J Physiol Pharmacol. 1977;55(1):48–51.
Houchens DP, Ovejera AA, Riblet SM, et al. Human brain tumor xenografts in nude mice as a chemotherapy model. Eur J Cancer Clin Oncol. 1983;19(6):799–805.
Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253(5022):905–9.
Sabatini DM, Erdjument-Bromage H, Lui M, et al. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. 1994;78(1):35–43.
Brown EJ, Albers MW, Shin TB, et al. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369(6483):756–8.
Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12(1):21–35.
Lipton JO, Sahin M. The neurology of mTOR. Neuron. 2014;84(2):275–91.
Osborne JP, Fryer A, Webb D. Epidemiology of tuberous sclerosis. Ann N Y Acad Sci. 1991;615:125–7.
Bourneville DM. Sclerose tubereuse des circonvolutions cerebrales: idiotie et epilepsie hemiplegique. Arch Neurol. 1880;1:81–91.
Northrup H, Krueger DA, G. International Tuberous Sclerosis Complex Consensus. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49(4):243–54.
Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355(13):1345–56.
Chu-Shore CJ, Major P, Camposano S, et al. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia. 2010;51(7):1236–41.
van Slegtenhorst M, de Hoogt R, Hermans C, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997;277(5327):805–8.
European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell. 1993;75(7):1305–15.
Tee AR, Fingar DC, Manning BD, et al. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci U S A. 2002;99(21):13571–6.
Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–93.
Inoki K, Li Y, Xu T, et al. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17(15):1829–34.
Sarbassov DD, Ali SM, Sengupta S, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22(2):159–68.
Goh S, Butler W, Thiele EA. Subependymal giant cell tumors in tuberous sclerosis complex. Neurology. 2004;63(8):1457–61.
Roth J, Roach ES, Bartels U, et al. Subependymal giant cell astrocytoma: diagnosis, screening, and treatment. Recommendations from the International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2012;49(6):439–44.
Adriaensen ME, Schaefer-Prokop CM, Stijnen T, et al. Prevalence of subependymal giant cell tumors in patients with tuberous sclerosis and a review of the literature. Eur J Neurol. 2009;16(6):691–6.
de Ribaupierre S, Dorfmuller G, Bulteau C, et al. Subependymal giant-cell astrocytomas in pediatric tuberous sclerosis disease: when should we operate? Neurosurgery. 2007;60(1):83–9. discussion 89–90.
Franz DN, Leonard J, Tudor C, et al. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 2006;59(3):490–8.
Koenig MK, Butler IJ, Northrup H. Regression of subependymal giant cell astrocytoma with rapamycin in tuberous sclerosis complex. J Child Neurol. 2008;23(10):1238–9.
Krueger DA, Care MM, Holland K, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363(19):1801–11.
•• Franz DN, Belousova E, Sparagana S, et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2013;381(9861):125–32. Randomized, placebo-controlled trial demonstrating efficacy of everolimus in the treatment of subependymal giant cell astrocytomas in TSC.
Franz DN, Belousova E, Sparagana S, et al. Everolimus for subependymal giant cell astrocytoma in patients with tuberous sclerosis complex: 2-year open-label extension of the randomised EXIST-1 study. Lancet Oncol. 2014;15(13):1513–20.
Krueger DA, Northrup H. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49(4):255–65.
Bissler JJ, Kingswood JC. Renal angiomyolipomata. Kidney Int. 2004;66(3):924–34.
Rakowski SK, Winterkorn EB, Paul E, et al. Renal manifestations of tuberous sclerosis complex: incidence, prognosis, and predictive factors. Kidney Int. 2006;70(10):1777–82.
Shepherd CW, Gomez MR, Lie JT, et al. Causes of death in patients with tuberous sclerosis. Mayo Clin Proc. 1991;66(8):792–6.
Ewalt DH, Diamond N, Rees C, et al. Long-term outcome of transcatheter embolization of renal angiomyolipomas due to tuberous sclerosis complex. J Urol. 2005;174(5):1764–6.
Moss J, Avila NA, Barnes PM, et al. Prevalence and clinical characteristics of lymphangioleiomyomatosis (LAM) in patients with tuberous sclerosis complex. Am J Respir Crit Care Med. 2001;164(4):669–71.
Costello LC, Hartman TE, Ryu JH. High frequency of pulmonary lymphangioleiomyomatosis in women with tuberous sclerosis complex. Mayo Clin Proc. 2000;75(6):591–4.
Bissler JJ, McCormack FX, Young LR, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358(2):140–51.
•• Bissler JJ, Kingswood JC, Radzikowska E, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. 2013;381(9869):817–24. Randomized, placebo-controlled trial demonstrating efficacy of everolimus in the treatment of renal angiomyolipomas in TSC.
Haemel AK, O’Brian AL, Teng JM. Topical rapamycin: a novel approach to facial angiofibromas in tuberous sclerosis. Arch Dermatol. 2010;146(7):715–8.
Song MG, Park KB, Lee ES. Resurfacing of facial angiofibromas in tuberous sclerosis patients using CO2 laser with flashscanner. Dermatol Surg. 1999;25(12):970–3.
Boixeda P, Sanchez-Miralles E, Azana JM, et al. CO2, argon, and pulsed dye laser treatment of angiofibromas. J Dermatol Surg Oncol. 1994;20(12):808–12.
Hofbauer GF, Marcollo-Pini A, Corsenca A, et al. The mTOR inhibitor rapamycin significantly improves facial angiofibroma lesions in a patient with tuberous sclerosis. Br J Dermatol. 2008;159(2):473–5.
Koenig MK, Hebert AA, Roberson J, et al. Topical rapamycin therapy to alleviate the cutaneous manifestations of tuberous sclerosis complex: a double-blind, randomized, controlled trial to evaluate the safety and efficacy of topically applied rapamycin. Drugs R D. 2012;12(3):121–6.
Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet. 2008;372(9639):657–68.
Weiner HL, Carlson C, Ridgway EB, et al. Epilepsy surgery in young children with tuberous sclerosis: results of a novel approach. Pediatrics. 2006;117(5):1494–502.
Ma TS, Elliott RE, Ruppe V, et al. Electrocorticographic evidence of perituberal cortex epileptogenicity in tuberous sclerosis complex. J Neurosurg Pediatr. 2012;10(5):376–82.
Ruppe V, Dilsiz P, Reiss CS, et al. Developmental brain abnormalities in tuberous sclerosis complex: a comparative tissue analysis of cortical tubers and perituberal cortex. Epilepsia. 2014;55(4):539–50.
Zeng LH, Xu L, Gutmann DH, et al. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol. 2008;63(4):444–53.
•• Krueger DA, Wilfong AA, Holland-Bouley K, et al. Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann Neurol. 2013;74(5):679–87. Case series describing the efficacy of everolimus for refractory epilepsy in TSC. The findings in this trial allowed for the development of a randomized, placebo-controlled trial of everolimus in TSC.
•• French JA, Lawson JA, Yapici Z, et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet. 2016. doi:10.1016/S0140-6736(16)31419-2. A randomized, placebo-controlled trial of everolimus for refractory epilepsy in TSC, which demonstrated superiority of everolimus versus placebo.
Ehninger D, Silva AJ. Rapamycin for treating tuberous sclerosis and autism spectrum disorders. Trends Mol Med. 2011;17(2):78–87.
Numis AL, Major P, Montenegro MA, et al. Identification of risk factors for autism spectrum disorders in tuberous sclerosis complex. Neurology. 2011;76(11):981–7.
Vignoli A, La Briola F, Peron A, et al. Autism spectrum disorder in tuberous sclerosis complex: searching for risk markers. Orphanet J Rare Dis. 2015;10:154.
Richards C, Jones C, Groves L, et al. Prevalence of autism spectrum disorder phenomenology in genetic disorders: a systematic review and meta-analysis. Lancet Psychiatry. 2015;2(10):909–16.
Jeste SS, Varcin KJ, Hellemann GS, et al. Symptom profiles of autism spectrum disorder in tuberous sclerosis complex. Neurology. 2016;87(8):766–72.
• Ehninger D, Han S, Shilyansky C, et al. Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat Med. 2008;14(8):843–8. Study in an animal model of TSC showing that affected mice demonstrate deficits in learning and memory, and that brief treatment with rapamycin may ameliorate the cognitive dysfunction and behavioral deficits seen in affected mice. This study shows promise for similar applications of rapamycin derivatives in humans, and the potential for the treatment of cognitive and behavioral symptoms in affected patients.
Sato A, Kasai S, Kobayashi T, et al. Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat Commun. 2012;3:1292.
Poduri A, Evrony GD, Cai X, et al. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron. 2012;74(1):41–8.
Orlova KA, Parker WE, Heuer GG, et al. STRADalpha deficiency results in aberrant mTORC1 signaling during corticogenesis in humans and mice. J Clin Invest. 2010;120(5):1591–602.
Lim JS, Kim WI, Kang HC, et al. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med. 2015;21(4):395–400.
Lee JH, Huynh M, Silhavy JL, et al. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet. 2012;44(8):941–5.
Cheung KM, Lam CW, Chan YK, et al. Atypical focal cortical dysplasia in a patient with Cowden syndrome. Hong Kong Med J. 2014;20(2):165–7.
Baulac S, Ishida S, Marsan E, et al. Familial focal epilepsy with focal cortical dysplasia due to DEPDC5 mutations. Ann Neurol. 2015;77(4):675–83.
Ishida S, Picard F, Rudolf G, et al. Mutations of DEPDC5 cause autosomal dominant focal epilepsies. Nat Genet. 2013;45(5):552–5.
Hernandez RN, Feinberg RL, Vaurio R, et al. Autism spectrum disorder in fragile X syndrome: a longitudinal evaluation. Am J Med Genet A. 2009;149a(6):1125–37.
Gross C, Hoffmann A, Bassell GJ, et al. Therapeutic strategies in fragile X syndrome: from bench to bedside and back. Neurotherapeutics. 2015;12(3):584–608.
Hagerman PJ, Stafstrom CE. Origins of epilepsy in fragile X syndrome. Epilepsy Curr. 2009;9(4):108–12.
Darnell JC, Van Driesche SJ, Zhang C, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146(2):247–61.
• Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33(2):67–75. Study demonstrating increased phosphorylation of the mTOR substrates S6K1 and eIF4E in brain tissue and peripheral blood lymphocytes of patients with fragile X syndrome (FXS). This study provides a mechanistic link between FXS and disorders such as TSC and also provides new therapeutic targets for FXS.
Verkerk AJ, Pieretti M, Sutcliffe JS, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65(5):905–14.
Sharma A, Hoeffer CA, Takayasu Y, et al. Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci. 2010;30(2):694–702.
Hoeffer CA, Sanchez E, Hagerman RJ, et al. Altered mTOR signaling and enhanced CYFIP2 expression levels in subjects with fragile X syndrome. Genes Brain Behav. 2012;11(3):332–41.
Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362(4):329–44.
Li X, Alafuzoff I, Soininen H, et al. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer’s disease brain. FEBS J. 2005;272(16):4211–20.
Davies SW, Turmaine M, Cozens BA, et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90(3):537–48.
Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36(6):585–95.
Williams A, Sarkar S, Cuddon P, et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat Chem Biol. 2008;4(5):295–305.
Rensing N, Han L, Wong M. Intermittent dosing of rapamycin maintains antiepileptogenic effects in a mouse model of tuberous sclerosis complex. Epilepsia. 2015;56(7):1088–97.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Anna Jeong declares no conflict of interest.
Michael Wong is a site PI for a Novartis-sponsored clinical trial on the effects of everolimus on refractory seizures in tuberous sclerosis patients.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Pediatric Neurology
Rights and permissions
About this article

Cite this article
Jeong, A., Wong, M. mTOR Inhibitors in Children: Current Indications and Future Directions in Neurology. Curr Neurol Neurosci Rep 16, 102 (2016). https://doi.org/10.1007/s11910-016-0708-8
Published:
DOI: https://doi.org/10.1007/s11910-016-0708-8