
Abstract
Purpose of Review
Herein, we summarize recent advances which provide new insights into the role of the autonomic nervous system in the control of blood flow and blood pressure during hyperinsulinemia. We also highlight remaining gaps in knowledge as it pertains to the translation of findings to relevant human chronic conditions such as obesity, insulin resistance, and type 2 diabetes.
Recent Findings
Our findings in insulin-sensitive adults show that increases in muscle sympathetic nerve activity with hyperinsulinemia do not result in greater sympathetically mediated vasoconstriction in the peripheral circulation. Both an attenuation of α-adrenergic-receptor vasoconstriction and augmented β-adrenergic vasodilation in the setting of high insulin likely explain these findings. In the absence of an increase in sympathetically mediated restraint of peripheral vasodilation during hyperinsulinemia, blood pressure is supported by increases in cardiac output in insulin-sensitive individuals.
Summary
We highlight a dynamic interplay between central and peripheral mechanisms during hyperinsulinemia to increase sympathetic nervous system activity and maintain blood pressure in insulin-sensitive adults. Whether these results translate to the insulin-resistant condition and implications for long-term cardiovascular regulation warrants further exploration.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Blood glucose levels increase following consumption of a meal. Increases in blood glucose promote release of insulin from the pancreas, which is essential for peripheral glucose uptake. In addition to its metabolic actions, insulin elicits profound vasodilatory effects in the peripheral circulation, promoting the delivery of insulin and glucose to tissues such as skeletal muscle [1•, 2,3,4,5,6]. Indeed, insulin-stimulated skeletal muscle vasodilation plays an important role in glycemic control [3, 5,6,7]. Consistent with this, impairments in insulin-mediated blood flow in adults with insulin resistance parallel with decrements in glucose disposal [1•].
At the level of the vascular endothelium, insulin binds the insulin receptor (IR) and activates downstream IR substrate 1/2/PI3K signaling, resulting in phosphorylation of endothelial nitric oxide (NO) synthase [8,9,10]. During hyperinsulinemia, the vasodilatory effects of insulin in skeletal muscle must be opposed by countercurrent vasoconstrictor mechanisms in order to preserve blood pressure. This is analogous to exercise-induced hyperemia, where Loring Rowell once stated, “skeletal muscle is a sleeping giant whose blood flow must be under tonic vasoconstrictor constraint if hypotension is to be averted” [11]. Insulin promotes endothelin-1-mediated vasoconstriction via activation of ERK1/2. Endothelin-1 is one key countercurrent mechanism to maintain blood pressure [8, 12, 13], but is not the sole opposing force to insulin-induced, NO-dependent vasodilation. Hyperinsulinemia also elicits an increase in the activity of the sympathetic nervous system [14,15,16,17,18,19,20]. In this regard, inhibition of local sympathetic vascular control (via regional sympathectomy) was shown to elicit a more rapid NO-mediated vasodilation in response to insulin compared to control [21]. Using an isolated forearm model, Lembo and colleagues further supported a crosstalk between insulin and the sympathetic nervous system [22, 23].
The insulin-mediated increase in sympathetic nervous system activity directed towards the skeletal muscle (muscle sympathetic nerve activity, MSNA) in insulin-sensitive individuals is gradual, occurring over approximately 30–60 min. This time course has been primarily attributed to the amount of time needed for insulin to transfer across the blood brain barrier [24,25,26]. In the brain, insulin increases sympathetic nervous system activity via actions within the arcuate nucleus [27,28,29,30,31,32]. Increases in central insulin also enhance the gain of the arterial baroreflex control of sympathetic nervous system activity [33, 34]. Recent evidence further supports a role for both the arterial baroreceptors [35,36,37,38,39] and carotid chemoreceptors [40] in insulin-mediated sympathoexcitation.
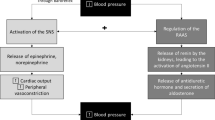
The purpose of this brief review is to summarize recent work by our group, in context of known findings, to provide new insights into the role of the autonomic nervous system in the control of blood pressure during hyperinsulinemia and consequent peripheral vasodilation (Fig. 1). We also highlight remaining gaps in knowledge as it pertains to the translation of findings to relevant human chronic conditions such as obesity, insulin resistance, and type 2 diabetes.

Role of the autonomic nervous system in the hemodynamic response to hyperinsulinemia. Our findings in insulin-sensitive adults do not support the role of sympathetically mediated vasoconstriction as a principal mechanism to restrain insulin-induced skeletal muscle vasodilation for the maintenance of blood pressure during hyperinsulinemia. Both insulin-induced attenuation of α-adrenergic vasoconstriction and augmented β-adrenergic vasodilation likely explain these findings. Due to the lack of sympathetically mediated restraint of peripheral vasodilation during hyperinsulinemia, healthy individuals must heavily rely on increases in cardiac output to support blood pressure. Together, these results support a dynamic interplay between central and peripheral mechanisms during hyperinsulinemia to increase muscle sympathetic nerve activity and maintain blood pressure in insulin-sensitive adults. BP, blood pressure; Endo, endothelium; SMC, smooth muscle cells; IR, insulin receptor; i, insulin
Role of Sympathetic Nervous System Activation in Restraining Insulin-Stimulated Vasodilation
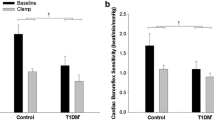
As noted above, hyperinsulinemia elicits a prominent increase in MSNA in insulin-sensitive adults [14,15,16,17,18,19,20]. Activation of the sympathetic nervous system stimulates norepinephrine release from sympathetic nerve terminals, which ultimately binds α-adrenergic receptors and promotes vasoconstriction. With this, increases in MSNA during hyperinsulinemia have the potential to limit insulin-induced vasodilation via greater α-adrenergic mediated vasoconstriction [16, 20, 41]. Considering this scenario, we hypothesized that blunting MSNA would elicit a greater increase in femoral blood flow and vascular conductance during hyperinsulinemia compared to baseline [42••]. Using neck suction (to acutely unload the arterial baroreceptors and decrease MSNA), we observed no effect of hyperinsulinemia on the skeletal muscle blood flow, vascular conductance, nor total peripheral resistance response to acute MSNA inhibition [42••]. Consistent with this, blocking α-adrenergic receptor-mediated vasoconstriction (with prazosin) was similarly shown to have no effect on insulin-stimulated blood flow [41]. Thus, we concluded that the role of the sympathetic nervous system in restraining peripheral vasodilation, particularly in healthy adults, is not augmented during hyperinsulinemia. On the other hand, neck suction did promote a greater decrease in blood pressure during hyperinsulinemia and this response was accompanied by greater decreases in cardiac output [42••]. Combined with the observation that cardiac output was markedly increased during insulin infusion, these results support the idea that neurally mediated increases in cardiac output, rather than restraint of peripheral vasodilation, may be critical to the preservation of systemic blood pressure during hyperinsulinemia (Fig. 1).
Given the lack of an effect of hyperinsulinemia on the peripheral vascular response to an acute reduction in MSNA, our understanding of the “net effect” of insulin on sympathetic control of blood flow and blood pressure remained incomplete. We therefore sought to extend these findings by examining the effect of hyperinsulinemia on sympathetic transduction (i.e., the dynamic vascular and/or blood pressure response to MSNA) [43•]. To do this, we employed a signal-averaging technique [44] to characterize changes in blood pressure and total vascular conductance following spontaneous bursts of MSNA at rest and during hyperinsulinemia. Consistent with our results using neck suction [42••], we observed no significant differences in the peak blood pressure nor vascular response to bursts of MSNA between rest and hyperinsulinemia (i.e., “net” sympathetic transduction was unchanged) [43•]. However, after taking into account the increase in MSNA burst height that occurs with hyperinsulinemia (presumably associated with greater efferent sympathetic activity), we found that sympathetic transduction for a given burst amplitude was blunted during insulin infusion [43•]. These findings lend further support to the idea that during hyperinsulinemia, a given amount of sympathetic activity is less capable of restraining insulin-stimulated vasodilation. Indeed, we went on to show that in young healthy men and women, acute sympathetic activation (via a cold pressor test) elicited less vasoconstriction in the setting of high insulin compared to control [45••]—however, contributing mechanisms remained unclear.
Role of Hyperinsulinemia in Attenuating Sympathetically Mediated Vasoconstriction
We were initially surprised to observe that insulin-mediated increases in skeletal muscle blood flow were not under considerable sympathetically mediated vasoconstrictor restraint [42••]. With this, we hypothesized that insulin may attenuate α-adrenergic receptor-mediated vasoconstriction [22, 23, 46,47,48,49]. In isolated aortic rings, we showed that insulin exposure blunted α1-adrenergic-mediated vasoconstriction [42••]. We further demonstrated that the ability of insulin to blunt α1-adrenergic-mediated vasoconstriction requires the following: (1) an intact endothelium, and (2) an unrestricted PI3K/Akt insulin-signaling pathway. Specifically, endothelium denudation and pharmacological inhibition of NO synthase or the upstream enzyme PI3K [50•, 51] abolished the insulin-induced suppression of α1-adrenergic vasoconstriction. Similarly, insulin-induced suppression of α1-adrenergic vasoconstriction was lost in aortic rings from insulin-resistant db/db mice [42••]. Accordingly, vascular insulin signaling is capable of attenuating sympathetically mediated vasoconstriction and that “sympatholytic” factor appears to be endothelial-derived and NO-dependent.
Norepinephrine release from sympathetic nerve terminals binds α-adrenergic receptors to elicit vasoconstriction; however, norepinephrine can also bind β-adrenergic receptors—which promote vasodilation. Crosstalk between insulin and β-adrenergic receptors was previously supported by the literature, particularly in the heart [52,53,54,55,56]. Thus, we hypothesized that insulin-induced attenuation of sympathetic vasoconstriction may be due to a potentiation of β-adrenergic receptor-mediated NO production [57]. In accordance, we found epinephrine and norepinephrine-induced vasoconstriction to be attenuated in isolated mouse arteries exposed to insulin. Importantly, the ability of insulin to suppress epinephrine and norepinephrine-induced vasoconstriction was lessened with propranolol, a non-specific β-adrenergic receptor antagonist [57]. In alignment with this observation, insulin-induced vasodilation within the human forearm is attenuated with intra-arterial infusion of propranolol [58]. Notably, we also showed that insulin enhanced β-adrenergic-mediated vasodilation in an endothelium and NO-dependent manner. These observations may be mediated via crosstalk between the insulin receptor and β-adrenergic receptors, as has been shown in cardiac tissue [56]. Together, these findings support the link between insulin and β-adrenergic signaling in the modulation of vascular tone (Fig. 1).
Role of Arterial Baroreceptors in Mediating Insulin-Induced Sympathoexcitation
Given the observed ability of hyperinsulinemia to attenuate sympathetically mediated vasoconstriction, the question arose as to the mechanism by which insulin-mediated sympathoexcitation is achieved. In addition to insulin’s actions within the arcuate nucleus to increase efferent sympathetic activity [27,28,29,30,31,32], the peripheral vasodilator effect of insulin has been postulated to contribute to baroreflex-mediated increases in MSNA during hyperinsulinemia [35,36,37,38,39]. We thus reasoned that rescuing peripheral resistance with co-infusion of the vasoconstrictor phenylephrine would attenuate the MSNA response to hyperinsulinemia [59••]. Consistent with this hypothesis, we found that co-infusion with phenylephrine returned MSNA and total peripheral resistance to baseline levels [59••]. Notably, a role of insulin-stimulated vasodilation and response of the arterial baroreflex in insulin-mediated increases in MSNA has been discounted previously on the basis that in healthy adults, blood pressure is unchanged during systemic insulin infusion [20]. In addition, increases in MSNA during hyperinsulinemia are observed prior to increases in leg blood flow [36]. Based on these data, the authors concluded that the ability of insulin to stimulate MSNA is dissociated from its acute hemodynamic action [36]. Similarly, Anderson and colleagues [20] argued that during hyperinsulinemia, the change in MSNA per unit reduction in blood pressure was much greater than what was observed with another peripheral vasodilator—again going against a peripheral baroreflex mechanism. Important to these conclusions is the assumption that the peripheral vasculature retains its sensitivity to sympathetic activation during insulin exposure; however, as discussed above, sympathetically mediated vasoconstriction is attenuated during hyperinsulinemia [42••, 43•, 45••]. In the context of prior findings, we propose that the peripheral vasodilator effect of insulin contributes, at least in part, to baroreflex-mediated increases in MSNA during hyperinsulinemia in insulin-sensitive adults [59••].
Role of Peripheral Chemoreceptors in Mediating Insulin-Induced Sympathoexcitation
A novel role for the carotid chemoreceptors in insulin-mediated activation of the sympathetic nervous system and glucose regulation has been recently proposed [40, 60,61,62,63,64]. Using a combination of acute hyperoxia [65] and low-dose intravenous dopamine [66, 67] (to acutely attenuate carotid chemoreceptor activity), we sought to examine the contribution of the carotid chemoreceptors to insulin-mediated increases in MSNA in healthy, insulin-sensitive individuals [68•]. Consistent with the work of others [69, 70], we observed a limited role for the carotid body chemoreceptors in basal MSNA in healthy adults. We further found no consistent, measurable effect of acute hyperoxia or dopamine on MSNA during hyperinsulinemia [68•]. We interpreted these results to mean that the carotid body chemoreceptors do not contribute to insulin-mediated increases in MSNA in young, healthy, insulin-sensitive adults. Additionally, the glucose infusion rate required to maintain euglycemia was unaffected by interventions targeting the peripheral chemoreceptors—further supporting the idea that the carotid body chemoreceptors do not contribute to insulin-mediated increases in MSNA and/or insulin sensitivity in young, healthy adults. However, through an exploratory post hoc analysis, we found that, in some individuals, hyperoxia may have had a greater effect on MSNA during insulin than at baseline. The individuals where this occurred were the least insulin-sensitive (lowest glucose infusion rate) and lend support to the idea that the carotid chemoreceptors’ role in sympathetic regulation during hyperinsulinemia may be altered with progressive disease (e.g., obesity, insulin resistance, type 2 diabetes).
Perspectives
The increased prevalence of obesity worldwide has resulted in an increase in obesity-related disorders including hypertension, insulin resistance, and type 2 diabetes. Indeed, insulin resistance affects nearly one-third of the US population and precedes the development of type 2 diabetes. As shown by our group [71, 72] and others [73, 74], MSNA is nearly two times greater in obese, insulin-resistant adults compared to lean counterparts. Higher MSNA in obesity is directly associated with higher fasting insulin (i.e., insulin resistance) [75,76,77,78,79]. With this information, one might anticipate that the MSNA response to hyperinsulinemia is further augmented in obesity. However, the majority of currently available data refute the notion that MSNA responses to acute hyperinsulinemia are greater in adults with obesity compared to normal weight controls [74, 80,81,82,83]. Disconnect between the effect of chronic (i.e., insulin resistance) and acute increases in circulating insulin (i.e., a meal) on MSNA may be due to impairments in cellular insulin action within the peripheral vasculature in human insulin resistance (i.e., reduced insulin-induced vasodilation and/or augmented α-adrenergic-mediated constriction) [74, 80, 84]. Indeed, it is well established that adults with insulin resistance exhibit impaired insulin-stimulated skeletal muscle vasodilation [1•, 2, 7, 85]. Based on current understanding, one may speculate that obese, insulin-resistant individuals would not exhibit further elevation in MSNA during acute hyperinsulinemia due, in part, to these impairments in the peripheral vasodilatory response to hyperinsulinemia [74, 80, 84, 86] and consequently reduced stimulus on the arterial baroreflex. Of note, work from Straznicky and colleagues [83] refutes the notion that a blunted MSNA response in insulin-resistant adults is related to the magnitude of insulin-stimulated blood flow. Given that insulin attenuates sympathetically mediated vasoconstriction [45••], and this may be absent in the setting of insulin resistance [42••], future experiments directly addressing these questions in a cohort of insulin-resistant individuals are warranted. Furthermore, whether human obesity alters the efficiency with which insulin enters the central nervous system [87, 88] and/or the sensitivity of important brain and/or peripheral (i.e., carotid chemoreceptors) regions to insulin stimulation remains untested.
In addition to a lack of clear understanding of the presence of central (brain) insulin resistance in obesity, it is not well known whether sex differences exist in the neural and/or vascular responses to hyperinsulinemia. Reviewed in detail elsewhere [89••, 90], insulin-mediated increases in MSNA occur similarly in normal weight male and female rats and humans (unpublished observations from our group). In contrast, recent data suggest that obesity prone male rats have a tenfold greater increase in sympathetic nervous system activity with insulin exposure, a phenomena not observed in obesity-prone female rats [91]. In agreement with these data, our group [92] and others [93, 94] have shown that obesity induced by Western diet feeding increases blood pressure in male animals only. Together, these data suggest that men may be sensitized to the action of insulin within in the brain, and this differs from the response in women; however, definitive data in humans are lacking.
Conclusion
In aggregate, our findings in insulin-sensitive adults do not support the role of sympathetically mediated vasoconstriction as a principal mechanism to restrain insulin-induced skeletal muscle vasodilation for the maintenance of blood pressure during hyperinsulinemia. Both insulin-induced attenuation of α-adrenergic vasoconstriction and augmented β-adrenergic vasodilation likely explain these findings. Due to the lack of sympathetically mediated restraint of peripheral vasodilation during hyperinsulinemia, healthy individuals must heavily rely on increases in cardiac output to support blood pressure. Together, these results support a dynamic interplay between central and peripheral mechanisms during hyperinsulinemia to increase muscle sympathetic nerve activity and maintain blood pressure in insulin-sensitive adults. Future research translating results to obesity and insulin resistance is warranted in order to enhance our understanding of the pathophysiology of obesity and diabetes, which will be necessary prior to the development of new therapeutic interventions.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Park LK, et al. Skeletal muscle microvascular insulin resistance in type 2 diabetes is not improved by eight weeks of regular walking. J Appl Physiol (1985). 2020;129(2):283–96. This report provides evidence that in sedentary adults with type 2 diabetes, insulin-stimulated increases in leg vascular conductance and capillary perfusion in skeletal muscle are blunted.
Reynolds LJ, et al. Obesity, type 2 diabetes, and impaired insulin-stimulated blood flow: role of skeletal muscle NO synthase and endothelin-1. J Appl Physiol (1985). 2017;122(1):38–47.
Baron AD, et al. Mechanism of insulin resistance in insulin-dependent diabetes mellitus: a major role for reduced skeletal muscle blood flow. J Clin Endocrinol Metab. 1991;73(3):637–43.
Baron AD, et al. Insulin-mediated skeletal muscle vasodilation contributes to both insulin sensitivity and responsiveness in lean humans. J Clin Invest. 1995;96(2):786–92.
Barrett EJ, et al. The vascular actions of insulin control its delivery to muscle and regulate the rate-limiting step in skeletal muscle insulin action. Diabetologia. 2009;52(5):752–64.
Barrett EJ, et al. Insulin regulates its own delivery to skeletal muscle by feed-forward actions on the vasculature. Am J Physiol Endocrinol Metab. 2011;301(2):E252–63.
Laakso M, et al. Impaired insulin-mediated skeletal muscle blood flow in patients with NIDDM. Diabetes. 1992;41(9):1076–83.
Kim JA, et al. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation. 2006;113(15):1888–904.
Steinberg HO, et al. Insulin-mediated skeletal muscle vasodilation is nitric oxide dependent. A novel action of insulin to increase nitric oxide release. J Clin Invest. 1994;94(3):1172–9.
Yu Q, Gao F, Ma XL. Insulin says NO to cardiovascular disease. Cardiovasc Res. 2011;89(3):516–24.
Rowell LB. Ideas about control of skeletal and cardiac muscle blood flow (1876–2003): cycles of revision and new vision. J Appl Physiol (1985). 2004;97(1):384–92.
Eringa EC, et al. Physiological concentrations of insulin induce endothelin-mediated vasoconstriction during inhibition of NOS or PI3-kinase in skeletal muscle arterioles. Cardiovasc Res. 2002;56(3):464–71.
Eringa EC, et al. Selective resistance to vasoactive effects of insulin in muscle resistance arteries of obese Zucker (fa/fa) rats. Am J Physiol Endocrinol Metab. 2007;293(5):E1134–9.
Young CN, et al. Insulin enhances the gain of arterial baroreflex control of muscle sympathetic nerve activity in humans. J Physiol. 2010;588(Pt 18):3593–603.
Vollenweider P, et al. Differential effects of hyperinsulinemia and carbohydrate metabolism on sympathetic nerve activity and muscle blood flow in humans. J Clin Investig. 1993;92(1):147–54.
Vollenweider L, et al. Insulin-induced sympathetic activation and vasodilation in skeletal muscle. Effects of insulin resistance in lean subjects. Diabetes. 1995;44(6):641–5.
Scherrer U, Sartori C. Insulin as a vascular and sympathoexcitatory hormone: implications for blood pressure regulation, insulin sensitivity, and cardiovascular morbidity. Circulation. 1997;96(11):4104–13.
Lembo G, et al. Abnormal sympathetic overactivity evoked by insulin in the skeletal muscle of patients with essential hypertension. J Clin Invest. 1992;90(1):24–9.
Berne C, et al. The sympathetic response to euglycaemic hyperinsulinaemia. Evidence from microelectrode nerve recordings in healthy subjects. Diabetologia. 1992;35(9):873–9.
Anderson EA, et al. Hyperinsulinemia produces both sympathetic neural activation and vasodilation in normal humans. J Clin Investig. 1991;87(6):2246–52.
Sartori C, et al. Effects of sympathectomy and nitric oxide synthase inhibition on vascular actions of insulin in humans. Hypertension. 1999;34(4 Pt 1):586–9.
Lembo G, et al. Insulin reduces reflex forearm sympathetic vasoconstriction in healthy humans. Hypertension. 1993;21(6 Pt 2):1015–9.
Lembo G, et al. Insulin blunts sympathetic vasoconstriction through the alpha 2-adrenergic pathway in humans. Hypertension. 1994;24(4):429–38.
Woods SC, et al. Insulin and the blood-brain barrier. Curr Pharm Des. 2003;9(10):795–800.
Margolis RU, Altszuler N. Insulin in the cerebrospinal fluid. Nature. 1967;215(5108):1375–6.
Banks WA. The source of cerebral insulin. Eur J Pharmacol. 2004;490(1–3):5–12.
Ward KR, et al. Sympathetic response to insulin is mediated by melanocortin 3/4 receptors in the hypothalamic paraventricular nucleus. Hypertension. 2011;57(3):435–41.
Luckett BS, Frielle JL, Wolfgang L, Stocker SD. Arcuate nucleus injection of an anti-insulin affibody prevents the sympathetic response to insulin. Am J Physiol Heart Circ Physiol. 2013;304(11):H1538–46.
Cassaglia PA, et al. Insulin acts in the arcuate nucleus to increase lumbar sympathetic nerve activity and baroreflex function in rats. J Physiol. 2011;589(Pt 7):1643–62.
Bardgett ME, McCarthy JJ, Stocker SD. Glutamatergic receptor activation in the rostral ventrolateral medulla mediates the sympathoexcitatory response to hyperinsulinemia. Hypertension. 2010;55(2):284–90.
Cassaglia PA, Shi Z, Brooks VL. Insulin increases sympathetic nerve activity in part by suppression of tonic inhibitory neuropeptide Y inputs into the paraventricular nucleus in female rats. Am J Physiol Regul Integr Comp Physiol. 2016;311(1):R97–103.
Dampney RA. Arcuate nucleus - a gateway for insulin’s action on sympathetic activity. J Physiol. 2011;589(Pt 9):2109–10.
Pricher MP, Freeman KL, Brooks VL. Insulin in the brain increases gain of baroreflex control of heart rate and lumbar sympathetic nerve activity. Hypertension. 2008;51(2):514–20.
Azar AS, Brooks VL. Impaired baroreflex gain during pregnancy in conscious rats: role of brain insulin. Hypertension. 2011;57(2):283–8.
Scherrer U, et al. Suppression of insulin-induced sympathetic activation and vasodilation by dexamethasone in humans. Circulation. 1993;88(2):388–94.
Spraul M, Ravussin E, Baron AD. Lack of relationship between muscle sympathetic nerve activity and skeletal muscle vasodilation in response to insulin infusion. Diabetologia. 1996;39(1):91–6.
Muntzel MS, et al. Mechanisms of insulin action on sympathetic nerve activity. Clin Exp Hypertens. 1995;17(1–2):39–50.
Lu H, et al. The co-existence of insulin-mediated decreased mean arterial pressure and increased sympathetic nerve activity is not mediated by the baroreceptor reflex and differentially by hypoglycemia. Clin Exp Hypertens. 1998;20(2):165–83.
Fagius J, Berne C. Rapid resetting of human baroreflex working range: insights from sympathetic recordings during acute hypoglycaemia. J Physiol. 1991;442:91–101.
Ribeiro MJ, Sacramento JF, Gonzalez C, Guarino MP, Monteiro EC, Conde SV. Carotid body denervation prevents the development of insulin resistance and hypertension induced by hypercaloric diets. Diabetes. 2013;62(8):2905–16.
Randin D, et al. Effects of adrenergic and cholinergic blockade on insulin-induced stimulation of calf blood flow in humans. Am J Physiol. 1994;266(3 Pt 2):R809–16.
Limberg JK, et al. Sympathetically mediated increases in cardiac output, not restraint of peripheral vasodilation, contribute to blood pressure maintenance during hyperinsulinemia. Am J Physiol Heart Circ Physiol. 2020;319(1):H162–70. Results support the idea that during hyperinsulinemia, a sympathetically mediated increase in cardiac output, rather than restraint of peripheral vasodilation, is the principal contributor to the maintenance of systemic blood pressure.
Young B, Padilla J, Johnson B, Curry T, Fadel P, Limberg J. Sympathetic transduction during euglycemic-hyperinsulinemia in humans. FASEB J. 2021;35. https://doi.org/10.1096/fasebj.2021.35.S1.03500. These preliminary findings suggest that during hyperinsulinemia, sympathetic transduction for a given MSNA burst amplitude is attenuated.
Young BE, et al. Sympathetic transduction in humans: recent advances and methodological considerations. Am J Physiol Heart Circ Physiol. 2021;320(3):H942–53.
Limberg JK, et al. Hyperinsulinemia blunts sympathetic vasoconstriction: a possible role of beta-adrenergic activation. Am J Physiol Regul Integr Comp Physiol. 2021;320(6):R771–9. These findings support the idea that sympathetic vasoconstriction can be attenuated during systemic hyperinsulinemia in the leg vasculature of both men and women and that this phenomenon may be in part mediated by potentiation of β-adrenergic vasodilation neutralizing α-adrenergic vasoconstriction.
Sakai K, et al. Intra-arterial infusion of insulin attenuates vasoreactivity in human forearm. Hypertension. 1993;22(1):67–73.
Lembo G, et al. The crosstalk between insulin and the sympathetic nervous system: possible implications in the pathogenesis of essential hypertension. Blood Press Suppl. 1996;1:38–42.
Lembo G, et al. Insulin modulation of vascular reactivity is already impaired in prehypertensive spontaneously hypertensive rats. Hypertension. 1995;26(2):290–3.
Fujishima S, et al. Effects of intra-arterial infusion of insulin on forearm vasoreactivity in hypertensive humans. Hypertens Res. 1995;18(3):227–33.
Olver TD, et al. Persistent insulin signaling coupled with restricted PI3K activation causes insulin-induced vasoconstriction. Am J Physiol Heart Circ Physiol. 2019;317(5):H1166–72. Results demonstrate that insulin-induced vasoconstriction is a pathophysiological phenomenon that can be recapitulated when sustained insulin signaling is coupled with depressed PI3K activation.
Montagnani M, et al. Inhibition of phosphatidylinositol 3-kinase enhances mitogenic actions of insulin in endothelial cells. J Biol Chem. 2002;277(3):1794–9.
Wang Q, et al. Inhibiting insulin-mediated beta2-adrenergic receptor activation prevents diabetes-associated cardiac dysfunction. Circulation. 2017;135(1):73–88.
Karoor V, et al. Insulin stimulates sequestration of beta-adrenergic receptors and enhanced association of beta-adrenergic receptors with Grb2 via tyrosine 350. J Biol Chem. 1998;273(49):33035–41.
Jonsson C, et al. Insulin and beta-adrenergic receptors mediate lipolytic and anti-lipolytic signalling that is not altered by type 2 diabetes in human adipocytes. Biochem J. 2019;476(19):2883–908.
Fu Q, et al. Insulin inhibits cardiac contractility by inducing a Gi-biased beta2-adrenergic signaling in hearts. Diabetes. 2014;63(8):2676–89.
Fu Q, Wang Q, Xiang YK. Insulin and beta adrenergic receptor signaling: crosstalk in heart. Trends Endocrinol Metab. 2017;28(6):416–27.
Gros R, Borkowski KR, Feldman RD. Human insulin-mediated enhancement of vascular beta-adrenergic responsiveness. Hypertension. 1994;23(5):551–5.
Creager MA, Liang CS, Coffman JD. Beta adrenergic-mediated vasodilator response to insulin in the human forearm. J Pharmacol Exp Ther. 1985;235(3):709–14.
McMillan NJ, Soares RN, Harper JL, Shariffi B, Moreno-Cabañas A, Curry TB, et al. Increased muscle sympathetic nerve activity with acute hyperinsulinemia: role of the arterial baroreflex response to insulin-stimulated peripheral vasodilation. Am J Physiol Endocrinol Metab. https://doi.org/10.1152/ajpendo.00391.2021. Using three separate protocols in humans, results show increases in both MSNA and cardiac output during hyperinsulinemia can be attributed to the baroreflex response to peripheral vasodilation induced by insulin.
Conde SV, et al. Insulin resistance: a new consequence of altered carotid body chemoreflex? J Physiol. 2017;595(1):31–41.
Conde SV, Sacramento JF, Guarino MP. Carotid body: a metabolic sensor implicated in insulin resistance. Physiol Genomics. 2018;50(3):208–14.
Gao L, et al. Glucose sensing by carotid body glomus cells: potential implications in disease. Front Physiol. 2014;5:398.
Joyner MJ, et al. Role of the carotid body chemoreceptors in glucose homeostasis and thermoregulation in humans. J Physiol. 2018;596(15):3079–85.
Limberg JK, et al. Is insulin the new intermittent hypoxia? Med Hypotheses. 2014;82(6):730–5.
Dejours P. Control of respiration by arterial chemoreceptors. Ann N Y Acad Sci. 1963;109:682–95.
Niewinski P, et al. Consequences of peripheral chemoreflex inhibition with low-dose dopamine in humans. J Physiol. 2014;592(Pt 6):1295–308.
Limberg JK, Johnson BD, Holbein WW, Ranadive SM, Mozer MT, Joyner MJ. Interindividual variability in the dose-specific effect of dopamine on carotid chemoreceptor sensitivity to hypoxia. J Appl Physiol. 2016;120(2):138–47.
Limberg JK, et al. Role of the carotid chemoreceptors in insulin-mediated sympathoexcitation in humans. Am J Physiol Regul Integr Comp Physiol. 2020;318(1):R173–81. Data suggest that the carotid chemoreceptors do not contribute to acute insulin-mediated increases in MSNA in young, healthy adult humans.
Stickland MK, et al. Carotid chemoreceptor modulation of blood flow during exercise in healthy humans. J Physiol. 2011;589(Pt 24):6219–30.
Stickland MK, Morgan BJ, Dempsey JA. Carotid chemoreceptor modulation of sympathetic vasoconstrictor outflow during exercise in healthy humans. J Physiol. 2008;586(6):1743–54.
Limberg JK, et al. Altered neurovascular control of the resting circulation in human metabolic syndrome. J Physiol. 2012;590(Pt 23):6109–19.
Limberg JK, et al. Neural control of blood flow during exercise in human metabolic syndrome. Exp Physiol. 2014;99(9):1191–202.
Huggett RJ, et al. Sympathetic neural activation in nondiabetic metabolic syndrome and its further augmentation by hypertension. Hypertension. 2004;44(6):847–52.
Vollenweider P, et al. Impaired insulin-induced sympathetic neural activation and vasodilation in skeletal muscle in obese humans. J Clin Investig. 1994;93(6):2365–71.
Grassi G, et al. Effect of central and peripheral body fat distribution on sympathetic and baroreflex function in obese normotensives. J Hypertens. 2004;22(12):2363–9.
Monroe MB, et al. Relation of leptin and insulin to adiposity-associated elevations in sympathetic activity with age in humans. Int J Obes Relat Metab Disord. 2000;24(9):1183–7.
Grassi G, et al. Sympathetic neural abnormalities in type 1 and type 2 diabetes: a systematic review and meta-analysis. J Hypertens. 2020;38(8):1436–42.
Huggett RJ, et al. Sympathetic nerve hyperactivity in non-diabetic offspring of patients with type 2 diabetes mellitus. Diabetologia. 2006;49(11):2741–4.
Smith MM, Minson CT. Obesity and adipokines: effects on sympathetic overactivity. J Physiol. 2012;590(Pt 8):1787–801.
Spraul M, et al. Muscle sympathetic nerve activity in response to glucose ingestion. Impact of plasma insulin and body fat. Diabetes. 1994;43(2):191–6.
Straznicky NE, et al. Neuroadrenergic dysfunction along the diabetes continuum: a comparative study in obese metabolic syndrome subjects. Diabetes. 2012;61(10):2506–16.
Curry TB, et al. Relationship of muscle sympathetic nerve activity to insulin sensitivity. Clin Auton Res. 2014;24(2):77–85.
Straznicky NE, et al. Blunted sympathetic neural response to oral glucose in obese subjects with the insulin-resistant metabolic syndrome. Am J Clin Nutr. 2009;89(1):27–36.
Laakso M, et al. Decreased effect of insulin to stimulate skeletal muscle blood flow in obese man. A novel mechanism for insulin resistance. J Clin Invest. 1990;85(6):1844–52.
Lteif A, et al. Endothelin limits insulin action in obese/insulin-resistant humans. Diabetes. 2007;56(3):728–34.
Vollenweider P, et al. Impaired insulin-induced sympathetic neural activation and vasodilation in skeletal muscle in obese humans. J Clin Invest. 1994;93(6):2365–71.
Kaiyala KJ, et al. Obesity induced by a high-fat diet is associated with reduced brain insulin transport in dogs. Diabetes. 2000;49(9):1525–33.
Kern W, et al. Low cerebrospinal fluid insulin levels in obese humans. Diabetologia. 2006;49(11):2790–2.
Shi Z, Wong J, Brooks VL. Obesity: sex and sympathetics. Biol Sex Differ. 2020;11(1):10. Obesity increases sympathetic nerve activity in men, but not women. This comprehensive review explores current evidence suggesting that sexually dimorphic sympathoexcitatory responses to insulin may contribute.
Brooks VL, et al. Obesity-induced increases in sympathetic nerve activity: sex matters. Auton Neurosci. 2015;187:18–26.
Shi Z, et al. Sites and sources of sympathoexcitation in obese male rats: role of brain insulin. Am J Physiol Regul Integr Comp Physiol. 2020;318(3):R634–48.
Grunewald ZI, et al. TRAF3IP2 (TRAF3 interacting protein 2) mediates obesity-associated vascular insulin resistance and dysfunction in male mice. Hypertension. 2020;76(4):1319–29.
Galipeau D, Verma S, McNeill JH. Female rats are protected against fructose-induced changes in metabolism and blood pressure. Am J Physiol Heart Circ Physiol. 2002;283(6):H2478–84.
Galipeau DM, Yao L, McNeill JH. Relationship among hyperinsulinemia, insulin resistance, and hypertension is dependent on sex. Am J Physiol Heart Circ Physiol. 2002;283(2):H562–7.
Funding
The work summarized herein was largely supported by the National Heart, Lung, and Blood Institute grants R00 HL130339 (J.K.L.), R01 HL153523 (J.K.L.), and R01 HL137769 (J.P.), as well as AHA15SDG25080095 (JKL).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Human and Animal Rights
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Obesity
Rights and permissions
About this article

Cite this article
Limberg, J.K., Soares, R.N. & Padilla, J. Role of the Autonomic Nervous System in the Hemodynamic Response to Hyperinsulinemia—Implications for Obesity and Insulin Resistance. Curr Diab Rep 22, 169–175 (2022). https://doi.org/10.1007/s11892-022-01456-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11892-022-01456-1