
Abstract
Pulmonary arterial hypertension (PAH) is a rare, progressively worsening disease characterized by dysfunction among endothelial and smooth muscle cells within the pulmonary vasculature with a resultant increase in pulmonary vascular resistance, right ventricular maladaptation and failure, and ultimately early death. The three major therapeutic classes of medications available to treat PAH act as either prostacyclin analogs or endothelin receptor antagonists (ERAs) or by increasing local nitric oxide (NO) levels by means of phosphodiesterase type 5 inhibitors. Several recent trials have investigated the use of oral prostanoid therapy, next-generation ERAs, and soluble guanylate cyclase stimulators (to increase NO levels) as well as novel formulations of pre-existing therapies. The goal of this manuscript is to briefly review established therapies and then discuss recent developments and practical considerations in each of the major drug classes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
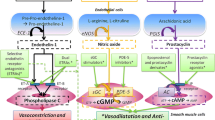
Pulmonary hypertension is a rare disease with a complex pathobiology that involves progressive endothelial and smooth muscle cell dysfunction within the pulmonary vasculature [1] resulting in right ventricular (RV) maladaptation, RV failure, and ultimately death. The disease process is thought to result from an imbalance of vasoconstriction, vascular remodeling, and cellular proliferation within the lung [1]. Current pulmonary arterial hypertension (PAH) therapies target these processes at distinct biochemical pathways involving activation of the prostacyclin pathway, endothelin receptor antagonism, and inhibition of phosphodiesterase-5. Without PAH-specific therapies available, initial survival estimates were quite poor with a 1-, 3-, and 5-year mortality of 68, 48, and 34 %, respectively [2]. Recent data suggest improvement with 1- and 2-year survival reaching as high as 97 and 91 %, respectively [2]. The aim of this paper is to provide clinical insight and a brief review of the literature published regarding current and new therapeutic options that have clinical data for the treatment of PAH (Table 1).
Prostacyclin Pathway
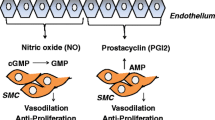
Prostacyclin is an important signaling molecule involved in vasodilation and the inhibition of platelet aggregation, inflammation, and proliferation of vascular smooth muscle cells. Prostanoid therapy exists in the following forms: intravenous, subcutaneous, inhaled, and oral.
Established Therapies
Epoprostenol (Intravenous)
Epoprostenol, an intravenously (IV) delivered prostanoid, is the oldest and most established drug of this class with demonstrated functional, hemodynamic, and survival benefits in PAH patients. The pivotal prospective, randomized controlled study illustrating these benefits enrolled 81 New York Heart Association (NYHA) functional class III and IV patients and determined outcomes at 12 weeks [3]. In 2002, two long-term registry-based studies demonstrated survival, functional, and hemodynamic benefits over a 9–10-year period [4, 5]. These studies found a 1-, 2-, and 3-year survival of 85, 70, 62.8 % and 87.8, 76.3, 63 %, respectively [4, 5]. Practically, epoprostenol presents challenging management considerations given its short half-life of 3–5 min and chemical instability at room temperature [6]. Despite this, epoprostenol (Flolan®) maintains its place in the long-term treatment of patients with severe NYHA functional class (FC) III/IV disease (Table 1).
A more convenient formulation of epoprostenol is now available. A phase IV exploratory study involving epoprostenol-naïve patients, Epoprostenol for Injection in Pulmonary Arterial Hypertension (EPITOME-1), evaluated the safety and tolerability of Veletri®, a room-temperature stable intravenous formulation of epoprostenol, versus Flolan® and found it to be well tolerated and with a similar adverse effect profile [7•]. A subsequent 41-patient open-label, phase IIIb study (EPITOME-2), a transition study of the above two epoprostenol formulations, demonstrated no change in efficacy and no new safety or tolerability concerns [8•]. Additionally, a small, 8-patient, open-label, phase IIIb study in Japan (EPITOME4) also switched from Flolan® to Veletri® and found maintenance of pulmonary hemodynamics and improvement in treatment satisfaction at 12 weeks [9•].
Treprostinil (Subcutaneous, Intravenous, Inhaled)
Treprostinil is a room-temperature stable formulation, with a half-life of approximately 4 h and is administered subcutaneously (SC) and intravenously (IV). A 2002 12-week, double-blind, placebo-controlled trial with SC treprostinil demonstrated a dose-dependent improvement in exercise capacity, improvement in hemodynamics, and a reduction in Borg dyspnea index [10]. Long-term survival data from center registries and open-label trial data have found similar survival rates among patients receiving SC treprostinil versus IV epoprostenol [10, 11]. For further support of its efficacy, a phase IV small multicenter study proved that a safe transition can be done between SC treprostinil and IV epoprostenol; however, some patients did not tolerate the downtitration or required reinstitution of their previous dose of SC therapy [12].
With equivalent bioavailability compared with SC treprostinil, IV treprostinil is now also available. Two investigator-initiated trials proved the safety of transition from epoprostenol to IV treprostinil [13, 14] and the efficacy of the IV formulation in patients starting a prostacyclin ‘de novo’ [15]. A placebo-controlled trial of IV treprostinil, the Treprostinil for Untreated Symptomatic PAH trial (TRUST) demonstrated marked median improvement in a 6-min walk distance (83 m) [16]. Lastly, the Treprostinil Sodium in Patients with Severe Pulmonary Arterial Hypertension (TRIUMPH I) trial evaluated the safety and efficacy of inhaled treprostinil on a background therapy of either a PDE-5 inhibitor or ERA and found improvements in 6MWD and quality of life measures in FC III PAH patients [17].
Iloprost (Inhaled)
The first inhaled prostacyclin approved, iloprost, is an alternative to continuous infusion prostanoid therapy. Aerosolized iloprost has demonstrated improved hemodynamics, exercise capacity, and symptoms after 12 weeks in patients with idiopathic PAH, connective tissue disease-associated and anorexigen-associated, and FC III or IV patients [18]. In a subsequent study with up to a 5-year follow-up, many patients were unable to be maintained on inhaled iloprost monotherapy and required intensification of therapy or lung transplant [19]. As such, monotherapy with inhaled iloprost has a limited role in management and has been relegated as a second-line or add-on agent [20].
Emerging Prostanoid Therapies
Beraprost (Oral, Twice-Daily Dosing)
Beraprost is an oral prostacyclin, approved in Japan, that demonstrated improvements in 3- and 6-month 6MWD; however, this benefit diminished at 12 months compared with placebo [21, 22]. This drug never received approval in the USA or Europe, but a multicenter, double-blind, randomized, placebo-controlled phase III study with a new beraprost formulation looking at beraprost add-on to inhaled treprostinil is currently underway (BEAT (Beraprost-314d Added-on to Tyvaso®) Study; NCT01908699).
Oral Treprostinil (Three Times Daily Dosing)
Approved by the FDA in December 2013, oral treprostinil has been evaluated as both a monotherapy and combination therapy agent with mixed results. The Oral Treprostinil as Monotherapy for the Treatment of PAH (FREEDOM-M), a trial comparing oral treprostinil to placebo without background therapy, found a significant, but small, 23-m increase in 6MWD at 12 weeks [23•]. Two studies evaluating combination therapy (FREEDOM-C, FREEDOM-C2) [24, 25] with oral treprostinil therapy on a background of ERA or phosphodiesterase type 5 (PDE-5) inhibitors did not meet its primary endpoint of improvement in 6MWD at 16 weeks, perhaps due to the trail design and dosing regimens. The trial, however, did provide additional insight on titration and dosing of this oral formulation; similar findings were observed in FREEDOM-C2. Notably, there was a discontinuation rate of 22 % in the treprostinil group presumably due to a higher incidence of side effects [24]. An international, phase III, multicenter trial involving early randomization (between 30 days and 1 year) of oral treprostinil to patients on a background of ERA or PDE-5 inhibitors is currently in process (FREEDOM-Ev; NCT01560624) with primary outcome measures of time to first clinical worsening event and change in 6MWD at 24 weeks; enrollment is estimated at 858 with study completion estimated by August 2016. A phase III, international, multicenter, open-label, long-term study of oral treprostinil (NCT01560637), involving PAH patients, designed to assess long-term safety and effects on exercise capacity is currently recruiting patients. Lastly, an open-label, phase III extension trial (FREEDOM-EXT; NCT01027949) has been underway as of 2006 with estimated enrollment of 900, and a completion date of December, 2020 will evaluate 6MWD and long-term safety as its primary endpoint.
Selexipag (Oral Prostacyclin Analog—Twice-Daily Dosing)
Selexipag is an orally available, selective prostacyclin receptor (IP receptor) agonist currently under active evaluation as a treatment for PAH. A 2012 phase II trial with 43 symptomatic PAH patients on a background therapy of ERA or PDE-5 inhibitor found that patients randomized to selexipag had a 30.3 % decrease in pulmonary vascular resistance at 17 weeks and was well tolerated [26•]. A phase III, multicenter, double-blind, placebo-controlled study Prostacyclin (PGI2) Receptor Agonist in Pulmonary Arterial Hypertension (GRIPHON; NCT01106014) with primary outcome measures of time to first morbidity or mortality event has recently been completed [27]. Secondary outcome measures included 6MWD and Borg dyspnea index with an enrollment of 1156 patients. As of the writing of this paper, a June 16th press release by Actelion regarding the GRIPHON trial found that in meeting its primary efficacy endpoint, selexipag reduced the risk of time to first morbidity or mortality event versus placebo by 39 % (p < 0.0001) with patients being treated for up to 4.3 years [27]. A single-arm, long-term, open-label study assessing the safety and tolerability of selexipag is also underway (NCT01112306).
Discussion
Continuous infusion prostacyclin therapy is still recommended in FC IV patients. Some individuals believe that it may be possible to treat aggressively upfront with continuous infusion and then switch to oral therapies while others believe that a combined oral and inhaled therapeutic approach may be best. These therapeutic questions will need prospective studies to determine the best management strategy. Oral prostacyclin and prostacyclin analogs are attractive based on infection risk, however, may be difficult to tolerate (GI side-effect profile) and currently cannot be taken by mouth in a “crushed” form if a patient cannot tolerate oral intake. This is a safety concern, as these patients will need to be treated temporarily with a continuous infusion and then resumed at a lower oral dose and re-uptitrated. This, in addition to the current multidosing regimens with multiple pills, and cost will likely limit its use.
Endothelin Receptor Antagonist Pathway
Endothelin is a potent vasoconstrictor and smooth muscle cell mitogen produced in vascular endothelial cells and has been shown to circulate at elevated levels in patients with pulmonary hypertension [28, 29]. Data suggest that patients with PAH have elevated endothelin due to increased synthesis and decreased clearance, which contributes to disease progression [28–30]. In 2001, endothelin receptor antagonists (ERA) were the first orally administered FDA-approved medication for PAH.
Established Therapies
Bosentan (Oral, Twice-Daily Dose)
Bosentan, a nonselective endothelin A and B receptor antagonist, was the first ERA approved by the FDA in 2001 for patients with PAH and FC III or IV. In a double-blind, placebo-controlled study, bosentan was shown to increase 6MWD and improve Borg dyspnea index, World Health Organization functional class (FC), and time to clinical worsening after 16 weeks of treatment [31]. A subsequent study has shown a favorable response in patients with FC II disease [32]. However, many patients still progress on monotherapy, requiring prostanoid therapy on follow-up, up to 23–44 % of the time [33]. In addition, bosentan requires monthly monitoring of liver function tests due to its dose-dependent and sporadic long-term increase in aminotransferases that has led to acute liver failure [34]. Bosentan also requires monthly pregnancy testing (as do all ERAs) and hemoglobin measures for its potential incidence of anemia while on therapy [35]. The side effects include nasal congestion and edema.
Ambrisentan (Oral, Daily Dose)
In 2007, the FDA approved ambrisentan, a selective endothelin A receptor antagonist. Like bosentan, it improved exercise capacity in PAH patients but without a significant risk of aminotransferase abnormalities [36, 37]. After the mandated post-marketing oversight illustrated significantly reduced risk of liver dysfunction, the black box warning requiring monthly liver function testing in order to receive drug shipment was discontinued. Its advantages include once daily instead of twice daily administration and no FDA requirement to monitor liver enzymes; the side effects include nasal congestion and edema [36].
Macitentan (Oral, Daily Dose)
Macitentan is the most recent endothelin receptor antagonist approved this past year by the Study with an Endothelin Receptor Antagonist in Pulmonary Arterial Hypertension to Improve Clinical Outcome (SERAPHIN) trial, a double-blind, randomized control trial of 742 PAH patients that showed a 43 % reduction in the occurrence of the composite primary endpoint of morbidity and mortality [38••]. The study compared two doses of macitentan (3 and 10 mg) to placebo in PAH patients not receiving IV or SC prostanoid therapy and found an absolute risk reduction of 8.4 and 15 % for the composite primary endpoint for 3- and 10-mg doses, respectively. This trial is the first reported phase III PAH trial utilizing a composite endpoint as the primary outcome, whereas prior studies have primarily used measures of functional status and exercise tolerance as the primary endpoint with time to clinical worsening as a secondary endpoint.
This trial required significantly more patients to be enrolled compared with previous phase III PAH trials. The trial enrolled patients from many countries who had not participated in PAH trials. This allowed for subgroup analyses by geographic region. Surprisingly, the evaluation of the North American study patients did not demonstrate statistical significance for the primary endpoint. In addition, unexpectedly, unlike previous studies, exercise capacity did not improve significantly in SERAPHIN. There was no significant difference in transaminase levels between the treatment and placebo arms, and thus, mandatory monthly testing was not FDA mandated.
Macitentan is currently being evaluated for other PH groups including patients with pre- and post-capillary pulmonary hypertension and inoperable chronic thromboembolic pulmonary hypertension (NCT02021292, NCT02070991). Currently recruiting, SYMPHONY, A Study of Macitentan in Pulmonary Arterial Hypertension to validate the PAH-SYMPACT is a prospective, multicenter, open-label, single-arm phase IIIb psychometric validation study (NCT01841762) for a new quality of life questionnaire for PAH patients. Enrollment is estimated at 275 with projected completion by January 2015.
Discussion
ERA oral therapy continues to be utilized for PAH patients both as monotherapy and in combination with other PAH agents. The lack of functional capacity improvement with macitentan was not expected and is of unclear significance. The other ERAs have improved time to clinical worsening as secondary endpoints in placebo-controlled patient-naïve trials; a head to head trial of the agents is not clinically feasible in this orphan disease. The use of bosentan because of liver toxicity will likely decrease, although it will become generic shortly. The AMBITION study (A Study of First-line Ambrisentan and Tadalafil Combination Therapy in Patients with Pulmonary Arterial Hypertension; NCT01178073), an upfront combination study of ambrisentan and tadalafil, will be reported soon and may support the initiation of two therapies at diagnosis compared to a sequential approach.
Nitric Oxide Pathway
Nitric oxide is a powerful, short-acting vasodilatory molecule that is produced by endothelial cells. It acts on vascular smooth muscle cells by stimulating the enzyme guanylyl cyclase to produce cyclic guanosine monophosphate (cGMP), which causes smooth muscle relaxation and vasodilatation within the pulmonary vasculature. cGMP in the lung is rapidly broken down by phosphodiesterase type 5 (PDE5) enzyme.
Established Therapies
Sildenafil (Oral)
Sildenafil is a PDE5 inhibitor approved in 2005 for PAH treatment based on the results from the Sildenafil Use in Pulmonary Arterial Hypertension (SUPER-1) trial [39]. The study compared placebo with 20, 40, and 80 mg three times daily of sildenafil in WHO functional class II and III patients. Sildenafil improved exercise capacity (6MWD), WHO functional class, and hemodynamics at 12 weeks and at 1 year of therapy versus placebo with all three doses [39]. The FDA approved the 20-mg dose; however, the long-term open-label study utilized the 80 mg thrice daily [40]. Side effects of this medication include headache (relieved with pre-dose Tylenol® for a short time with initiation) dyspepsia, flushing, and diarrhea.
Tadalafil (Oral)
The Pulmonary Arterial Hypertension and Response to Tadalafil (PHIRST) study using tadalafil improved exercise capacity, quality of life measures, and reduced time to clinical worsening [41]. The study evaluated multiple doses ranging from 2.5 to 40 mg daily. Only the 40-mg dose met the primary endpoint of improved 6MWD. Patients were either treatment-naïve or on ERA background therapy with bosentan. The changes in WHO functional class were not statistically significant [41]. The side-effect profile of tadalafil is similar to sildenafil with the convenience advantage of once daily dosing. However, only the 40-mg dose is approved limiting uptitration.
Riociguat (Guanylatecyclase Activator) (Oral, Three Times Daily Dosing)
Riociguat is the first soluble guanylate cyclase stimulator approved for PAH. It exerts its effects by both working synergistically with NO and independently to increase cGMP levels [42]. Two phase III trials involving PAH and chronic thromboembolic PH (CTEPH) patients, The Pulmonary Arterial Hypertension Soluble Guanylate Cyclase-Stimulator Trial 1 (PATENT-1) and Chronic Thromboembolic Pulmonary Hypertension Soluble Guanylate Cyclase-Stimulator Trial 1 (CHEST-1), had positive results. In PATENT-1, a randomized study of riociguat on a background therapy (ERAs or prostanoids) or treatment-naïve patients yielded a significant improvement in the primary endpoint of exercise capacity (6MWD) and secondary endpoints including hemodynamics (pulmonary vascular resistance (PVR)), laboratory markers of right ventricular stress (N-terminal pro brain naturetic peptide), and functional class [43••]. Of note, PATENT-2, a long-term extension study of PATENT-1, is ongoing (NCT00863681). CHEST-1, the first randomized study of CTEPH patients, had a significant improvement in both exercise capacity (6MWD) and hemodynamics (PVR) [44••]. The FDA approved riociguat for both indications in October of 2013. The medication is approved in multiple doses and requires uptitration and blood pressure measurements in the outpatient setting.
The Exposure Registry, an open-label observational study of riociguat in patients with CTEPH (EXPERT; NCT02092818), is actively recruiting patients and will evaluate the number of adverse events and all-cause mortality up to a period of 4 years. The estimated enrollment is 900 patients and should be completed by September 2018.
Emerging Therapies
Vardenafil (Oral)
Vardenafil is currently under investigation for treatment in PAH patients and does not have FDA approval as of the publishing date of this article. A small randomized double-blind placebo trial of 66 PAH patients improved both exercise capacity and hemodynamics at 12 weeks [45•]. A recent mechanistic study found that vardenafil reduced oxidative stress while improving PA pressures [46], illustrating the need to investigate the role of oxidative stress pathways in PAH as a possible therapeutic target [46].
NO (Inhaled, for Home Use)
A phase II, 16-week placebo-controlled clinical trial of inhaled NO as an add-on therapy in symptomatic PAH patients is currently enrolling (NCT01457781). The primary endpoint is PVR with multiple secondary endpoints including change in time to clinical worsening, 6MWD, FC, and the CAMPHOR questionnaire.
Discussion
PDE-5 inhibitors are well tolerated and utilized as monotherapy or in combination with ERAs and prostacyclin agents. In clinical practice, it is common for even compliant patients to miss doses of sildenafil based on its dosing schedule of thrice daily. Tadalafil is often used now due to its convenient dosing, but with only the 40-mg dose approved, physicians cannot uptitrate. The use of a PDE-5 inhibitor with a guanylate cyclase stimulator is contraindicated due to significant hypotension as was seen in the Evaluation of the Pharmacodynamic Effect of the Combination of Sildenafil and Riociguat on Blood Pressure and Other Safety Parameters (PATENT PLUS) study [47]. Riociguat, the first approved agent for CTEPH, illustrated that some PAH-specific medications may be efficacious for other PH etiologies; however, formal testing is required. Similar to other PAH therapies (sildenafil, oral treprostinil), the three times daily dosing plus the uptitration schema add some complexity to its use. Specialized pharmacies clinically assess these patients prior to physician-determined uptitration of the medication.
Conclusions: Where Does a Physician Start?
Clinical and basic research investigations continue to explore novel mechanistic pathways as potential therapeutic targets for PAH. With many new options, it is still too early to determine where agents are best suited in the treatment algorithm. However, it remains that the sickest of patients with syncope, hypotension, and evidence of end organ damage should be started emergently on infusion therapies in the hospital [48–50]. Each patient needs to be evaluated by multiple objective measures including a catheterization at diagnosis and then the patient and the physician should devise a plan that best suits him/her. The multiple treatment guidelines recently published help [48–50] to form a systematic approach, but in truth, much of our understanding of personalized medicine for PAH is still largely unknown. Individual patient phenotyping must be explored to better understand combination therapies in addition to treatment strategy-based clinical trials.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351:1655–65.
Waxman AB, Zamanian RT. Pulmonary arterial hypertension: new insights into the optimal role of current and emerging prostacyclin therapies. Am. J. Cardiol. 2013;111:1A–16A; quiz 17A–19A.
Barst RJ, Rubin LJ, Long WA, McGoon MD, Rich S, Badesch DB, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med. 1996;334:296–301.
Sitbon O, Humbert M, Nunes H, Parent F, Garcia G, Hervé P, et al. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol. 2002;40:780–8.
McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension the impact of epoprostenol therapy. Circulation. 2002;106:1477–82.
Agarwal R, Gomberg-Maitland M. Current therapeutics and practical management strategies for pulmonary arterial hypertension. Am Heart J. 2011;162:201–13.
Chin KM, Badesch DB, Robbins IM, Tapson VF, Palevsky HI, Kim NH, et al. Two formulations of epoprostenol sodium in the treatment of pulmonary arterial hypertension: EPITOME-1 (epoprostenol for injection in pulmonary arterial hypertension), a phase IV, open-label, randomized study. Am Heart J. 2014;167:218–225.e1.
Sitbon O, Delcroix M, Bergot E, Boonstra AB, Granton J, Langleben D, et al. EPITOME-2: an open-label study assessing the transition to a new formulation of intravenous epoprostenol in patients with pulmonary arterial hypertension. Am Heart J. 2014;167:210–7.
Tamura Y, Ono T, Fukuda K, Satoh T, Sasayama S. Evaluation of a new formulation of epoprostenol sodium in Japanese patients with pulmonary arterial hypertension (EPITOME4). Adv Ther. 2013;30:459–71. The Epitome-1, 2, and 4 trials demonstrate that Veletri is similar in efficacy and safety when compared to Flolan and has higher treatment satisfaction. These studies may motivate providers to transition patients from Flolan, one of the most established treatments in PAH, to Veletri.
Simonneau G, Barst RJ, Galie N, Naeije R, Rich S, Bourge RC, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med. 2002;165:800–4.
Barst RJ, Galie N, Naeije R, Simonneau G, Jeffs R, Arneson C, et al. Long-term outcome in pulmonary arterial hypertension patients treated with subcutaneous treprostinil. Eur Respir J. 2006;28:1195–203.
Minai OA, Parambil J, Dweik RA, Davila GH, Peterson L, Rollins KD, et al. Impact of switching from epoprostenol to IV treprostinil on treatment satisfaction and quality of life in patients with pulmonary hypertension. Respir Med. 2013;107:458–65.
Gomberg-Maitland M, Tapson VF, Benza RL, McLaughlin VV, Krichman A, Widlitz AC, et al. Transition from intravenous epoprostenol to intravenous treprostinil in pulmonary hypertension. Am J Respir Crit Care Med. 2005;172:1586–9.
Sitbon O, Manes A, Jais X, Pallazini M, Humbert M, Presotto L, et al. Rapid switch from intravenous epoprostenol to intravenous treprostinil in patients with pulmonary arterial hypertension. J Cardiovasc Pharmacol. 2007;49:1–5.
Tapson VF, Gomberg-Maitland M, McLaughlin VV, Benza RL, Widlitz AC, Krichman A, et al. Safety and efficacy of IV treprostinil for pulmonary arterial hypertension: a prospective, multicenter, open-label, 12-week trial. Chest J. 2006;129:683–8.
Hiremath J, Thanikachalam S, Parikh K, Shanmugasundaram S, Bangera S, Shapiro L, et al. Exercise improvement and plasma biomarker changes with intravenous treprostinil therapy for pulmonary arterial hypertension: a placebo-controlled trial. J Heart Lung Transplant. 2010;29:137–49.
McLaughlin VV, Benza RL, Rubin LJ, Channick RN, Voswinckel R, Tapson VF, et al. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: a randomized controlled clinical trial. J Am Coll Cardiol. 2010;55:1915–22.
Olschewski H, Simonneau G, Galiè N, Higenbottam T, Naeije R, Rubin LJ, et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;347:322–9.
Opitz CF, Wensel R, Winkler J, Halank M, Bruch L, Kleber F-X, et al. Clinical efficacy and survival with first-line inhaled iloprost therapy in patients with idiopathic pulmonary arterial hypertension. Eur Heart J. 2005;26:1895–902.
Wu Y, O’Callaghan DS, Humbert M. An update on medical therapy for pulmonary arterial hypertension. Curr Hypertens Rep. 2013;15:614–22.
Galiè N, Humbert M, Vachiéry J-L, Vizza CD, Kneussl M, Manes A, et al. Effects of beraprost sodium, an oral prostacyclin analogue, in patients with pulmonary arterial hypertension: a randomized, double-blind, placebo-controlled trial. J Am Coll Cardiol. 2002;39:1496–502.
Barst RJ, McGoon M, McLaughlin V, Tapson V, Rich S, Rubin L, et al. Beraprost therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2003;41:2119–25.
Jing Z-C, Parikh K, Pulido T, Jerjes-Sanchez C, White RJ, Allen R, et al. Efficacy and safety of oral treprostinil monotherapy for the treatment of pulmonary arterial hypertension: a randomized, controlled trial. Circulation. 2013;127:624–33. This trial demonstrated a modest but significant improvement in 6MWD, its primary endpoint. Additional trials are currently underway to assess the efficacy of oral treprostinil when used in combination with other agents.
Tapson VF, Torres F, Kermeen F, Keogh AM, Allen RP, Frantz RP, et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients on background endothelin receptor antagonist and/or phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C study): a randomized controlled trial. Chest. 2012;142:1383–90.
Tapson VF, Jing Z-C, Xu K-F, Pan L, Feldman J, Kiely DG, et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients receiving background endothelin receptor antagonist and phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C2 study): a randomized controlled trial. Chest. 2013;144:952–8.
Simonneau G, Torbicki A, Hoeper MM, Delcroix M, Karlócai K, Galiè N, et al. Selexipag: an oral, selective prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. Eur Respir J. 2012;40:874–80. This small, phase II trial demonstrated the efficacy and safety of oral selexipag and motivated the larger phase III GRIPHON trial that has recently been completed and still awaits publication. Preliminary results show that the primary endpoint was met, which may lead to an FDA approval and potentially a new popular therapeutic option.
Selexipag meets primary endpoint in pivotal phase III GRIPHON outcome study in patients with pulmonary arterial hypertension <ATLN.VX>. Reuters [Internet]. 2014 Jun 15 [cited 2014 Jul 10]; Available from: http://www.reuters.com/article/2014/06/15/idUSnHUGn8QMsa+1cc+ONE20140615.
Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411–5.
Giaid A, Yanagisawa M, Langleben D, Michel RP, Levy R, Shennib H, et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med. 1993;328:1732–9.
Rubens C, Ewert R, Halank M, Wensel R, Orzechowski H-D, Schultheiss H-P, et al. Big endothelin-1 and endothelin-1 plasma levels are correlated with the severity of primary pulmonary hypertension*. Chest J. 2001;120:1562–9.
Channick RN, Simonneau G, Sitbon O, Robbins IM, Frost A, Tapson VF, et al. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet. 2001;358:1119–23.
Galiè N, Rubin L, Hoeper M, Jansa P, Al-Hiti H, Meyer G, et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. Lancet. 2008;371:2093–100.
McLaughlin VV, Sitbon O, Badesch DB, Barst RJ, Black C, Galiè N, et al. Survival with first-line bosentan in patients with primary pulmonary hypertension. Eur Respir J. 2005;25:244–9.
Humbert M, Segal ES, Kiely DG, Carlsen J, Schwierin B, Hoeper MM. Results of European post-marketing surveillance of bosentan in pulmonary hypertension. Eur Respir J. 2007;30:338–44.
Commissioner O of the Safety Information—Tracleer (bosentan) tablets [Internet]. [cited 2014 Jul 16]. Available from: http://www.fda.gov/Safety/MedWatch/SafetyInformation/ucm327930.htm.
Ben-Yehuda O, Pizzuti D, Brown A, Littman M, Gillies H, Henig N, et al. Long-term hepatic safety of ambrisentan in patients with pulmonary arterial hypertension. J Am Coll Cardiol. 2012;60:80–1.
Galiè N, Olschewski H, Oudiz RJ, Torres F, Frost A, Ghofrani HA, et al. Ambrisentan for the treatment of pulmonary arterial hypertension results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation. 2008;117:3010–9.
Pulido T, Adzerikho I, Channick RN, Delcroix M, Galiè N, Ghofrani H-A, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369:809–18. A landmark trial that used morbidity and mortality as a composite primary endpoint. The results led to the approval of macitentan.
Galiè N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353:2148–57.
Rubin LJ, Badesch DB, Fleming TR, Galiè N, Simonneau G, Ghofrani HA, et al. Long-term treatment with sildenafil citrate in pulmonary arterial hypertension: the super-2 study. Chest J. 2011;140:1274–83.
Galiè N, Brundage BH, Ghofrani HA, Oudiz RJ, Simonneau G, Safdar Z, et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation. 2009;119:2894–903.
Lang M, Kojonazarov B, Tian X, Kalymbetov A, Weissmann N, Grimminger F, et al. The soluble guanylate cyclase stimulator riociguat ameliorates pulmonary hypertension induced by hypoxia and SU5416 in rats. PLoS One. 2012;7:e43433.
Ghofrani H-A, Galiè N, Grimminger F, Grünig E, Humbert M, Jing Z-C, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013;369:330–40.
Ghofrani H-A, D’Armini AM, Grimminger F, Hoeper MM, Jansa P, Kim NH, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med. 2013;369:319–29. The above two trials by Ghofrani et al. demonstrated the efficacy of Riociguat, a therapy in a novel class, in both PAH and CTEPH. It is the only therapy approved by the FDA for the treatment of inoperable CTEPH.
Jing Z-C, Yu Z-X, Shen J-Y, Wu B-X, Xu K-F, Zhu X-Y, et al. Vardenafil in pulmonary arterial hypertension: a randomized, double-blind, placebo-controlled study. Am J Respir Crit Care Med. 2011;183:1723–9. This small study demonstrated the efficacy of Vardenafil. Additional studies are needed to demonstrate if Vardenafil is more efficacious than existing PDE-5 inhibitors.
Fan Y-F, Zhang R, Jiang X, Wen L, Wu D-C, Liu D, et al. The phosphodiesterase-5 inhibitor vardenafil reduces oxidative stress while reversing pulmonary arterial hypertension. Cardiovasc Res. 2013;99:395–403.
Nazzareno Galie, Dieter Neuser, Katharina Muller, Andrea-Viviana Scalise, Ekkehard Grunig. A placebo-controlled, double-blind phase ii interaction study to evaluate blood pressure following addition of riociguat to patients with symptomatic pulmonary arterial hypertension (pah) receiving sildenafil (PATENT PLUS). B98 Clin. TRIALS Pulm. Hypertens. [Internet]. American Thoracic Society; 2013 [cited 2014 Jul 22]. p. A3530–A3530. Available from: http://www.atsjournals.org/doi/abs/10.1164/ajrccm-conference.2013.187.1_MeetingAbstracts.A3530.
Taichman DB, Ornelas J, Chung L, Klinger J, Lewis S, Mandel J, et al. Pharmacological therapy for pulmonary arterial hypertension in adults: CHEST guideline. CHEST J. [Internet]. 2014 [cited 2014 Jul 25]; Available from: doi:10.1378/chest.14-0793.
Galié N, Corris PA, Frost A, Girgis RE, Granton J, Jing ZC, et al. Updated treatment algorithm of pulmonary arterial hypertension. J. Am. Coll. Cardiol. [Internet]. 2013 [cited 2014 Jul 25];62. Available from: doi:10.1016/j.jacc.2013.10.031.
Galié N, Manes A. New treatment strategies for pulmonary arterial hypertension hopes or hypes? J Am Coll Cardiol. 2013;62:1101–2.
Compliance with Ethics Guidelines
Conflict of Interest
Aaron M. Wolfson and Nathaniel Steiger declare no conflict of interest. Mardi Gomberg-Maitland has received funding for clinical trials from Actelion, Gilead, Medtronic, Novartis, Lung Biotechnology, Reata, and Ventripoint; she has served as a consultant for Actelion, Bayer, Gilead, Medtronic, Merck, Bellerophon, and United Therapeutics; she has received honoraria for CME from Medscape and ABComm.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Pulmonary Hypertension
Rights and permissions
About this article

Cite this article
Wolfson, A.M., Steiger, N. & Gomberg-Maitland, M. New Pharmacotherapies for Pulmonary Hypertension: Where Do They Fit in?. Curr Hypertens Rep 16, 496 (2014). https://doi.org/10.1007/s11906-014-0496-y
Published:
DOI: https://doi.org/10.1007/s11906-014-0496-y