
Abstract
Pulmonary arterial hypertension (PAH) is characterized by molecular and pathologic alteration to the pulmonary circulation, resulting in increased pulmonary vascular resistance, right ventricular failure, and eventual death. Pharmacologic treatment of PAH consists of use of a multitude of pulmonary vasodilators, sometimes in combination. PAH has been associated with increased thrombosis and disrupted coagulation and fibrinolysis, making anticoagulation an attractive and frequently employed therapeutic modality. Observational studies have provided some insight into the therapeutic potential of anticoagulation in idiopathic PAH, but there is a distinct lack of well-controlled prospective trials. Due to the conflicting evidence, there is a large amount of heterogeneity in the application of therapeutic anticoagulation in PAH and further well-controlled prospective trials are needed to clarify its role in treating PAH.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
It has been 125 years since Romberg described the first reported case of pulmonary arterial hypertension (PAH): a young patient who died suddenly, identifying both right ventricular and pulmonary artery sclerosis without apparent cause [1]. Since then, particularly in the last two decades, the pathobiology of PAH has been the subject of intense study, revealing foundational cellular and molecular mechanisms of the disease that have led to the availability of four classes of PAH-targeted therapeutics [2]. While available therapies have demonstrated efficacy at improving functionality and quality of life, PAH continues to be a progressive disease that results in eventual right ventricular failure and death. This poor prognosis heightens the necessity for additional disease-modifying therapies. One pathobiologic aspect of PAH that has been recognized from the early pathologic descriptions is the frequent presence of in situ thrombus within the pulmonary arteries of individuals with PAH [3]. This, coupled with modern knowledge of dysregulated coagulation, antithrombotic homeostasis, and endothelial dysfunction in PAH, has led to efforts at using systemic anticoagulation in the disease in an effort to improve outcomes.
However, anticoagulation therapy in patients with World Health Organization (WHO) group 1 has not been the subject of a randomized controlled trial to determine efficacy, leading to immense variability in clinical practice. With the changing demographic of the disease, the advent of effective PAH-specific therapies, and recently published studies involving anticoagulation in the modern PAH patient, a critical reappraisal of the data is needed. The goals of this review will be to review the biologic plausibility for anticoagulation in PAH, summarize the historical uncontrolled data, and finally to integrate the more recent highest-quality evidence into expert recommendations in an effort to provide clinicians with the most evidenced-based approach to anticoagulation in PAH.
Pathophysiologic Rationale for Anticoagulation in PAH
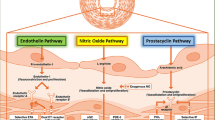
The pulmonary vascular remodeling in PAH involves a complex milieu of excessive cell growth, inflammation, a metabolic shift towards aerobic glycolysis, and loss of potent antiproliferative and vasodilatory mediators [4]. Current approved therapies primarily target a single aspect of this multifaceted pathobiology—modulating the nitric oxide, endothelin, and prostacyclin vasoactive pathways [5]. While these therapies have proved beneficial for patient outcomes, there is ongoing interest in targeting other facets of the disease process.
Autopsy Observations of In Situ Thrombus
The initial use of anticoagulation therapy in PAH was initially based on postmortem observational studies that demonstrated a high prevalence of in situ thrombosis within the pulmonary vasculature of patients with PAH (Fig. 1). In 1966, Storstein et al. described the characteristic pathology of PAH, with the comment of “occasional complete occlusion of the lumen by organized thrombi” [3]. Further, Fuster et al., in their 1984 landmark paper, characterized lung tissue obtained at autopsy from 56 pulmonary hypertension (PH) patients. This revealed two major histologic patterns: plexogenic arteriopathy in 18 patients (32 %) and chronic thromboembolism in 32 (57 %). Interestingly, in many of the plexogenic arteriopathy specimens, they noted that in the characteristic focal lesions, it was “not infrequent” to see fresh platelet-fibrin thrombi and organized arterial thrombi.

In situ thrombus within plexiform lesions in IPAH patients. a Platelet aggregates (arrow) within a plexiform lesion (arrowhead). b Recent thrombus (arrow) within a pulmonary artery with associated plexiform lesion (circled)
Circulating Markers of a Prothrombotic State
Since these autopsy-based descriptive studies, it is increasingly recognized that patients with PAH have dysregulated coagulation and antithrombotic homeostasis, which may contribute to a prothrombotic state. Endothelial cells (EC) are a fundamental contributor to fibrinolytic homeostasis through the synthesis and secretion of factors such as tissue plasminogen activator (tPA), tissue factor (TF), and plasminogen activator inhibitor (PAI)-1, and there is considerable evidence that endothelial cell dysfunction inherent to PAH may contribute to disordered coagulation and thrombolysis [6].
Human studies of circulating markers of thrombogenesis and fibrinolysis in PAH have been conflicting. It has been demonstrated that in PAH, there are increased circulating microparticles containing active TF and higher amounts of EC-derived microparticles in occluded pulmonary artery blood than peripheral blood. Moreover, the quantity of these TF-bearing microparticles correlated with the patient’s functional class and 6-min-walk distance [7]. Another study suggesting a relative prothrombotic state measured euglobulin clot lysis time (ECLT) and peripheral blood levels of tPA and PAI-1 before and after a peripheral venous occlusion test in patients with both PAH and secondary PH. They found that patients (relative to healthy controls) had significantly prolonged ECLT, a blunted tPA response to vascular occlusion, and increased levels and activity of PAI-1 [8]. Similarly, a separate analysis of 16 patients with PAH found that relative to healthy controls there was elevated venous and arterial von Willebrand factor antigen, and increased levels of PAI-1, most pronounced in the arterial circulation—suggesting possible intrapulmonary production [9]. In contrast, Kopec et al. studied 27 patients with hemodynamically confirmed IPAH and 16 controls, finding significantly increased serum levels of profibrinolytic markers tPA and plasmin-antiplasmin. However, they did not find increased levels of thrombogenic markers PAI-1, thombin-antithrombin, or prothrombin fragments 1 + 2 [10]. These studies strongly suggest dysregulated thrombolytic homeostasis, but definitive causation and a deeper mechanistic understanding remain to be determined.
Cellular and Animal Models
There has been limited work utilizing experimental models of PH to investigate effects of anticoagulation therapies on the pathobiology of PH. Aspirin, when given to the monocrotaline rat model of PH, reduced right ventricular systolic pressure (RVSP) and right ventricular (RV) hypertrophy. Interestingly, this correlated with a reduction in serotonin, a recognized contributor to the pathobiology of PAH [11]. A separate study, also utilizing the monocrotaline rat model of PH, examined pretreatment with rivaroxaban (a direct factor Xa inhibitor), enoxaparin, warfarin, or placebo. The animals treated with rivaroxaban had a significant reduction in right ventricular (RV) systolic pressure (RVSP) and RV hypertrophy, whereas warfarin only resulted in reduced RVSP, and enoxaparin did not affect either parameter [12]. A mechanistic understanding of these findings is still lacking, but they warrant further investigation given the therapeutic potential in PAH.
Molecular and cellular work is similarly limited but hints at potential mechanisms and targeting of dysregulated coagulation in PAH. Increased activity of Rho (Ras homologous) GTP-binding proteins has been implicated in numerous foundational aspects of the pathobiology of PAH, including cellular proliferation, hypertrophy, vasoconstriction, and inflammation [13]. Interestingly, Rho kinase activity (via RhoA and Rac) has been demonstrated to increase expression of both tissue factor and thromboxane A2-induced platelet activation and aggregation [14, 15]. Additionally, Rac in combination with thrombin is associated with increased activity of nuclear factor-kappa B-dependent tissue factor expression and activity—thus contributing to a prothrombotic state [16].
In conglomeration, there is considerable autopsy, human biomarker, and translational data that suggest dysregulated coagulation and thrombolytic homeostasis occur in PAH (summarized in Fig. 2). Further, with multiple pharmacologic agents readily available that target the discussed pathways, this represents a novel and attractive area for therapeutic intervention beyond the traditional vasodilatory agents.

Summary of proposed pathways by which the deranged pulmonary endothelium in PAH may lead to a prothrombotic state
Anticoagulation in PAH: Clinical Data
Early Studies of Anticoagulation in PAH
Prior to the development of modern pulmonary vasodilator therapies, the cornerstone of management consisted of diuretics, digoxin therapy, and warfarin therapy. The early use of warfarin therapy in PAH was based primarily on biologic plausibility described above and two landmark observational studies. A 1966 cohort study published by Storstein et al. included 17 IPAH patients and found no benefit in the 10 patients receiving anticoagulation [3]. In 1984, Fuster et al. published the first large retrospective study that supported anticoagulation in PAH, including 120 patients with IPAH (referred to at the time as primary pulmonary hypertension) and found via multivariate analysis that pulmonary artery saturation and anticoagulation were significant predictors of prognosis [17•]. Almost a decade later, Rich et al. performed an uncontrolled prospective study of calcium channel blocker therapy in IPAH, finding in secondary analysis that anticoagulation was associated with improved survival in a subset of those patients receiving anticoagulation due to “nonuniformity” on ventilation/perfusion scanning [18•]. The benefit of anticoagulation was independent of their response to calcium channel blockers. These two observational studies set into motion the widespread adoption of anticoagulation in IPAH.
In 2006, Johnson et al. published the first systematic review of seven single-center observational studies looking at the effect of anticoagulation on mortality in PAH [19]. A summary of these studies and all subsequent observational data is listed in Table 1. While a meta-analysis could not be performed due to study heterogeneity, five of seven studies demonstrated a benefit to anticoagulation with the conclusion that anticoagulation in PAH may be beneficial, but called for more rigorous study. Further, some of the positive studies were confounded by the variable use of other therapeutics (i.e., Ogata et al. combined anticoagulation with either nifedepine or isoproterenol), limiting conclusions regarding the efficacy of anticoagulation [20]. Other positive studies were conflicted by a skewed patient population, as demonstrated by Frank et al., where 70 % of participants had PH due to aminorex, a specific anorexigen. The survival time of aminorex-exposed warfarin-treated or naïve patients was 7.5 years on average—much longer than typical IPAH survival at that time. Further, survival benefit associated with warfarin was isolated to anorexigen-exposed patients; there was no survival difference in subgroup analysis of IPAH patients receiving warfarin [21•]. During this era, clinical trials reported heterogenous use of anticoagulation in PAH (ranging from 51 to 86 %), reflecting guideline recommendations that called for consideration of anticoagulation in patients with IPAH or anorexigen-induced PAH [28].
Recent and Future Studies of Anticoagulation in PAH
In the last five years, there has been an additional systematic review and four observational studies on the subject. While still observational in nature, these studies have included patients on more modern PAH-specific therapies, utilized larger sample sizes with multicenter experiences, and relied on more stringent methodological characteristics. Further, because of the current shift toward early recognition, diagnosis, and treatment of patients using highly effective PAH-specific therapies along with changing patient demographics, it has become increasingly recognized that the efficacy of anticoagulation must be studied within this context. Two studies specifically addressed patients with connective tissue disease-associated PAH (CTD-PAH), which had not been examined previously. Ngian et al. used a multicenter Australian cohort of 117 patients with CTD-PAH (∼84 % with SSc-PAH) to evaluate predictors of mortality [25]. After adjusting for multiple covariates, anticoagulation (∼30 % of cohort) was found to be protective with an adjusted hazard ratio of 0.2 (0.05–0.78, p = 0.02). Conversely, Johnson et al. utilized a single-center Canadian cohort of 275 SSc-PAH patients, of which 28 % were anticoagulated, to determine the probability that anticoagulation improves survival [24]. They matched treated and untreated patients using propensity scoring and then utilized a Bayesian logistic regression model to determine that there was no difference in 3-year survival between the two groups (61 % warfarin-unexposed vs 58 % warfarin-exposed). Additionally, they found that the probability of improving survival by 6 months or more with warfarin was only 23.5 %. In other words, there was a 76.5 % chance of no benefit or harm at 6 months. Interestingly, there did not appear to be an increase in bleeding in CTD-PAH patients who received anticoagulation in either study compared to non-anticoagulated CTD-PAH patients.
Additional insight into anticoagulation in PAH patients has been garnered from large prospective registry studies, including the European database Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA) and the US-based longitudinal Registry to Evaluate Early and Long-term PAH Disease Management (REVEAL). COMPERA included 1283 patients with PAH, 62 % of whom had IPAH, finding that 66 % of PAH patients were treated with anticoagulation while 43 % of those with other forms of PAH were treated with anticoagulation. Survival difference at 3 years was found to be significantly increased in IPAH patients subjected to anticoagulation, even following rigorous Cox multivariable regression analysis. In contrast, in those with alternative forms of PAH, there was no survival advantage seen with anticoagulation [26•]. The recent REVEAL Registry analysis of anticoagulation longitudinally studied patients with IPAH and systemic sclerosis-associated PAH (SSc-PAH). In contrast to COMPERA, it was found that there was no survival difference in IPAH with anticoagulation; however, in SSc-PAH, there was noted to be increased mortality in those receiving warfarin in the previous year or at any time post-baseline when compared to matched warfarin-naïve patients [27•]. Recent systematic reviews include a contribution by Caldeira et al. that included two prospective and seven retrospective observational studies. In a random effect meta-analysis of general PAH patients, it was found that oral anticoagulation was associated with a 31 % mortality risk reduction [29].
In addition to the conflicted results of the efficacy of anticoagulation in PAH, there remains concern for study bias in all the aforementioned studies, and thus, there remains heterogeneity of anticoagulation use in PAH. Of the nine studies summarized in Table 1, when stratifying studies before and after 2006, there is no difference in rates of PAH patients (excluding CTD-PAH) being subjected to anticoagulation before and after 2006 (61 vs 60 %, respectively). However, in two of the largest recently published randomized controlled trials in PAH, SERAPHIN (macitentan), and AMBITION (combination ambrisentan and tadalafil), only 51 and 31 % of patients were anticoagulated (respectively), indicating a more recent trend towards using less anticoagulation [30, 31]. This controversy emphasizes the need for rigorous randomized controlled trials of anticoagulation in PAH. This effort has been hampered by the generic status of warfarin and tight National Institutes of Health funding lines. An alternative approach might involve a trial of the novel oral anticoagulants, though it remains unknown if industry sources would be interested in funding a trial for a rare disease such as PAH. Additionally, given the efficacy of currently approved PAH-specific therapies and careful exclusion of patients with CTEPH, it may prove difficult to enroll enough patients to detect a clinically significant difference in outcome.
Conclusion
Given the uncertainty regarding the role of anticoagulants in PAH, our approach has been to address the issue on a case-by-case basis with shared physician-patient decision-making. Careful exclusion of patients that may be placed at risk by anticoagulation should be exercised, excluding patients with history of adherence issues, participation in high-risk activities, or prior major bleeding complications. Clinical judgment must be used to identify patients who may be at increased risk for thrombosis, such as those with impaired cardiac output, chronic central venous access, and immobility. Ultimately, providers must rely on clinical judgment and critical application of the somewhat contradictory and inconclusive data available. Investigation of anticoagulation in well-designed clinical studies remains an important unmet need in PAH.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Romberg E. Ueber Sklerose der Lungen arterie. Dtsch Archiv Klin Med. 1891;:197–206.
Galie N, Corris PA, Frost A, Girgis RE, Granton J, Jing Z-C, et al. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62:D60–72.
Storstein O, Efskind L, Müller C, Rokseth R, Sander S. Primary pulmonary hypertension with emphasis on its etiology and treatment. Acta Med Scand. 1966;179:197–212.
Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2012;122:4306–13.
Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med. 2004;351:1425–36.
Budhiraja R, Tuder RM, Hassoun PM. Endothelial dysfunction in pulmonary hypertension. Circulation. 2004;109:159–65.
Bakouboula B, Morel O, Faure A, Zobairi F, Jesel L, Trinh A, et al. Procoagulant membrane microparticles correlate with the severity of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;177:536–43.
Altman R, Scazziota A, Rouvier J, Gurfinkei E, Favaloro R, Perrone S, et al. Coagulation and fibrinolytic parameters in patients with pulmonary hypertension. Clin Cardiol. Wiley Periodicals, Inc; 1996;19:549–54.
Hoeper MM, Sosada M, Fabel H. Plasma coagulation profiles in patients with severe primary pulmonary hypertension. Eur Respir J. 1998;12:1446–9.
Kopeć G, Moertl D, Steiner S, Stępień E, Mikołajczyk T, Podolec J, et al. Markers of thrombogenesis and fibrinolysis and their relation to inflammation and endothelial activation in patients with idiopathic pulmonary arterial hypertension. PLoS ONE. 2013;8:e82628–8.
Shen L, Shen J, Pu J, He B. Aspirin attenuates pulmonary arterial hypertension in rats by reducing plasma 5-hydroxytryptamine levels. Cell Biochem Biophys. 2011;61:23–31.
Delbeck M, Nickel KF, Perzborn E, Ellinghaus P, Strassburger J, Kast R, et al. A role for coagulation factor Xa in experimental pulmonary arterial hypertension. Cardiovasc Res. 2011;92:159–68.
Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–69.
Nagata K, Ishibashi T, Sakamoto T, Ohkawara H, Shindo J, Yokoyama K, et al. Rho/Rho-kinase is involved in the synthesis of tissue factor in human monocytes. Atherosclerosis. 2002;163:39–47.
Akbar H, Kim J, Funk K, Cancelas JA, SHANG X, Chen L, et al. Genetic and pharmacologic evidence that Rac1 GTPase is involved in regulation of platelet secretion and aggregation. J Thromb Haemost. Blackwell Publishing Ltd; 2007;5:1747–55.
Djordjevic T, Hess J, Herkert O, Görlach A, Belaiba RS. Rac regulates thrombin-induced tissue factor expression in pulmonary artery smooth muscle cells involving the nuclear factor-kappaB pathway. Antioxid Redox Signal. 2004;6:713–20.
Fuster V, Steele PM, Edwards WD, Gersh BJ, McGoon MD, Frye RL. Primary pulmonary hypertension: natural history and the importance of thrombosis. Circulation. 1984;70:580–7. Classic observational study highlighting the frequency of in situ thrombosis and therapeutic potential of anticoagulation.
Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med. 1992;327:76–81. One of the earliest prospective efforts to study the effects of anticoagulation in PAH; subgroup analysis revealed that patients on vitamin K antagonists had reduced mortality.
Johnson SR, Mehta S, Granton JT. Anticoagulation in pulmonary arterial hypertension: a qualitative systematic review. Eur Respir J. 2006;28:999–1004.
Ogata M, Ohe M, Shirato K, Takishima T. Effects of a combination therapy of anticoagulant and vasodilator on the long-term prognosis of primary pulmonary hypertension. Jpn Circ J. 1993;57:63–9.
Frank H, Mlczoch J, Huber K, Schuster E, Gurtner HP, Kneussl M. The effect of anticoagulant therapy in primary and anorectic drug-induced pulmonary hypertension. Chest. 1997;112:714–21. This retrospective cohort study found a survival advantage with vitamin K antagonist therapy in patients with anorectic-induced PAH, but not with idiopathic disease.
Roman A, Rodés-Cabau J, Lara B, Bravo C, Monforte V, Pallissa E, et al. Clinico-hemodynamic study and treatment of 44 patients with primary pulmonary hypertension. Med Clin (Barc). 2002;118:761–6.
Kawut SM, Horn EM, Berekashvili KK, Garofano RP, Goldsmith RL, Widlitz AC, et al. New predictors of outcome in idiopathic pulmonary arterial hypertension. Am J Cardiol. 2005;95:199–203.
Johnson SR, Granton JT, Tomlinson GA, Grosbein HA, Le T, Lee P, et al. Warfarin in systemic sclerosis-associated and idiopathic pulmonary arterial hypertension. A Bayesian approach to evaluating treatment for uncommon disease. J Rheumatol. 2012;39:276–85.
Ngian G-S, Stevens W, Prior D, Gabbay E, Roddy J, Tran A, et al. Predictors of mortality in connective tissue disease-associated pulmonary arterial hypertension: a cohort study. Arthritis Res Ther. 2012;14:R213–3.
Olsson KM, Delcroix M, Ghofrani HA, Tiede H, Huscher D, Speich R, et al. Anticoagulation and survival in pulmonary arterial hypertension: results from the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA). Circ. Lippincott Williams & Wilkins; 2014;129:57–65. This large prospective cohort study found survival benefit with anticoagulation in IPAH.
Preston IR, Roberts KE, Miller DP, Sen GP, Selej M, Benton WW, et al. Effect of warfarin treatment on survival of patients with pulmonary arterial hypertension (PAH) in the Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL). Circulation. 2015;132:2403–11. This study, similar to Olsson et al., is a large prospective cohort study but in contrast found no survival benefit with anticoagulation in PAH. This contradiction highlights the need for a large, placebo-controlled therapeutic trial of anticoagulation in PAH.
Galie N, Seeger W, Naeije R, Simonneau G, Rubin LJ. Comparative analysis of clinical trials and evidence-based treatment algorithm in pulmonary arterial hypertension. JAC. 2004;43:81S–8S.
Caldeira D, Loureiro MJ, Costa J, Pinto FJ, Ferreira JJ. Oral anticoagulation for pulmonary arterial hypertension: systematic review and meta-analysis. Can J Cardiol. 2014;30:879–87.
Pulido T, Adzerikho I, Channick RN, Delcroix M, Galie N, Ghofrani HA, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369:809–18.
Galie N, Barberà JA, Frost AE, Ghofrani HA, Hoeper MM, McLaughlin VV, et al. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med. 2015;373:834–44.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Dr. Badesch reports grants and personal fees from Actelion, Gilead, Bayer, Belleraphon, United Therapeutics, and Lung LLC, and grants from Reatta, Eiger, and NIH. Drs. Robinson, Pugliese, and Fox declare no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Pulmonary Hypertension
Rights and permissions
About this article

Cite this article
Robinson, J.C., Pugliese, S.C., Fox, D.L. et al. Anticoagulation in Pulmonary Arterial Hypertension. Curr Hypertens Rep 18, 47 (2016). https://doi.org/10.1007/s11906-016-0657-2
Published:
DOI: https://doi.org/10.1007/s11906-016-0657-2