
Abstract
The etiology of colon cancer is complex, yet it is undoubtedly impacted by intestinal microbiota. Whether the contribution to colon carcinogenesis is generated through the presence of an overall dysbiosis or by specific pathogens is still a matter for debate. However, it is apparent that interactions between microbiota and the host are mediated by a variety of processes, including signaling cascades, the immune system, host metabolism, and regulation of gene transcription. To fully appreciate the role of microbiota in colon carcinogenesis, it will be necessary to expand efforts to define populations in niche environments, such as colonic crypts, explore cross talk between the host and the microbiota, and more completely define the metabolomic profile of the microbiota. These efforts must be pursued with appreciation that dietary substrates and other environmental modifiers mediate changes in the microbiota, as well as their metabolism and functional characteristics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Although screening improves our ability to identify and remove colon adenomas at a lower grade, many individuals do not undergo screening, which contributes to the fact that colon cancer is the second leading cause of cancer deaths in the US [1]. Importantly, the incidence of colon cancer has been projected to increase by 52 % by the year 2030, largely due to an expansion of our aging population [2]. Therefore, cancer chemoprevention is critical if we are to reduce the contribution of colon cancer to the national health-care crisis we are currently facing. To effectively intercede in colon carcinogenesis, it is imperative that we identify the environmental factors that contribute to the disease and/or serve to promote colon health.
The human intestinal microbiota consists of as many as 100 trillion commensal single-cell organisms that benefit overall host physiology, metabolism, development, and immune homeostasis [3]. Because of the preponderant role of intestinal microbiota in gut development and pathology, especially inflammatory bowel disease (IBD; a risk factor for colorectal cancer [CRC]) [4], early efforts have focused on elucidating the multifactorial role of gut microbes in CRC [5, 6]. Recent findings indicate that antibiotic use, dietary fiber intake, and other host factors that modify the luminal microbe environment could contribute to bacterial “dysbiosis”—that is, alter the normal homeostasis between the gut bacteria and the host [7–9]. With regard to potential pathways linking microbiota and the onset of CRC, it is now appreciated that the metabolism of dietary substrates by select groups of intestinal microbiota can lead to the production of mutagens (fecapentaenes, diacylglycerols, sulphide, secondary bile acids, and reactive oxygen species [ROS]) that promote DNA damage [5]. This paradigm-shifting body of literature indicates that microbial dysbiosis may contribute to the etiology of CRC [8–12]. Therefore, in this review, we will briefly discuss recent advances in our understanding of the role of intestinal microbiota in healthy versus disease states. In addition, we will describe some of the mechanisms whereby select environmental factors influence gut microbial populations and their metabolism and how these factors modulate colon homeostasis, possibly impacting tumorigenesis.
Microbial Ecology
It is well documented that interactions between the intestinal mucosal cellular phenotypes and commensal bacterial populations protect the host by establishing a homeostatic balance—that is, “tolerance” to luminal bacteria. This precludes the induction of overt colonic inflammation in response to the commensals [13]. However, inappropriate interactions between bacteria and the host may contribute to colon carcinogenesis through the induction of chronic inflammation and the promotion of immune evasion. These nonhomeostatic interactions can occur when the number or virulence of pathogenic species exceeds a set point, resulting in the development of a pathological inflammatory state. Although we are still early in the process of developing an appreciation for what constitutes a “normal” microbiota, recent efforts have begun to identify perturbations that are indicative of some disease states [14]. For example, analyses of colon tumors and uninvolved tissues have revealed elevated numbers of S. bovis, Bacteroides fragilis, and Escherichia coli at involved sites [13, 15]. Similar perturbations in microbial populations exist when normal mucosa is compared with inflamed tissues in patients with IBD [16]. S. bovis promotes expression of proliferating cell nuclear antigen and polyamines and elevates IL-8 production. This may contribute to the induction of early preneoplastic lesions of colon cancer (aberrant crypt foci) and eventual tumor development in animal models [10]. From a mechanistic perspective, it has been demonstrated that pathological inflammatory states promote a procarcinogenic environment by inducing ROS and nitric oxide (NO) release, which contribute to DNA damage and eventual mutations [13]. ROS and NO are produced in response to elevated IL-8 levels induced by S. bovis, which suggests a link between specific bacterial populations and the induction of mutagenesis [10].
An intriguing novel hypothesis for the induction of colon cancer by microbiota implicates single keystone pathogens that may exist at very low levels in the colon [17••]. In this scenario, “alpha bugs” harboring certain virulence traits are able to initiate colon cancer development. For example, fragilysin-producing Bacteroides spp. are associated with relapses of IBD and the induction of colon tumors in Apc/Min mice. This toxin induces TH17 cell-dependent cleavage of E-cadherin, thereby activating STAT3 and NF-κB and promoting the expression of proinflammatory cytokines. Together, these effects compromise barrier function, promote β-catenin nuclear signaling leading to enhanced proliferation, and stimulate migration of neutrophils, all of which promote pathogenesis [17••]. It is thought that the resulting changes in the colon luminal environment contribute to a dysbiosis, thereby promoting oncogenic potential. Although this theory requires additional study, it is an intriguing concept that could explain the development of colon cancer in certain individuals and not in others, when gross microbial populations are not significantly different.
In order to establish a clearer view of how microbial populations may influence cancer initiation/progression in the colon, it is necessary to further define regional niches within the intestine. For example, stem cells residing at the base of colon crypts are likely targets for cancer-initiating mutations [18, 19]. Therefore, if microbiota are involved in the transformative processes, it will be necessary to characterize the bacteria in these discrete locations. Indeed, results from a recent study suggest that intestinal-crypt-specific microbial populations contribute to a unique environment adjacent to stem cells [20••]. These observations reflect earlier reports that the microbiota within the lumen may be distinct from populations present within the mucous layer lining the colon [21]. The alpha bug theory may have even greater potential for explaining enhanced colon cancer risk if experiments designed to evaluate this theory are conducted within relevant niche environments in the colon.
Signaling Between Bacteria and Colon Mucosa and Inflammatory Responses
Commensal microbes (bacteria, viruses, parasites, and archaea) and potentially pernicious commensals (pathobionts) have resulted in the development of host sensing mechanisms for general microbial motifs [22]. Intestinal microbiota and the host can directly interact through binding of microbe-associated molecular patterns (MAMPs) to the multiple pattern recognition receptors (PRRs) on intestinal epithelial and innate immune cells [23]. PRR-induced signaling mediates the cross talk that exists between the microbiota and host physiology. In addition to direct interaction with bacteria, compounds released by the microbes are able to penetrate mucous barriers in the intestine and interact with the PRRs on colonocytes and macrophages within the colonic mucosa [24]. The PRRs include the highly characterized toll-like receptors (TLRs) found on cell membranes, as well as endosomal membrane components, where both external and internalized MAMPs are detected and processed. The nucleotide oligomerization domain (NOD)-like receptors (NLRs) present in the cytoplasm recognize many different bacterial toxins and ligands. The third class of PRRs are the retinoic acid inducible gene I (RIG-I)-like receptors, which are also cytoplasmic, and their functions include recognition of viral RNAs [25].
Signaling through PRRs contributes to maintenance of the innate defense system and allows the host to develop tolerance for the presence of commensal bacteria. These interactions produce other beneficial effects, such as the promotion of epithelial cell proliferation, IgA production, mucin gene expression, goblet cell differentiation, and the maintenance of tight junctions that promote barrier function in colonocytes [26, 27]. Normal interactions of bacteria with the PRR induce the activation of NF-κB, leading to the transcription of inflammatory cytokines, chemokines, and antimicrobial genes [13, 26]. However, in a state of dysbiosis or when PRR expression is perturbed, as occurs in subjects with IBD [28] and colon carcinogenesis [29], overstimulation of the PRR pathways contributes to elevated IL-6 expression [13]. This is noteworthy because IL-6 appears to play a key role in epithelial cell malignant transformation, in part through its activation of the Stat3 pathway and subsequent activation of proliferation, suppression of apoptosis, and stimulation of angiogenic pathways [13].
The site of PRR activation is key to the appropriate homeostatic response. For example, activation of TLR9 receptors on the apical surface of intestinal epithelial cells initiates a tolerant homeostatic response. In contrast, if the binding occurs on the basolateral side of the same cells, IκB is degraded and the canonical NF-κB pathway is activated, resulting in the release of many proinflammatory cytokines and other signaling molecules to protect against bacterial translocation and invasion [30]. In the context of colon health, the inflammasome-forming NLRs are generally considered protective, whereas TLRs may be procarcinogenic [31–33]. Thus, perturbations in these microbe-initiated signaling pathways promote active inflammation and may contribute to colonic pathology.
Because host and microbe interactions through PRR signaling are critical for modulating host responses, it is possible that the presence of PRRs on host cells influences the microbiota. For example, mice deficient in NOD-2 pattern recognition receptors develop a dysbiosis that promotes colonic epithelial dysplasia and inflammation, a condition that improves with antibiotic treatment [33]. Interestingly, it appears that the elevated disease risk in the NOD-2-deficient mice is communicable, since microbe transplantation from wild-type (WT) mice to NOD-2-deficient mice reduces disease and contributes to a long-term change in the intestinal microbiota. Similarly, transplantation in the opposite direction enhances disease in WT mice. The progression and severity of colitis-associated cancer in NOD-2-deficient animals is mediated by IL-6, and NOD-2 expression has been directly linked to the microbial ecology of the intestine [33]. Moreover, ablation of the IL-6 signaling pathway restores NOD-2-dependent control over the colonic microbiota and reduces tumor progression. The absence of NOD-2 has been shown to contribute to a disruption in the epithelial barrier function, making the loss-of-function mutations in NOD-2 found in Crohn’s disease and colon cancer a potential clinically relevant therapeutic target. The fact that changes in NOD-2 status and IL-6 antagonism can alter the intestinal microflora suggests that future interventions should target the restoration of beneficial interactions between the microbiota and the host.
With respect to adaptive immune system responses to bacteria, it has been demonstrated that barrier defects and microbial products drive CD4+ T-helper cell IL-23/IL-17-mediated tumor growth [34, 35]. Interestingly, IL-17-producing CD4+ T effector (Th17) cells are increased in tumor microenvironments, and IL-23 and its downstream target IL-17A promote growth of tumors through induction of local inflammatory promoters [36–38]. Therefore, at least in some contexts, impaired intestinal barrier function facilitates the translocation of microbial components, resulting in the production of cytokines that facilitate angiogenesis and promote tumor progression in part via an IL-17/IL-6-Stat3 pathway [39]. On the other hand, IL-17 can promote antitumor cytotoxic T cell responses, thereby inhibiting tumor progression by stimulating tumor immunity [40]. Collectively, these findings suggest that IL-17-producing T cells play a dual role in antitumor immune responses [41, 42].
Bacterial Metabolism
The intestinal microbiota are able to inhabit the colon because nutritive residues from the diet not digested in the small intestine provide most of the substrates necessary to support their growth. Although a major class of undigested nutrients used by the microbiota includes nonstarch polysaccharides, luminal prokaryotes are also able to utilize a wide range of substrates provided to them, including dietary polyphenols [6]. Because of the relative quantities of carbohydrates present in the fecal stream, much of the microbial metabolism in the proximal colon is saccharolytic in nature, resulting in the production of short-chain fatty acids (SCFAs) [43]. The microbiota both produce and utilize SCFAs as a nutrient source. SCFAs are also absorbed by colonocytes, which preferentially utilize butyrate as a metabolic substrate [44].
When carbohydrates are limiting, bacteria utilize other nutrients, including proteins, lipids, and xenobiotics. Products of protein fermentation include SCFAs and branched-chain fatty acids, as well as potentially toxic metabolites such as ammonia, sulfur-containing compounds, amines, phenolics, and indoles [45, 46]. Urine levels of p-cresol and phenol, among other unique bacterial metabolites from protein and phenolic substrates, can be used as an indicator of protein fermentation in the colon, which appears to increase with aging [46]. High levels of hydrogen sulfide, another metabolite from sulfur-containing amino acids, can promote DNA damage and also impair the oxidation of butyrate, due to inhibition of cytochrome c oxidase [47]. Dietary xenobiotics affect the microbial populations present in the gut and, more important, may modulate their physiology and gene expression patterns [48••]. Although the effect of dietary polyphenols on microbial metabolism has been historically less well studied, we now know that these putative substrates alter the metabolic profile of microbiota [49•, 50]. Microbial metabolites of phenolic molecules, including α-hydroxybutrate, hippurate, and 3-OH-hippurate, have been reported to be early biomarkers of insulin resistance and glucose intolerance in nondiabetic individuals [51, 52]. Subjects with low levels of these compounds in circulation are more likely to demonstrate depressed glucose tolerance. This suggests an intriguing mechanistic link between obesity and an elevated risk of colon cancer [53]. Observations such as these point out the critical need not only to characterize microbial populations present in the gut, but also to gain an understanding of the physiological and functional characteristics of the bacteria present.
Metabolomic screening using blood or urine is a valuable tool that can be used to monitor microbial and host metabolic interactions [6]. Efforts are currently underway that utilize targeted metabolomic approaches to more thoroughly characterize microbial metabolites of dietary components, including polyphenols [54]. However, more comprehensive and exploratory metabolomic analysis of the feces is needed to characterize the composite metabolic activity of the microbiota. Considering the likely implications of first-pass metabolism of microbial metabolites by the colon epithelium, it is necessary to evaluate the fecal stream to fully appreciate whether and/or how microbial xenobiotic metabolites mitigate or enhance colon carcinogenesis. As we learn more about the interactions between the metabolites derived from microbial metabolism and how these influence host physiology, it should be possible to develop strategies that impact disease development by manipulating the microbiota to produce a more desirable metabolic profile [55, 56].
Metabolite Actions in Colonocytes
Epithelial Cell Cycle Regulation
Butyrate produced by microbial fermentation contributes to energy homeostasis in colonocytes, and the loss of this metabolic substrate in gnotobiotic mice impairs intermediary metabolism, which promotes autophagy from activation of p27kip1 [57•, 58]. In addition to having depressed levels of TCA cycle enzymes, colonocytes from gnotobiotic animals also have reduced NADH/NAD + and ATP levels. Colonizing gnotobiotic mice with butyrate-producing Butyrivibrio fibrisolvens essentially restores oxidative phosphorylation and reduces autophagy to levels observed in conventionally raised mice. Utilization of an inhibitor of oxidative phosphorylation impairs recovery, suggesting that butyrate suppresses autophagy as a result of its influence on metabolism. Restoration of mitochondrial homeostasis and suppression of autophagy in germ-free animals by butyrate demonstrates that it is possible to “correct the physiology of energy-deprived colonocytes” [58]. Butyrate absorption and metabolism are also depressed in ulcerative colitis [44], which may contribute to the increased risk of colon cancer in colitis patients.
Butyrate also contributes to apoptosis induction in colonocytes, a response that is dependent upon the dietary lipid source [59]. For example, when butyrate is used in diets containing fish oil, there is an upregulation of apoptosis, but when butyrate is provided in diets containing corn oil, there is no effect on apoptosis, and early lesions of colon cancer are increased [60]. Microbial metabolism also contributes to the conversion of procarcinogens to carcinogens, including the bile acid metabolites deoxycholic acid (DCA) and chenodeoxycholic acid (CDCA). Recently, these secondary bile acid metabolites were found to create oxidative stress through the activation of membrane-associated NAD(P)H oxidases in colonocytes and to induce apoptosis in colon cancer cells [61]. Barrasa et al. [61] also demonstrated that the induction of apoptosis is dependent upon cell sensitivity to butyrate-induced apoptosis. For example, cells resistant to butyrate-induced apoptosis exhibit elevated levels of Bcl-2 expression, and this confers protection against the induction of apoptosis through the intrinsic mitochondrial pathway by bile acid metabolites. The provision of corn oil with a fermentable fiber (pectin/butyrate source) in the diet enhances expression of Bcl-2 and does not induce apoptosis [62]. Together, these results suggest that perturbations in the normal apoptotic cascade in colon cancer cells are due, in part, to altered butyrate responses, which also contribute to resistance to other apoptosis-inducing agents, such as bile acids and chemotherapeutic agents [61].
Regulation of Colonocyte Gene Transcription
Cell cycle activity is impacted by microbial metabolites that influence gene expression in colonocytes. Many investigators have reported that changes in expression of specific cell cycle regulators, such as p21cip1, p27kip1, and Bcl-2, occur in response to microbial metabolites [60, 63, 64]. In addition, some have reported global changes in colonocyte transcriptional profiles following exposure to butyrate and long chain fatty acids [59, 65, 66].
Changes in gene expression may result from epigenetic modifications to the genome and/or chromatin structure. A long-recognized effect of butyrate is its ability to serve as a histone deacetylase (HDAC) inhibitor [60, 67, 68]. Recent evidence demonstrates that HDAC inhibition can occur in response to a variety of microbial metabolites, including seleno-α-keto acids, small molecule thiols, indoles, and polyphenols [68]. Alterations in histone acetylation status, induced by either HDAC inhibitors or histone acetyltransferase (HAT) promoters [57•], has a direct effect on gene transcription [69], and it is through this mechanism that microbial metabolites are thought to influence the expression of key genes, such as p21, that regulate colonocyte proliferation [70].
Other epigenetic regulators of transcription include histone methylation, phosphorylation, and ubiquitination [68], as well as DNA methylation [64]. Butyrate, in combination with fish oil, was recently shown to enhance Bcl-2 promoter methylation in tumors from animals in which apoptosis was also increased [64]. This response was tumor specific and did not occur in the uninvolved tissues.
Diet and Other Environmental Effects on Microbiota, Microbial Function, and GI Health Outcomes
Although researchers have not identified a consensus group of bacteria associated with colon cancer, novel studies using germ-free mice have conclusively demonstrated that the intestinal endogenous bacterial community represents a risk factor [71, 72]. This would suggest that modifiers of luminal and tissue-adherent microbes in the gut (e.g., diet) affect cancer initiation/progression via production of microbial-derived tumorigenic metabolites.
Dietary Fiber
Colon cancer development is not uniform in all populations; those in developed countries such as the U.S. have high incidence rates, whereas native Africans have much lower rates of colon cancer [45]. It is thought that the high-carbohydrate and high-fiber diets in Africa support bacterial populations that protect against cancer development. However, our understanding of how a high-fiber diet influences the microbiota and microbial metabolism in healthy individuals, as compared with those with colon adenomas, is minimal. A recent cross-sectional case–control study found that a high-fiber diet in healthy individuals resulted in elevated butyrate concentrations in the feces, as well as an increase in butyrate-producing bacteria, relative to those in the healthy group consuming a low-fiber diet or those with adenomas consuming a high-fiber diet [73]. The greatest differences in bacterial populations were between patients with adenomas who had lower levels of Clostridium, Roseburia, and Eubacterium spp. and higher Enterococcus and Streptococcus spp., as compared with healthy controls who were consuming the higher fiber diets. Because these bacteria are from two classes of the Firmicute phylum that produce butyrate, a decrease in their numbers likely contributed to the lower fecal levels of butyrate in the adenoma group. The consumption of fiber-rich diets can also enhance microbial methanogenesis, which contributes to a reduction in hydrogen-producing bacteria [45]. This is noteworthy because excess hydrogen in the colon impairs NAD regeneration.
Disease-promoting responses to a Western diet are, in part, mitigated by consumption of resistant starches [74]. These starches reach the colon, where they are utilized by the microbiota and, thus, impact luminal metabolic profiles. It has been recently demonstrated that consumption of soluble fibers in healthy men induces a beneficial shift in the gut microbiota, including an increase in Faecalibacterium prausnitzii, a bacterium, which exhibits antiinflammatory properties [75]. Interestingly, Chen et al. [73] have demonstrated that elevated consumption of fiber favorably alters gut microbiota (elevation in butyrate-producing Clostridium and Roseburia spp.) in patients with advanced colorectal adenoma. Mounting evidence suggests that the immunomodulatory activities of fiber are mediated by microbial metabolites [76].
Lipids
In addition to dietary fiber, dietary lipids (both amount and type) can also influence the intestinal microbiota. For example, fish oil, containing omega-3 polyunsaturated fatty acids (PUFA), attenuates omega-6 PUFA induced dysbiosis [77], potentially modulating interactions between microbiota, intestinal mucosal immune responses, and neoplastic changes. This may partly explain why fish oil and fiber combination consumption reduces colon cancer risk [78–80]. A recent study of monozygotic twins (males and females) was conducted to determine the effect of diet, health status, and body mass index on microbial populations [81]. Higher energy intake in the twins contributed to a reduction in numbers of Bacteroides spp. and a small increase in Bifidobacteria, when compared with those with lower energy intake. In addition, specific lipid classes influenced bacterial growth, with monounsaturated fatty acids reducing the numbers of Bifidobacteria, omega-3 PUFA increasing numbers of Lactobacillus group bacteria, and omega-6 PUFA reducing Bifidobacteria numbers. Even within co-twins, those having disparate consumption of saturated fatty acids (SFAs) had the lowest similarity in the number of Bacteriodes spp., suggesting that SFA levels and/or proportion in the diet affects the diversity of this bacterial group.
Probiotics
Recent reviews of the potential for probiotics (live organisms that, when consumed in adequate amounts, confer health benefits to the host) to prevent colon tumorigenesis describe a multitude of pathways that may be involved [82, 83]. Probiotics may mitigate colon tumorigenesis through their ability to compete for nutrients and, thus, reduce promutagenic species. In addition, probiotic species are able to stimulate host immunity, interfere with bacterial mucosal adherence and invasion, and stimulate epithelial barrier function, thereby suppressing the translocation of bacteria into the host [13, 84]. The metabolic and inflammatory modifications resulting from probiotics include the enhancement of SCFA production, reduction in luminal pH, and inhibition of prostaglandin synthase-2 (COX-2) and inducible nitric oxide synthase (iNOS) [13, 84]. One recent study reported that exposure to probiotics may delay the transition from an inflammatory state to cancer in a model of colitis-associated colon cancer [85]. Even though the probiotic mixture used induced an increase in nonspecific inflammation and did not reduce the overall inflammatory state of the colon, it significantly reduced the number of carcinomas [85]. Additional studies are needed to clarify the role of probiotics in modulating colon cancer risk.
Antibiotics
Early-life exposures to antibiotics can influence the colon microbial ecology. For example, both the distribution and functional characteristics of intestinal microbiota can be impacted [86]. Antibiotic exposure increases the relative proportions of Firmicutes to Bacteriodetes. Antibiotics also alter the copy number of genes involved in SCFA production, including Butyryl CoA Transferase (BCoAT) and Formyltetrahydrofolate synthetase (FTHFS). As a result, the level of butyrate and the ratio of butyrate to acetate are elevated with certain antibiotic exposures [86]. Early-life antibiotic exposures are therefore capable of influencing taxonomic distributions, the expression of genes involved in metabolism, and thus the metabolic capabilities of the bacteria present.
Radiation
Although little is known about the effects of radiation on intestinal microbiota, early studies in the 1950s and 1960s reported that germ-free animals were more protected against radiation, as compared with animals harboring conventional microbial populations. For example, germ-free mice have an elevated survival rate when exposed with differing doses of x-rays, as compared with conventional mice or germ-free mice inoculated with a single bacterium (E. coli) [87]. More recently, it has been reported that germ-free animals exposed to 10–22 Gy of 137Cs showed no signs of radiation enteritis induced by injury to the small intestine, as compared with conventional (containing a normal microbiota) or conventionalized (germ-free mice inoculated with bacterial mixtures) mice, both of which presented typical injury responses [88]. Importantly, restoring gut microbiota with single-bacterium cultures resulted in levels of radioprotection similar to those found in germ-free mice, indicating that the lethal radiosensitivity induced by microbiota in the intestine is strain dependent [88]. These studies demonstrate that the extent of intestinal damage is predicated by bacterial populations residing in the colon and suggest that environmental exposures that perturb colonic microbiota (e.g., diet) may modulate host radiosensitivity. Along these lines, comparison of bacterial diversity in fecal samples from patients suffering from acute postradiotherapy diarrhea and those with no gastrointestinal distress after radiation treatment (total exposure of 4.3–5.4 Gy) revealed that microbial profiles of each group (control, no diarrhea, and diarrhea groups) clustered distinctly [89]. These data suggest that specific patterns of bacteria could be associated with risk or protection after radiation exposure. Subjects with clinical signs (i.e., diarrhea) exhibited a marked modification of their microbial profile, as compared with those who did not exhibit gastrointestinal complications. Additionally, it was reported that the proportion of Bacilli species and bacteria characterized in the Actinobacteria phylum were markedly higher in individuals with diarrhea, as compared with those without diarrhea, following radiation treatment [89].
Intestinal radiosensitivity is, in part, mediated by induction of inflammation that results from immune surveillance of intestinal bacterial populations. Commensal bacteria influence the response to intestinal injury through a number of mechanisms, including activation of the transcription factor NF-κB, which is activated downstream of signals generated by the immune surveillance pathways that utilize pattern recognition receptors (e.g., TLR pathways [26]). It has been reported that disruption of this pathway by knocking out IκB kinase, allowing IκB to permanently sequester NF-κB in the cytoplasm, triggers increased epithelial cell apoptosis in mice exposed to radiation [90].
Early-Life Exposures and Intestinal Microbiota
At birth, the intestinal tract is functionally immature and sterile. Consequently, the early neonatal period is a critical phase for both intestinal digestive development and establishment of gut microbiota [7, 91]. Dietary exposure profoundly impacts microbial populations that colonize the intestine. An obvious determinant of microbial species is the decision to provide breast milk or formula to the infant [92]. Breastfeeding exposes the infant to the mother’s microbiota and breast milk oligosaccharides. Because infants do not produce the enzyme necessary to digest breast milk oligosaccharides, these compounds become an important nutrient source for early microbial colonizers and, thereby, impact the resulting microbiome [93, 94]. Breast milk consumption produces a strong virulence-related microbial gene expression signal, which is likely responsible for the early training of the innate immune system [95•].
A recent in vitro study attempted to elucidate how energy density affects functional characteristics of the microbiota in fecal samples from two children. Although the number of stool samples used for the culturable populations was limited, the study demonstrated that high supplies of energy support strong butyrogenic microbiota populations, including elevated numbers of clostridia cluster XIVa [96]. When the energy load was reduced to either normal or low levels, the microbiota population was altered, and this reduced the overall metabolic activity in the cultures. Young et al. [97] suggested that the temporal changes in microbiota and their metabolism resulting from progressive modifications to substrate availability in weanlings also produces shifts in host transcriptional profiles in an attempt to maintain homeostasis.
Current observations suggest that postnatal intestinal microbiota also can be affected by fetal growth restriction. Fança-Berthon et al. [98] demonstrated that intrauterine growth retardation (IUGR) slows the expansion of cecocolonic microbiota numbers in neonatal rats, with a reduction in the numbers of Bifidobacterium spp. and bacteria in the clostridial clusters IV and XIVa. The implications of these changes with respect to host health require further study. However, our understanding that IUGR predisposes individuals to many long-term chronic diseases [99] suggests that early shifts in the intestinal microbiota contribute to disease risk.
In order to link changes in microbiota that occur throughout the life cycle with colon cancer risk, it will be necessary to utilize techniques that permit long-term monitoring of individuals. For these reasons, alternative and, especially, noninvasive molecular biology-based screening tests that assess the entire colon and rectum have great potential to enhance current screening methodology and reduce cancer incidence and mortality [100]. It is currently possible to use stool samples to monitor microbial populations, as well as (host) colonic gene expression profiles, in order to monitor discriminative molecular signatures that are apparent well before macroscopic tumors are apparent [52, 65]. Approaches such as this permit analysis of the multivariate structure underlying the microbiome and gut transcriptome over time, providing richer and fuller information content, as compared with analyses focusing on single data sets (e.g., only host transcriptome data or only microbiome data) and only single variables (e.g., gene by gene differential expression testing).
Conclusions
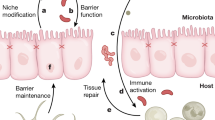
A growing body of literature indicates that microbial dysbiosis may contribute to the etiology of colorectal cancer [8, 9, 11, 82]. Although some of these changes may be induced very early in life, precisely how these various components influence intestinal development, microbiota composition, immunomodulatory responses, and the interplay between chronic inflammation and the initiation/progression of colon cancer over the long term is largely unknown. For the purpose of comparing normal, high-risk, and cancer patients, future experiments will need to utilize systems biology approaches to illuminate transgenomic cross talk between the host genome (signaling pathways) and the genomes of the microbiota and establish critical links between the functional state of the microbiota (e.g., metabolome) and host disease (Fig. 1). It is likely that these analyses will identify critical molecular biomarkers that define the relationship between intestinal microbiota and human colon cancer risk.

The contribution of microbiota to maintenance of gut health and prevention of colon cancer is multidimensional in nature. Complex microbial heterogeneity within the luminal environment, as well as the microenvironment(s) within colonic crypts, modulates colonocyte and immune cell processes. Many modifiers to the human superorganism exist, including diet, antibiotics, radiation, and early-life exposures. The net effects of these perturbations include changes in the luminal and/or crypt-specific microbiota and their functional characteristics, including metabolite generation. Host responses include changes in metabolism, inflammation, and regulation of gene expression
Since there is an exceptional amount of functional redundancy in the metabolic capabilities of bacteria present in the colon, much additional work needs to be done to determine microbial population differences between individuals and to evaluate temporal changes within individuals. Simple single-analyte approaches are likely not capable of discerning novel metabolites derived from microbial metabolism that may contribute to the mechanisms whereby diet influences colon cancer prevention. Recent evidence suggests that there are niche-/crypt-specific microbiota. Therefore, we may be missing the most highly relevant “localized” disease related changes in the microbiota due to the fact that they are not readily reflected in fecal stream populations. If we wish to determine which bacteria may be responsible for colonocyte transformation, it will be necessary to evaluate the potential for interactions within the stem cell niche located in the base of the crypt. Furthermore, it is important not only to identify the bacteria located in this niche, but also to functionally define the characteristics of those bacteria. Part of this characterization must include a thorough profiling of the metabolic products generated by the microbiota and how diet influences metabolite production.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011;61:212–36.
Smith BD, Smith GL, Hurria A, et al. Future of cancer incidence in the United States: burdens upon an aging, changing nation. J Clin Oncol. 2009;27:2758–65.
Kau AL, Ahern PP, Griffin NW, et al. Human nutrition, the gut microbiome and the immune system. Nature. 2011;474:327–36.
Man SM, Kaakoush NO, Mitchell HM. The role of bacteria and pattern-recognition receptor’s in Crohn’s disease. Nat Rev Gastroenterol Hepatol. 2011;8:152–68.
Candela M, Guidotti M, Fabbri A, et al. Human intestinal microbiota: cross talk with the host and its potential role in colorectal cancer. Crit Rev Micro. 2011;37:1–14.
Hamer HM, De Preter V, Windey K, Verbeke K. Functional analysis of colonic bacterial metabolism: relevant to health? Amer J Physiol Gastrointest Liver Physiol. 2012;302:G1–9.
Cho I, Blaser MJ. The human microbiome: at the interface of health and disease. Nat Rev Genet. 2012;13:260–70.
Shen XJ, Rawls JF, Randall T, et al. Molecular characterization of mucosal adherent bacteria and associations with colorectal adenomas. Gut Microbes. 2010;1:138–47.
Sobhani I, Tap J, Roudot-Thoraval F, et al. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS One. 2011;6:e16393.
Ellmerich S, Schöller M, Duranton B, et al. Promotion of intestinal carcinogenesis by Streptococcus bovis. Carcinogenesis. 2000;21:753–6.
Duncan CG, Leary RJ, Lin J, et al. Identification of microbial DNA in human cancer. BMC Med Genomics. 2009;2:22. doi:10.1186/1755-8794-2-22.
Fukata M, Abreau MT. Microflora in colorectal cancer: a friend to fear. Nat Med. 2010;16:639–41.
Compare D, Nardone G. Contribution of gut microbiota to colonic and extracolonic cancer development. Dig Dis. 2011;29:554–61.
Vipperia K, O’Keefe SJ. The microbiota and its metabolites in colonic mucosal health and cancer risk. Nutr Clin Practice. 2013;27:624–35.
Gold JS, Bayar S, Salem RR. Association of Streptococcus bovis bacteremia with colonic neoplasia and extracolonic malignancy. Arch Surg. 2004;139:760–5.
Bibiloni R, Mangold M, Madsen KL, et al. The bacteriology of biopsies differs between newly diagnosed, untreated, Crohn’s disease and ulcerative colitis patients. J Med Microbiol. 2006;55:1141–9.
•• Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nature Rev Microbiol. 2012;10:717–25. This paper proposes a new theory describing how low-abundance pathogens may be capable of causing inflammation.
McDonald SA, Preston SI, Lovell MJ, et al. Mechanisms of disease: from stem cells to colorectal cancer. Nat Clin Pract Gastroenterol Hepatol. 2006;3:267–74.
Willis ND, Przyborski SA, Hutchinson CJ, Wilson RG. Colonic and colorectal cancer stem cells: progress in the search of putative biomarkers. J Anat. 2008;213:59–65.
•• Pédron T, Mulet C, Dauga C, et al. A crypt-specific core microbiota resides in the mouse colon. mBIO. 2012;3(3):e00116–12. This paper points out the critical need to characterize specific niche microbial populations if we are going to fully understand the contribution of microbiota to colon cancer. It will be necessary to perform this type of work if we are going to identify chemoprotective interventions.
Macfarlane S, Woodmansey EJ, Macfarlane GT. Colonization of mucin by human intestinal bacteria and establishment of biofilm communities in a two-stage continuous culture system. Appl Environ Microbiol. 2005;71:7483–92.
Kosiewicz MM, Zirnheld AL, Alard P. Gut microbiota, immunity, and disease: A complex relationship. Front Microbiol. 2011;2:180. doi:10.3389/fmicb.2011.00180.
Wells JM, Rossi O, Meijerink M, van Baarlen P. Epithelial crosstalk at the microbiota-mucosal interface. Proc Natl Acad Sci USA. 2011;108 Suppl 1:4607–14.
Smythies LD, Shen R, Bimczok D, et al. Inflammation anergy in human intestinal macrophages is due to Smad-induced IκBα expression and NF-κB inactivation. J Biol Chem. 2010;285:19593–604.
Saleh M, Trinchieri G. Innate immune mechanisms of colitis and colitis-associated colorectal cancer. Nat Rev Immunol. 2011;11:9–20.
Abreu MT. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol. 2010;10:131–44.
Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, et al. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–41.
Brint EK, MacSharry J, Fanning A, et al. Differential expression of toll-like receptors in patients with irritable bowel syndrome. Am J Gastroenterol. 2011;106:329–36.
Pimentel-Nunes P, Gonçalves N, Boal-Carvalho I, et al. Decreased toll-interacting protein and peroxisome proliferator-activated receptor γ are associated with increased expression of toll-like receptors in colon carcinogenesis. J Clin Pathol. 2012;65:302–8.
Lee J, Mo J-H, Katakura K, et al. Maintenance of colonic homeostasis by distinctive apical TLR9 signalling in intestinal epithelial cells. Nat Cell Biol. 2006;8:1327–36.
Chen GY, Shaw MH, Redondo G, Núñez G. The innate immune receptor Nod1 protects the intestine from inflammation-induced tumorigenesis. Cancer Res. 2008;68:10060–7.
Arthur JC, Jobin C. The struggle within: Microbial influences on colorectal cancer. Inflamm Bowel Dis. 2011;17:396–409.
Couturier-Maillard A, Secher T, Rehman A, et al. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest. 2013;123:700–11.
Langowski JL, Zhang X, Wu L, et al. IL-23 promotes tumour incidence and growth. Nature. 2006;442:461–5.
Grivennikov SI, Wang K, Mucida D, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. 2012;491:254–8.
Wu S, Rhee K-J, Albesiano E, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15:1016–22.
Jobin C. Colorectal cancer: CRC – all about microbial products and barrier function? Nat Rev Gastroenterol Hepatol. 2012;9:694–6.
Tjalsma H, Boleij A, Marchesi JR, et al. A bacterial driver-passenger model for colorectal cancer: Beyond the usual suspects. Nat Rev Microbiol. 2012;10:575–82.
Wang L, Yi T, Zhang W, et al. IL-17 enhances tumor development in carcinogen-induced skin cancer. Cancer Res. 2010;70:10112–20.
Ji Y, Zhang W. Th17 cells: Positive or negative role in tumor? Cancer Immunol Immunother. 2010;59:979–87.
Murugaiyan G, Saha B. Protumor vs antitumor functions of IL-17. J Immunol. 2009;183:4169–75.
Yang S, Wang B, Guan C, et al. Foxp3 + IL-17+ T cells promote development of cancer-initiating cells in colorectal cancer. J Leuk Biol. 2011;89:85–91.
Macfarlane GT, Macfarlane S. Bacteria, colonic fermentation, and gastrointestinal health. J AOAC Int. 2012;95:50–60.
De Preter V, Arjis I, Windey K, et al. Imparied butyrate oxidation in ulcerative colitis is due to decreased butyrate uptake and a defect in the oxidation pathway. Infl Bowel Dis. 2012;18:1127–36.
O’Keefe SJD. Nutrition and colonic health: The critical role of microbiota. Curr Opin Gastroenterol. 2008;24:51–8.
Walton GE, van den Heuvel EGHM, Kosters MHW, et al. A randomized crossover study investigating the effects of galacto-oligosaccharides on the faecal microbiota in men and women over 50 years of age. Br J Nutr. 2012;107:1466–75.
Modis K, Coletta C, Erdelyi K, et al. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 2013;27:601–11.
•• Maurice CF, Haiser HJ, Turnbaugh PJ. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell. 2013;152:39–50. This paper demonstrates the importance of characterizing the functional changes in microbiota, in addition to determining microbial diversity when exposed to exogenous compounds.
• van Duynhoven J, Vaughan EE, Jacobs DM, et al. Metabolic fate of polyphenols in the human superorganism. Proc Natl Acad Sci. 2011;108:4531–8. This review provides an excellent overview of the multiple ways in which dietary polyphenols influence the dynamic relationship between microbiota and the host.
Cueva C, Sánchez–Patán F, Monagas M, et al. In vitro fermentation of grape seed flavan–3–ol fractions by human faecal microbiota: changes in microbial groups and phenolic metabolites. FEMS Microbiol Ecol. 2012;83:792–805.
Gall WE, Beebe K, Lawton KA, et al. α-hydroxybutyrate is an early biomarker of insulin resistance and glucose intolerance in a nondiabetic population. PLoS One. 2010;5:e10883.
Zhao C, Ivanov I, Dougherty ER, et al. Non-invasive detection of candidate molecular biomarkers in subjects with a history of insulin resistance and colorectal adenomas. Cancer Prev Res. 2009;2:590–7.
Yehuda-Shnaidman E, Schwartz B. Mechanisms linking obesity, inflammation and altered metabolism to colon carcinogenesis. Obesity Rev. 2012;13:1083–95.
Calani L, Dall’Asta M, Derlindati E, et al. Colonic metabolism of polyphenols from coffee, green tea, and hazelnut skins. J Clin Gastroenterol. 2012;46 Suppl 1:S95–9.
Holmes E, Kinross J, Gibson GR, et al. Therapeutic modulation of microbiota-host metabolic interactions. Sci Trans Med. 2012;4:137rv6.
Nicholson JK, Holmes E, Kinross J, et al. Host-gut microbiota metabolic interactions. Science. 2012;336:1262–7.
• Donohoe DR, Garge N, Zhang X, et al. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011;13:517–26. This paper describes a well-controlled set of experiments that identify the role of butyrate in colonocyte homeostasis.
Dumas M-E. The microbial-mammalian metabolic axis: Beyond simple metabolism. Cell Metab. 2011;13:489–90.
Cho Y, Kim H, Turner ND, et al. A chemoprotective fish oil- and pectin-containing diet temporally alters gene expression profiles in exfoliated rat colonocytes throughout oncogenesis. J Nutr. 2011;141:1029–35.
Crim KC, Sanders LM, Hong MY, et al. Upregulation of p21Waf1/Cip1 expression in vivo by butyrate administration can be chemoprotective or chemopromotive depending on the lipid component of the diet. Carcinogenesis. 2008;29:1415–20.
Barrasa JI, Santiago-Gómez A, Olmo N, et al. Resistance to butyrate impairs bile acid-induced apoptosis in human colon adenocarcinoma cells via up-regulation of Bcl-2 and inactivation of Bax. Biochim Biophys Acta. 2012;1823:2201–9.
Turk HF, Kolar SS, Fan YY, et al. Linoleic acid and butyrate synergize to increase Bcl-2 levels in colonocytes. Int J Cancer. 2011;128:63–71.
Cherbuy C, Honvo-Houeto E, Bruneau A, et al. Microbiota matures colonic epithelium through a coordinated induction of cell cycle-related proteins in gnotobiotic rat. Am J Physiol Gastrointest Liver Physiol. 2010;299:G348–57.
Cho Y, Turner ND, Davidson LA, et al. A chemoprotective fish oil/pectin diet enhances apoptosis via Bcl-2 promoter methylation in rat azoxymethane-induced carcinomas. Exp Biol Med. 2012;237:1387–93.
Davidson LA, Nguyen DV, Hokanson RM, et al. Chemopreventive n-3 polyunsaturated fatty acids reprogram genetic signatures during colon cancer initiation and progression in the rat. Cancer Res. 2004;64:6797–804.
Davidson LA, Wang N, Ivanov I, et al. Identification of actively translated mRNA transcripts in a rat model of early-stage colon carcinogenesis. Cancer Prev Res. 2009;2:984–94.
Wilson AJ, Chueh AC, Tögel L, et al. Apoptotic sensitivity of colon cancer cells to histone deacetylase inhibitors is mediated by an Sp1/Sp3-activated transcriptional program involving immediate-early gene induction. Cancer Res. 2010;70:609–20.
Rajendran P, Williams DE, Ho E, Dashwood RH. Metabolism as a key to histone deacetylase inhibition. Crit Rev Biochem Mol Biol. 2011;46:181–99.
Wu S, Li RW, Li W, Li C. Transcriptome characterization by RNA-seq unravels the mechanisms of butyrate-induced epigenomic regulation in bovine cells. PLoS One. 2012;7:e36940.
Wilson AJ, Byun DS, Nasser S, et al. HDAC4 promotes growth of colon cancer cells via repression of p21. Mol Biol Cell. 2008;19:4062–75.
Li Y, Kundu P, Seow SW, et al. Gut microbiota accelerate tumor growth via c-jun and STAT3 phosphorylation in APCMin/+ mice. Carcinogenesis. 2012;33:1231–8.
Arthur JC, Perez-Chanona E, Mühlbauer M, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120–3.
Chen H-M, Yu Y-N, Wang J-L, et al. Decreased dietary fiber intake and structural alteration of gut microbiota in patients with advanced colorectal adenoma. Am J Clin Nutr. 2013;97:1044–52.
Conlon MA, Kerr CA, McSweeney CS, et al. Resistant starches protect against colonic DNA damage and alter microbiota and gene expression in rats fed a Western diet. J Nutr. 2012;142:832–40.
Hooda S, Boler BMV, Serao MCR, et al. 454 pyrosequencing reveals a shift in fecal microbiota of healthy adult men consuming polydextrose or soluble corn fiber. J Nutr. 2012;142:1259–65.
Kuo S-M. The interplay between fiber and the intestinal microbiota in the inflammatory response. Adv Nutr. 2013;4:16–28.
Ghosh S, DeCoffe D, Brown K, et al. Fish oil attenuates omega-6 polyunsaturated fatty acid-induced dysbiosis and infectious colitis but impairs LPS dephosphorylation activity causing sepsis. PLoS One. 2013;8(2):e55468.
Chapkin RS, Seo J, McMurray DN, Lupton JR. Mechanisms by which docosahexaeonic acid and related fatty acids reduce colon cancer risk and inflammatory disorders of the intestine. Chem Phys Lipids. 2008;153:14–23.
Kolar SS, Barhoumi R, Callaway E, et al. Synergy between docosahexaenoic acid and butyrate elicits p53-independent apoptosis via mitochondrial Ca2+ accumulation in human colon cancer cells and primary cultures of rat colonic crypts. Am J Physiol Gastrointest Liver Physiol. 2007;293:G935–43.
Kolar SS, Barhoumi R, Jones CK, et al. Interactive effects of fatty acid and butyrate-induced mitochondrial Ca2+ loading and apoptosis in colonocytes. Cancer. 2011;117:5294–303.
Simoes CD, Maukonen J, Kaprio J, et al. Habitual dietary intake is associated with stool microbiota composition in monozygotic twins. J Nutr. 2013;143:417–23.
Azcárate-Peril MA, Sikes M, Bruno-Bárcena JM. The intestinal microbiota, gastrointestinal environment and colorectal cancer: A putative role for probiotics in prevention of colorectal cancer? Am J Physiol Gastrointest Liver Physiol. 2011;301:G401–24.
Zhu Y, Luo TM, Jobin C, Young HA. Gut microbiota and probiotics in colon tumorigenesis. Cancer Lett. 2011;309:119–27.
Gourbeyre P, Denery S, Bodinier M. Probiotics, prebiotics, and synbiotics: Impact on the gut immune system and allergic reactions. J Leukoc Biol. 2011;89:685–95.
Appleyard CB, Cruz ML, Isidro AA, et al. Pretreatemnt with the probiotic VSL#3 delays transition from inflammation to dysplasia in a rat model of colitis-associated cancer. Am J Physiol Gastrointest Liver Physiol. 2011;301:G1004–13.
Cho I, Yamanishi S, Cox L, et al. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature. 2012;488:621–6.
McLaughlin MM, Dacquisto MP, Jacobus DP, Horowitz RE. Effects of the germfree state on responses of mice to whole-body irradiation. Rad Res. 1964;23:333–49.
Crawford PA, Gordon JI. Microbial regulation of intestinal radiosensitivity. Proc Nat Acad Sci USA. 2005;102:13254–9.
Manichanh C, Varela E, Martinez C, et al. The gut microbiota predispose to the pathophysiology of acute postradiotherapy diarrhea. Am J Gastroenterol. 2008;103:1754–61.
Egan LJ, Eckmann L, Greten FR, et al. IκB-kinaseβ-dependent NF-κB activation provides radioprotection to the intestinal epithelium. Proc Nat Acad Sci USA. 2004;101:2452–7.
Palmer C, Bik EM, DiGiulio DB, et al. Development of the human infant intestinal microbiota. PLoS Biol. 2007;5:e177.
Donovan SM, Wang M, Li M, et al. Host-microbe interactions in the neonatal intestine: Role of human milk oligosaccharides. Adv Nutr. 2012;3:4505–55.
Marcobal A, Sonnenburg JL. Human milk oligosaccharide consumption by intestinal microbiota. Clin Microbiol Infect. 2012;18 Suppl 4:12–5.
Zivkovic AM, German JB, Lebrilla CB, Mills DA. Human milk glycobiome and its impact on the infant gastrointestinal microbiota. Proc Natl Acad Sci USA. 2011;108 Suppl 1:4653–8.
• Schwartz S, Friedberg I, Ivanov IV, et al. A metagenomic study of diet-dependent interaction between gut microbiota and host in infants reveals differences in immune response. Genome Biol. 2012;13:r32. doi:10.1186/gb-2012-13-4-r32. This paper describes the interplay between colonizing microbiota and gene expression in the developing neonatal intestinal tract.
Payne AN, Chassard C, Banz Y, Lacroix C. The composition and metabolic activity of child gut microbiota demonstrate differential adaptation to varied nutrient loads in an in vitro model of colonic fermentation. FEMS Microbiol Ecol. 2012;80:608–23.
Young W, Roy NC, Lee J, et al. Changes in bowel microbiota induced by feeding weanlings resistant starch stimulate transcriptomic and physiological responses. Appl Environ Microbiol. 2012;78:6656–64.
Fança-Berthon P, Hoebler C, Mouzet E, et al. Intrauterine growth restriction not only modifies the cecocolonic microbiota in neonatal rats but also affects its activity in young adult rats. J Ped Gastro Nutr. 2010;51:402–13.
Joss-Moore LA, Lane RH. The developmental origins of adult disease. Curr Opin Ped. 2009;21:230–4.
Ahlquist DA, Zou H, Domanico M, et al. Next-generation stool DNA accurately detects colorectal cancer and large adenomas. Gastroenterol. 2012;142:248–56.
Acknowledgment
Supported in part by USCP (HVM006-12 and R0002-11) and CDPB (PN 12-20) to N.D.T. and NIH U01CA162077 to R.S.C.
Compliance with Ethics Guidelines
ᇵ
Conflict of Interest
Nancy D. Turner, Lauren E. Ritchie, Robert S. Bresalier, and Robert S. Chapkin declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding authors
Additional information
This article is part of the Topical Collection on Inflammatory Bowel Disease
Rights and permissions
About this article
Cite this article
Turner, N.D., Ritchie, L.E., Bresalier, R.S. et al. The Microbiome and Colorectal Neoplasia: Environmental Modifiers of Dysbiosis. Curr Gastroenterol Rep 15, 346 (2013). https://doi.org/10.1007/s11894-013-0346-0
Published:
DOI: https://doi.org/10.1007/s11894-013-0346-0