
Abstract
The gastrointestinal system represents one of the largest interfaces between the human internal microenvironment and the external world. This system harbors trillions of commensal bacteria that reside in symbiosis with the host. Intestinal bacteria play a crucial role in maintaining systemic and intestinal immune and metabolic homeostasis because of their effect on nutrient absorption and immune development and function. Recently, altered gut bacterial composition (dysbiosis) was hypothesized to be involved in mechanisms through which islet autoimmunity is triggered. Evidence from animal models indicates that alterations in the gut bacterial composition precede disease onset, thus implicating a causal role for the gut microbiome in islet destruction. However, it remains unclear whether dysbiosis is directly linked to the mechanisms of human type 1 diabetes (T1D). In this review, we discuss data implicating the gut microbiota in disease progression with an emphasis on our recent studies performed in humans and in rodent models of T1D.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Type 1 diabetes (T1D) is a proinflammatory autoimmune disorder with a yet unidentified etiology. The disease leads to the specific loss of insulin-producing beta cells and subsequently causes dependency on daily insulin therapy for life [1]. In some cases, hyperglycemia may be preceded by a long prodromal autoimmune process that may last for many years and can be identified only by the presence of autoantibodies against β-cell autoantigens, such as GAD, islet antigen 2 (IA-2), insulin, and zinc transporter 8 (ZnT8A) [2].
More than 50 genes have been implicated in T1D mechanisms [3]. Of all the genetic factors linked with T1D, the HLA locus was found to have the strongest association with disease development, particularly DRB1 * 04-DQA1 * 03:01-B1 * 03:02(DR4-DQ8) and DRB1 * 03:01-DQ A1 * 05:01-B1 * 02:01 (DR3-DQ2), which are expressed in the majority of T1D patients who are diagnosed before 18 years of age [4]. Among other disease-susceptibility genes are insulin, cytotoxic T lymphocyte antigen (CTLA)-4, the interleukin (IL)-2 receptor, the tyrosine phosphatase PTPN22, and the intracellular viral RNA sensor MDA5 (Melanoma Differentiation-Associated protein 5) [5].
Environmental factors and changes in lifestyle over the last few decades have been hypothesized as major drivers of T1D [6••]. This possibility is supported by the rising incidence of T1D worldwide in industrialized nations, particularly in young children; this increase is occurring at a disease rate that cannot be explained based solely on genetic alterations [7]. Indeed, the annual increase in T1D incidence in European nations, such as Finland, Germany, and Norway, ranges between 2.4 and 3.3 % [7]. Strikingly, a more than 5-fold increase in the rate of new T1D cases in Finnish children younger than 15 years of age was recorded in 2006 compared to that observed in the 1950s (65 versus 12 cases per 100,000, respectively) [6••]. It was postulated that if the disease incidence continues to increase at the current pace, the global T1D incidence could double over the next decade [8]. The hypothesis that the environment is involved in diabetes progression is further supported by the observation that migration from countries with low incidence to countries with a high diabetes rate may increase the risk for diabetes [5]. Indeed, Pakistani children born to parents who migrated from Pakistan to the UK have a similar disease rate as the local population, which is strikingly 10-fold higher than the incidence in Pakistan [9]. Lastly, the differences in diabetes incidence between monozygotic twins further implicate the environment’s role in triggering the disease [10].
Viruses and T1D
Viruses have been implicated in the mechanism triggering human T1D, but a cause-and-effect relationship between microbial infections and disease progression has not yet been established [3, 5, 11]. Previous reports have demonstrated that viruses and virus-specific antibodies can be detected more frequently in individuals with recent diabetes onset compared to healthy control subjects [12–14]. Case reports and anecdotal data have implicated viruses, such as mumps, cytomegalovirus, rubella, Epstein-Barr virus, rotavirus, and varicella zoster virus, in human diabetes [reviewed in ref. 15]. Significant attention has been paid to the role of enterovirus, and Coxsackie B virus (CVB) in particular, in triggering beta cell destruction [16, 17]. Coxsackie B virus protein-1 was detected in islets from more than 50 % of newly diagnosed subjects compared to only a few healthy subjects [18]. Furthermore, islets from patients with T1D co-expressed the enterovirus-capsid protein and proinflammatory cytokines and chemokines and were infiltrated with T cells expressing CXCR3, the receptor for CXCL-10 [19, 20].
Evidence of an epidemic outbreak of T1D also points to the possibility that viruses could be a key component in disease pathogenesis [21]. A remarkable increase in diabetes incidence was observed in young children in Philadelphia in the first 6 months of 1993 [21]. Notably, a measles epidemic occurred in Philadelphia approximately 2 years prior to the T1D outbreak, establishing the hypothesis that the increased T1D rate might have been linked to the measles outbreak [21].
How viral infections lead to T1D is unknown. Viral infections may lead to diabetes via a number of mechanisms, including molecular mimicry, bystander activation of T cells, beta cell damage resulting in autoantigen release and activation of autoreactive T cells, and the induction of stress pathways in beta cells [5].
The Intestinal Microbiota
The intestine harbors ∼1014 microorganisms of more than 500 different species [reviewed in ref. 22]. The bacterial abundance is increased in the lower portions of the gastrointestinal tract, ranging from 100 to 1000 microorganisms per ml in the acidic microenvironment of the stomach to 1012 per ml in the colon. These microbes use complex polysaccharides and other mucosal compounds in addition to undigested plant fiber as energy sources leading to the production of vital metabolites, such as vitamin K, biotin, and short-chain fatty acids (SCFAs). The intestine contains 500–1000 bacterial species that belong to several major phyla [22]. The number of these bacterial groups varies between individuals, but Firmicutes, Bacteroidetes, Proteobacteria, and Actinobacteria are the most abundant phyla [22].
The gut bacterial composition can be altered as a result of environmental perturbations [23]. Antibiotic use is of the main factors that can modulate the gut microbiome, and it can lead to substantial long-term effects on the gut microbiota [6••]. Antibiotics also reduce the resistance to colonization, allowing foreign microbes to outgrow commensal bacteria, thus causing permanent changes in the structure of the microbiota [24].
The gut microbiota is required for the development of a normal immune system. Mice maintained in a germ-free environment from birth have altered intestinal morphology and function and aberrant lymphoid tissue organization and innate and adaptive immunity [25]. Emerging evidence supports the hypothesis that dysbiosis may be linked to the development of immune disorders [26]. For example, an increase in the abundance of the bacterial family Enterobacteriaceae and gram-negative anaerobes, such as Bacteroides, and a decrease in Bifidobacterium have been linked with inflammatory bowel disease in humans [27–29]. The intestinal microbiome has also been implicated in rheumatoid arthritis in humans [30] and in mouse models of autoimmune arthritis [31].
The hygiene hypothesis postulates that reduced exposure to microbes as a result of antibiotic use in early life results in dysregulated immunity; this hypothesis is a potential explanation for the dramatic increase in the frequency of immune-mediated diseases and T1D [32, 33]. Indeed, previous studies have demonstrated that antibiotic treatments can induce profound long-term alterations on gut bacterial composition [6••]. Antibiotic-induced alterations in the gut microbiome have been implicated in the global rise in diabetes incidence; however, there are currently no solid data linking antibiotic use in children with disease progression [34, 35].
The Immune System and the Intestinal Microbiome
Pattern-recognition receptors (PRRs), such as Toll-like receptors (TLRs) expressed by immune and non-immune cells, sense pathogen-associated molecular patterns (PAMPs) expressed by bacteria, viruses, and fungi [36–38]. The activation of TLRs mediated by signaling molecules, such as MyD88 or TIR domain-containing adapter protein inducing IFNβ (TRIF) [36–38], leads to a cascade of cellular signals involving mitogen-activated protein kinase (MAPK) activation and the expression of NF-kB and interferon-regulatory factor 3 (IRF3), culminating in the expression of proinflammatory cytokines [36–38].
Intestinal cells, such as Paneth and goblet cells, sense bacterial number and location via TLR pathways. TLR activation in these cells leads to the expression of the antimicrobial lectin regenerating islet-derived protein 3γ (REG3γ) [39, 40]. Adaptive immune mechanisms are also involved in defending the host from invading pathogens via immune cells present in the gut-associated lymphoid tissue (GALT), such as Peyer’s patches, Th17 cells [40], and immunoglobulin A [41]. Moreover, the microbiome itself serves as a barrier against potential pathogens [42]. The microbiome further promotes the maintenance of the intestinal barrier by promoting epithelial cell turnover and mucin production and by competing with pathogens for available nutrients and space [43].
The intestine is the largest organ of the immune system in the body and is inhabited with an enormous amount of microbes living in symbiosis with the host [44]. Recent studies have shed light on the interplay between the immune system and the intestinal microbiota that begin to interact with one another immediately after birth [44, 45]. A key element that allows a peaceful host-commensal bacteria mutualism is the separation of microbes from the host internal environment by barriers such as epithelial and mucosal layers and innate and adaptive immune mechanisms sensing and eliminating harmful pathogens [43]. Disrupting these mechanisms can lead to inflammation and autoimmunity [46].
A major function of the host intestinal immune system is to keep inflammatory responses in check to allow the survival of beneficial bacteria and simultaneously preserve the ability to fight potential pathogens [44]. Thus, interactions between commensal bacteria and host cells in the intestine are tightly regulated [42]. A key innate immune mechanism allowing the symbiosis between gut bacteria and the host immune system involves TLRs and NOD-like receptor (NLR) signaling pathways [42]. For example, the capsular polysaccharide A (PSA) of Bacteroides fragilis induces Treg cells that secrete the anti-inflammatory cytokine IL-10, thus preventing inflammation in the gut [47, 48]. Likewise, commensal bacteria, such as Bacteroides thetaiotaomicron, inhibit proinflammatory cytokine expression in the intestine by modulating nuclear factor-kB (NF-kB) [49]. Intestinal dendritic cells (DCs) contribute to the microbiome-host coexistence by sampling a small number of live commensal bacteria from the intestinal lumen to the mesenteric lymph nodes to induce a protective IgA response [50].
Rat Models of T1D and Virus-Induced Islet Destruction
The LEW1.WR1 and BioBreeding diabetes-resistant (BBDR) rat models of virus-induced T1D have a normal immune phenotype and do not develop T1D when housed in specific pathogen-free facilities [51–53]. However, infection with the parvovirus Kilham rat virus (KRV) leads to beta cell inflammation and destruction in 50 and 25 % of infected BBDR and LEW1.WR1 rats, respectively; this phenomenon is detectable 14–28 days following virus inoculation [51–53]. Another rat model of T1D is the diabetes-prone BioBreeding (BBDP) rat, which is severely lymphopenic. Unlike the BBDR and LEW1.WR1 rats, BBDP rats develop diabetes spontaneously [54, 55]. The disease in the rat is characterized by the specific loss of islet beta cells, glycosuria, ketonuria, and polyuria in a strain-specific manner [52, 53]. Susceptibility to virus-induced disease is dependent on the presence of class I Au and class II B/Du [52, 53]. We recently hypothesized that the innate immune system plays a key role in the mechanism triggering T1D in the BBDR and LEW1.WR1 rats [51, 56–58]. Indeed, activation of the innate immune system with TLR agonists, such as the viral mimic polyinosinic:polycytidylic acid (poly I:C) or CpG DNA, followed by infection with KRV substantially exacerbates T1D [59, 60]. Moreover, infection with KRV leads to a robust proinflammatory response associated with the up-regulation of a vast array of proinflammatory cytokines and chemokines in the spleen, pancreatic lymph nodes, and Peyer’s patches via mechanisms that involve TLR9 pathways [51, 58, 59]. Finally, modulation of virus-induced innate immunity with steroids [59], IL-1 receptor antagonists [57], and histone deacetylase inhibitors can prevent disease progression.
The Role of Gut Bacteria in Virus-Induced T1D
Recent evidence has suggested that the intestinal microbiome is involved in the pathogenesis of T1D in rat models. BBDP rats that developed T1D had reduced levels of Bacteroides compared to rats that remained diabetes-free and rats with antibiotic therapy-ameliorated T1D [61]. In addition, the transfer of Lactobacillus johnsonii N6.2 from the BBDR intestine to BBDP rats delayed disease development in a bacteria-specific manner [62]. Our own studies indicated that infection with KRV resulted in alterations in the gut microbiome reflected by increased abundances of the Bifidobacterium and Clostridium genera on day 5 following viral infection, prior to insulitis or hyperglycemia [51]. Furthermore, oral therapy with the broad-spectrum antibiotic sulfatrim (sulfamethoxazole plus trimethoprim) reversed virus-induced alterations in the gut microbiome and protected rats from insulitis and islet destruction [51]. The effect of sulfatrim on disease progression was specific because treatment with ampicillin, metronidazole, or neomycin sulfate did not alter the course of islet destruction. It remains unclear whether KRV-induced alterations in the gut microbiome are directly linked to diabetes progression. The observation that KRV alters the abundance of Bifidobacterium and Clostridium in the intestine prior to hyperglycemia may imply that infection with KRV results in conditions that favor the development of islet autoimmunity. This possibility requires testing in future studies.
The mechanism by which manipulation of the intestinal microbiota prevents islet destruction could be associated with interference with virus-induced adaptive and innate immunity [ref. 51 and Fig. 1]. Indeed, sulfatrim reduced the number of B lymphocytes in the pancreatic lymph nodes and Peyer’s patches and downregulated KRV-reactive T cells in the spleen [51]. Moreover, sulfatrim induced a reduction in the transcript levels of KRV-induced proinflammatory cytokines and chemokines in the pancreatic lymph nodes and Peyer’s patches [51]. The notion that the gut microbiome is involved in innate and adaptive immune responses outside the gut is perhaps not surprising because gut bacteria are involved in numerous physiological responses that can affect various organs and cells, such as nutrient adsorption, vitamins and hormone production, and the prevention of colonization by pathogens [63]. In any case, the evidence that manipulating the gut microbiome results in altered innate and adaptive immunity outside of the gastrointestinal tract and interferes with autoimmunity is consistent with previous observations from other immune disorders, such as allergies, asthma, diabetes, obesity, and cancer [63].

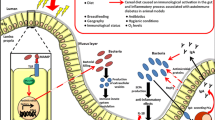
The potential role of the intestinal microbiome in T1D progression. Environmental and genetic factors play a key role in triggering type 1 diabetes. Virus infection leads to early inflammation in the periphery or intestine, which in turn leads to alterations in the gut microbiota towards a proinflammatory microbiome. Environmental risk factors, such as a high-fat diet and antibiotics, can also contribute to the development of dysbiosis. A shift in the abundance of the gut microbiome could result in altered functionality, such as a reduction in the production of anti-inflammatory SCFAs
The Role of TLRs and the Gut Microbiome and Diabetes
As discussed earlier, TLRs play a key role in innate immune recognition of pathogens and commensal bacteria and as such are a critical component in mechanisms that maintain gut homeostasis and control of the intestinal microbiome [64, 65]. Mice lacking the expression of TLR5, which binds bacterial flagellin, had alterations in their gut bacterial composition [66]; however, differences in the gut bacterial composition between wild-type and TLR-deficient mouse lines could be a result of divergence of the gut microbiota after long-term housing in isolation from one another [67].
The first observation pointing towards the role of TLR signaling and the gut microbiota in T1D occurred in the NOD mouse model in which gut bacteria were shown to play a protective role in disease development [68]. NOD mice with disrupted MyD88 signaling developed T1D when housed in specific pathogen-free conditions, but this effect was not observed in mice lacking TLR2, TLR3, and TLR4 [68]. The protective effect observed in these mice was hypothesized to be linked with altered responses to gut bacteria because MyD88-deficient NOD mice housed in germ-free conditions or treated with antibiotics had increased disease incidence [57].
The role of TLR signaling and the gut microbiome in T1D development was further addressed by us in RIP-B7.1 mice lacking the expression of critical TLR signaling molecules. RIP-B7.1 mice express the B7.1 costimulatory molecule under the control of the rat insulin promoter [69], and administering these mice Poly (I:C), which is a ligand of TLR3 and RIG-like helicases, results in insulitis and diabetes via mechanisms linked with type I interferon pathways [70] and the up-regulation of antigen presenting cells (APCs) and autoreactive T cells [71]. Data obtained from RIP-B7.1 mice with disrupted TLR pathways support the hypothesis that interactions between the innate immune system and the gut microbiota are involved in diabetes development. RIP-B7.1 mice with and without disrupted MyD88, TLR3, or TLR9 pathways housed under normal conditions remained diabetes-free [72]. In sharp contrast, wild-type RIP-B7.1 mice with intact TLR signaling or without TLR9 expression who were administered Poly (I:C) and oral sulfatrim developed diabetes, whereas RIP-B7.1 mice deficient in MyD88 and TLR3 treated under the same experimental conditions were protected from disease development [72].
Diabetes-susceptible TLR9-deficient mice had different intestinal microbiomes than those of diabetes-resistant TLR3- and MyD88-deficient RIP-B7.1 mice following treatment with sulfatrim plus Poly (I:C) [72]. This was reflected by an increase in the bacterial diversity in TLR9-deficient RIP-B7.1 mice compared to TLR3- and MyD88-deficient mice. Furthermore, the overall intestinal microbiome of TLR9-deficient mice either untreated or administered with sulfatrim plus Poly (I:C) was different than that of diabetes-resistant mice. Lastly, sulfatrim plus Poly (I:C) modulated the relative abundances of individual bacteria, including the Actinobacteria phylum and the Bifidobacterium, Lactobacillus, and Clostridium genera in TLR9-deficient versus disease-resistant mice. Together, these data support the possibility that the mechanism of diabetes in RIP-B7.1 mice is linked with altered gut bacterial composition. How Poly (I:C) alters commensal bacteria in the gut and how shifts in bacterial abundance lead to disease onset will require further investigation.
The Role of the Intestinal Microbiota in Human T1D
It is hypothesized that the dietary intake in developed nations, which has shifted to a high-fat, high-carbohydrate, low-fiber diet (“Western diet”), may have resulted in functional changes in the intestinal microbiota [73–76]. Such diet-induced alterations to gut bacterial communities are postulated to play a key role in the rising incidence of proinflammatory immune disorders in the developed world, including obesity, type 2 diabetes, and inflammatory bowel disease [77•]. SCFAs, such as butyric acid and acetic acid, produced by the fermentation of indigestible dietary plant fiber by the intestinal microbiota [reviewed in refs. 78•, 79] can enter the circulation (27) and regulate innate and adaptive immunity.
A number of recent studies implied that the increase in the incidence of T1D may be linked to alterations in the gut microbiome, particularly those associated with SCFA-producing bacterial species [ref. 66 and Fig. 1]. A metagenomics analysis performed in four cases and four controls from Finland revealed that the level of genes associated with carbohydrate metabolism, adhesions, motility, phages, sulfur metabolism, and stress responses was higher in cases [80]. The authors hypothesized that increased adhesion and flagella synthesis in autoimmune subjects may be linked with triggering islet autoimmunity. The 16S rRNA data from the same subjects suggested a higher proportion of butyrate-producing and mucin-degrading bacteria in controls compared to cases [80]. Another study performed in 18 seropositive subjects from Finland suggested a link between a low abundance of lactate-producing and butyrate-producing bacterial groups with islet autoimmunity [81]. Moreover, a study performed in 76 Finnish children, of whom 22 converted to seropositivity and later developed T1D, suggested an increase in the abundance of Bacteroides dorei in cases compared to individuals who did not convert to autoimmunity prior to seroconversion [82]. Data from a study that included 11 seropositive children from Finland and Estonia, of whom 4 progressed to T1D, demonstrated a reduction in bacterial diversity after autoantibody appearance prior to disease diagnosis [83]. Lastly, a study that included 28 diabetic children, of whom 18 were from Finland and the rest were from France, Greece, Estonia, and Lithuania, suggested that non-diabetic children have a more balanced microbiota and higher abundance of butyrate-producing bacterial groups [84].
We recently performed a cross-sectional study in stool samples from subjects living in Colorado [85]. The study included 35 new onset patients (up to 6 months following diagnosis), 21 first-degree relatives (FDRs) positive for at least one islet autoantibody, 32 seronegative FDRs, and 23 unrelated healthy subjects without a family history of T1D. The 16S rRNA data suggested that there is no clear bacterial signature of predominant bacterial communities in subjects with islet autoimmunity prior to or following disease diagnosis. However, the gut microbiomes of seropositive subjects and seronegative FDRs displayed an overall similarity to one another but were distinct from those of new onset patients and unrelated healthy controls [85]. Stratification of the seropositive cohort based on the expression of multiples versus one autoantibody further pointed towards an increase in the abundance of Bacteroides and a reduction in Prevotella and the phylum Firmicutes in the multiple autoantibody group. An increase in Bacteroides has been linked to the “Western diet”, which is characterized by a content high in protein and fat and low in plant fiber, whereas an increase in Prevotella has been linked to a diet rich in plant fibers [86]. Prevotella was found to be highly prevalent in African children with a diet rich in grains [87], and taxa from both Prevotella and Firmicutes can digest plant polysaccharides [86, 88] and promote the production of SCFAs, which are known for their anti-inflammatory properties [89].
Collectively, the available human data suggest that shifts in the gut microbial composition may occur in genetically susceptible individuals prior to seroconversion or hyperglycemia. However, it is difficult to assess whether these alterations represent an “autoimmune microbiome” with a causal role in disease progression, partially because of the variability observed between the various studies. The data variability could result at least in part from differences in sample size, methods of sample collection, geographical origins and climate, cultural differences, ethnicity, and data analysis. The mechanism by which the gut microbiota may promote islet autoimmunity is currently unclear. It is thought to be associated with its ability to modulate immunity and/or alter epithelial barrier function [35]. A recent study suggested that T cell cross-reactivity between islet antigens and microbes may be involved in the process of beta cell destruction [90]. Lastly, it was hypothesized that the translocation of intestinal bacteria to pancreatic lymph nodes and the subsequent activation of the nucleotide-binding oligomerization domain containing two pathways play a role in disease development [91].
Conclusions
It is becoming increasingly apparent that there is a link between the gut bacterial composition and various aspects of human health. Indeed, evidence from animal studies implicates the intestinal microbiome in the development of autoimmune disorders, including T1D. Studies conducted in individuals at risk for T1D have suggested that disease progression is associated with dysbiosis. Further longitudinal studies performed prior to and following seroconversion and disease diagnosis are required to establish a correlation between shifts in the intestinal microbiota and disease progression. Parallel studies performed in animal models of diabetes will be necessary to dissect the mechanisms by which intestinal microbes mediate disease and to develop microbiome-based therapies for disease prevention.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Campbell-Thompson M, Fu A, Kaddis JS, Wasserfall C, Schatz DA, Pugliese A, et al. Insulitis and β-cell mass in the natural history of type 1 diabetes. Diabetes. 2016;65(3):719–31. doi:10.2337/db15-0779.
Gianani R, Eisenbarth GS. The stages of type 1A diabetes: 2005. Immunol Rev. 2005;204:232–49.
de Beeck AO, Eizirik DL. Viral infections in type 1 diabetes mellitus—why the [beta] cells? Nat Rev Endocrinol. 2016;12(5):263–73. doi:10.1038/nrendo.2016.30.
Lernmark A, Larsson HE. Immune therapy in type 1 diabetes mellitus. Nat Rev Endocrinol. 2013;9(2):92–103.
Ghazarian L, Diana J, Simoni Y, Beaudoin L, Lehuen A. Prevention or acceleration of type 1 diabetes by viruses. Cell Mol Life Sci. 2012;70(2):239–55. doi:10.1007/s00018-012-1042-1.
Knip M, Siljander H. The role of the intestinal microbiota in type 1 diabetes mellitus. Nat Rev Endocrinol. 2016;12(3):154–67. doi:10.1038/nrendo.2015.218. This excellent review article discusses recent advance made in the area of the gut microbiome and T1D.
Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet. 2014;383(9911):69–82. doi:10.1016/S0140-6736(13)60591-7.
Patterson CC, Dahlquist GG, Gyürüs E, Green A, Soltész G. Incidence trends for childhood type 1 diabetes in Europe during 1989-2003 and predicted new cases 2005-20: a multicentre prospective registration study. Lancet. 2009;373(9680):2027–33. doi:10.1016/S0140-6736(09)60568-7.
Bodansky HJ, Staines A, Stephenson C, Haigh D, Cartwright R. Evidence for an environmental effect in the aetiology of insulin dependent diabetes in a transmigratory population. BMJ: Br Med J. 1992;304(6833):1020–2.
Hyttinen V, Kaprio J, Kinnunen L, Koskenvuo M, Tuomilehto J. Genetic liability of type 1 diabetes and the onset age among 22,650 young Finnish twin pairs: a nationwide follow-up study. Diabetes. 2003;52(4):1052–5. doi:10.2337/diabetes.52.4.1052.
Zipris D. Innate immunity and its role in type 1 diabetes. Curr Opin Endocrinol Diabetes Obes. 2008;15(4):326–31.
Dotta F, Censini S, van Halteren AGS, Marselli L, Masini M, Dionisi S, et al. Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc Natl Acad Sci U S A. 2007;104(12):5115–20.
King ML, Bidwell D, Voller A, Bryant J, Banatvala JE. Role of Coxsackie B viruses in insulin-dependent diabetes mellitus. Lancet. 1983;2(8355):915–6.
King ML, Shaikh A, Bidwell D, Voller A, Banatvala JE. Coxsackie-B-virus-specific IgM responses in children with insulin-dependent (juvenile-onset; type I) diabetes mellitus. Lancet. 1983;1(8339):1397–9.
Zipris D. Epidemiology of type 1 diabetes and what animal models teach us about the role of viruses in disease mechanisms. Clin Immunol. 2009;131(1):11–23.
Jun HS, Yoon JW. A new look at viruses in type 1 diabetes. Diabetes Metab Res Rev. 2003;19(1):8–31.
Jaeckel E, Manns M, von Herrath M. Viruses and diabetes. Ann N Y Acad Sci. 2002;958:7–25.
Chehadeh W, Weill J, Vantyghem M-C, Alm G, Lefebvre J, Wattre P, et al. Increased level of interferon-α in blood of patients with insulin-dependent diabetes mellitus: relationship with coxsackievirus B infection. J Infect Dis. 2000;181(6):1929–39.
Tanaka S, Nishida Y, Aida K, Maruyama T, Shimada A, Suzuki M, et al. Enterovirus infection, CXC chemokine ligand 10 (CXCL10), and CXCR3 circuit: a mechanism of accelerated beta-cell failure in fulminant type 1 diabetes. Diabetes. 2009;58(10):2285–91.
Uno S, Imagawa A, Saisho K, Okita K, Iwahashi H, Hanafusa T, et al. Expression of chemokines, CXC chemokine ligand 10 (CXCL10) and CXCR3 in the inflamed islets of patients with recent-onset autoimmune type 1 diabetes. Endocr J. 2010;57(11):991–6.
Lipman TH, Chang Y, Murphy KM. The epidemiology of type 1 diabetes in children in Philadelphia 1990–1994: evidence of an epidemic. Diabetes Care. 2002;25(11):1969–75.
Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol. 2014;14(10):667–85. doi:10.1038/nri3738.
Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL et al. The long-term stability of the human gut microbiota. Science. 2013;341(6141). doi:10.1126/science.1237439.
Blaser MJ, Falkow S. What are the consequences of the disappearing human microbiota? Nat Rev Microbiol. 2009;7(12):887–94.
Verdu EF, Galipeau HJ, Jabri B. Novel players in coeliac disease pathogenesis: role of the gut microbiota. Nat Rev Gastroenterol Hepatol. 2015;12(9):497–506. doi:10.1038/nrgastro.2015.90. http://www.nature.com/nrgastro/journal/v12/n9/abs/nrgastro.2015.90.html#supplementary-information.
Kamada N, Seo SU, Chen GY, Nunez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol. 2013;13(5):321–35.
Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13(9):R79-R. doi:10.1186/gb-2012-13-9-r79.
Tamboli CP, Neut C, Desreumaux P, Colombel JF. Dysbiosis in inflammatory bowel disease. Gut. 2004;53(1):1–4. doi:10.1136/gut.53.1.1.
Veenbergen S, Samsom JN. Maintenance of small intestinal and colonic tolerance by IL-10-producing regulatory T cell subsets. Curr Opin Immunol. 2012;24(3):269–76. doi:10.1016/j.coi.2012.03.004.
Vaahtovuo J, Munukka E, Korkeamaki M, Luukkainen R, Toivanen P. Fecal microbiota in early rheumatoid arthritis. J Rheumatol. 2008;35(8):1500–5.
Sinkorova Z, Capkova J, Niederlova J, Stepankova R, Sinkora J. Commensal intestinal bacterial strains trigger ankylosing enthesopathy of the ankle in inbred B10.BR (H-2(k)) male mice. Hum Immunol. 2008;69(12):845–50. doi:10.1016/j.humimm.2008.08.296.
Bach JF. Infections and autoimmune diseases. J Autoimmun. 2005;25(Supplement 1):74–80.
Liu AH, Leung DYM. Renaissance of the hygiene hypothesis. J Allergy Clin Immunol. 2006;117(5):1063–6. doi:10.1016/j.jaci.2006.03.027.
Bach J-F. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med. 2002;347(12):911–20. doi:10.1056/NEJMra020100.
de Souza HSP, Fiocchi C. Immunopathogenesis of IBD: current state of the art. Nat Rev Gastroenterol Hepatol. 2016;13(1):13–27. doi:10.1038/nrgastro.2015.186. http://www.nature.com/nrgastro/journal/v13/n1/abs/nrgastro.2015.186.html#supplementary-information.
Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4(7):499–511.
Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2(8):675–80. doi:10.1038/90609.
Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801.
Canfora EE, Jocken JW, Blaak EE. Short-chain fatty acids in control of body weight and insulin sensitivity. Nat Rev Endocrinol. 2015;11(10):577–91. doi:10.1038/nrendo.2015.128.
Salzman NH, Underwood MA, Bevins CL. Paneth cells, defensins, and the commensal microbiota: a hypothesis on intimate interplay at the intestinal mucosa. Semin Immunol. 2007;19(2):70–83. doi:10.1016/j.smim.2007.04.002.
Pabst O. New concepts in the generation and functions of IgA. Nat Rev Immunol. 2012;12(12):821–32.
Perez-Lopez A, Behnsen J, Nuccio S-P, Raffatellu M. Mucosal immunity to pathogenic intestinal bacteria. Nat Rev Immunol. 2016;16(3):135–48. doi:10.1038/nri.2015.17.
Schwabe RF, Jobin C. The microbiome and cancer. Nat Rev Cancer. 2013;13(11):800–12. doi:10.1038/nrc3610.
Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124(4):837–48. doi:10.1016/j.cell.2006.02.017.
Maynard CL, Elson CO, Hatton RD, Weaver CT. Reciprocal interactions of the intestinal microbiota and immune system. Nature. 2012;489(7415):231–41.
Vaarala O. Leaking gut in type 1 diabetes. Curr Opin Gastroenterol. 2008;24(6):701–6. doi:10.1097/MOG.0b013e32830e6d98.
Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008;453(7195):620–5. http://www.nature.com/nature/journal/v453/n7195/suppinfo/nature07008_S1.html.
Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, et al. The toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science. 2011;332(6032):974–7. doi:10.1126/science.1206095.
Kelly D, Campbell JI, King TP, Grant G, Jansson EA, Coutts AGP, et al. Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-[gamma] and RelA. Nat Immunol. 2004;5(1):104–12.
Macpherson AJ, Uhr T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science. 2004;303(5664):1662–5. doi:10.1126/science.1091334.
Hara N, Alkanani AK, Ir D, Robertson CE, Wagner BD, Frank DN, et al. Prevention of virus-induced type 1 diabetes with antibiotic therapy. J Immunol. 2012;189(8):3805–14.
Mordes JP, Guberski DL, Leif JH, Woda BA, Flanagan JF, Greiner DL, et al. LEW.1WR1 rats develop autoimmune diabetes spontaneously and in response to environmental perturbation. Diabetes. 2005;54(9):2727–33.
Mordes JP, Zipris D, Liu Z, Blankenhorn EP. Viruses and autoimmune diabetes in rats. In: Taylor K, Hyöty H, Toniolo A, Zuckerman JA, editors. Diabetes and viruses. New York: Springer; 2013. p. 57–70.
Crisa L, Greiner DL, Mordes JP, MacDonald RG, Handler ES, Czech MP, et al. Biochemical studies of RT6 alloantigens in BB/Wor and normal rats. Evidence for intact unexpressed RT6a structural gene in diabetes-prone BB rats. Diabetes. 1990;39(10):1279–88.
Rossini AA, Handler ES, Mordes JP, Greiner DL. Human autoimmune diabetes mellitus: lessons from BB rats and NOD mice--Caveat emptor. Clin Immunol Immunopathol. 1995;74(1):2–9.
Hara N, Alkanani A, Dinarello C, Zipris D. Histone deacetylase inhibitor suppresses virus-induced proinflammatory responses and type 1 diabetes. J Mol Med. 2013:1–10.
Hara N, Alkanani AK, Dinarello CA, Zipris D. Modulation of virus-induced innate immunity and type 1 diabetes by IL-1 blockade. Innate Immun. 2014;20(6):574–84.
Wolter TR, Wong R, Sarkar SA, Zipris D. DNA microarray analysis for the identification of innate immune pathways implicated in virus-induced autoimmune diabetes. Clin Immunol. 2009;132(1):103–15.
Londono P, Komura A, Hara N, Zipris D. Brief dexamethasone treatment during acute infection prevents virus-induced autoimmune diabetes. Clin Immunol. 2010;135(3):401–11.
Zipris D, Lien E, Xie JX, Greiner DL, Mordes JP, Rossini AA. TLR activation synergizes with Kilham rat virus infection to induce diabetes in BBDR rats. J Immunol. 2005;174(1):131–42.
Brugman S, Klatter FA, Visser JT, Wildeboer-Veloo AC, Harmsen HJ, Rozing J, et al. Antibiotic treatment partially protects against type 1 diabetes in the Bio-Breeding diabetes-prone rat. Is the gut flora involved in the development of type 1 diabetes? Diabetologia. 2006;49(9):2105–8.
Valladares R, Sankar D, Li N, Williams E, Lai KK, Abdelgeliel AS, et al. Lactobacillus johnsonii N6.2 mitigates the development of type 1 diabetes in BB-DP rats. PLoS One. 2010;5(5), e10507. doi:10.1371/journal.pone.0010507.
Gill N, Finlay BB. The gut microbiota: challenging immunology. Nat Rev Immunol. 2011;11(10):636–7.
Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118(2):229–41.
Larsson E, Tremaroli V, Lee YS, Koren O, Nookaew I, Fricker A, et al. Analysis of gut microbial regulation of host gene expression along the length of the gut and regulation of gut microbial ecology through MyD88. Gut. 2012;61(8):1124–31.
Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, et al. Metabolic syndrome and altered gut microbiota in mice lacking toll-like receptor 5. Science. 2010;328(5975):228–31.
Ubeda C, Lipuma L, Gobourne A, Viale A, Leiner I, Equinda M, et al. Familial transmission rather than defective innate immunity shapes the distinct intestinal microbiota of TLR-deficient mice. J Exp Med. 2012;209(8):1445–56.
Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature. 2008;455(7216):1109–13.
Guerder S, Picarella DE, Linsley PS, Flavell RA. Costimulator B7-1 confers antigen-presenting-cell function to parenchymal tissue and in conjunction with tumor necrosis factor alpha leads to autoimmunity in transgenic mice. Proc Natl Acad Sci U S A. 1994;91(11):5138–42.
Devendra D, Jasinski J, Melanitou E, Nakayama M, Li M, Hensley B, et al. Interferon-{alpha} as a mediator of polyinosinic:polycytidylic acid-induced type 1 diabetes. Diabetes. 2005;54(9):2549–56.
Wen L, Peng J, Li Z, Wong FS. The effect of innate immunity on autoimmune diabetes and the expression of Toll-like receptors on pancreatic islets. J Immunol. 2004;172(5):3173–80.
Alkanani AK, Hara N, Lien E, Ir D, Kotter CV, Robertson CE, et al. Induction of diabetes in the RIP-B7.1 mouse model is critically dependent on TLR3 and MyD88 pathways and is associated with alterations in the intestinal microbiome. Diabetes. 2014;63(2):619–31. doi:10.2337/db13-1007.
Hildebrandt MA, Hoffmann C, SherrilλGÇôMix SA, Keilbaugh SA, Hamady M, Chen Y, et al. High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology. 2009;137(5):1716–24.
Turnbaugh PJ, Bäckhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. 2008;3(4):213–23.
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–31. doi:10.1038/nature05414.
Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med. 2009;1(6):6ra14. doi:10.1126/scitranslmed.3000322.
David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505(7484):559–63. doi:10.1038/nature12820. This article describes the profound effect of diet on the gut bacterial composition.
Brestoff JR, Artis D. Commensal bacteria at the interface of host metabolism and the immune system. Nat Immunol. 2013;14(7):676–84. This article discusses how the gut microbiome influences the production of immunomodulatory, diet-dependent nutrients and metabolites and how these gut metabolites shape the immune system.
Wong JM, de Soure R, Kendall CW, Emam A, Jenkins DJ. Colonic health: fermentation and short chain fatty acids. J Clin Gastroenterol. 2006;40(3):235–43.
Brown CT, Davis-Richardson AG, Giongo A, Gano KA, Crabb DB, Mukherjee N, et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS One. 2011;6, e25792.
de Goffau MC, Luopajärvi K, Knip M, Ilonen J, Ruohtula T, Härkönen T, et al. Fecal microbiota composition differs between children with β-cell autoimmunity and those without. Diabetes. 2013;62(4):1238–44. doi:10.2337/db12-0526.
Davis-Richardson AG, Ardissone AN, Dias R, Simell V, Leonard MT, Kemppainen KM, et al. Bacteroides dorei dominates gut microbiome prior to autoimmunity in Finnish children at high risk for type 1 diabetes. Front Microbiol. 2014;5:678. doi:10.3389/fmicb.2014.00678.
Kostic Aleksandar D, Gevers D, Siljander H, Vatanen T, Hyötyläinen T, Hämäläinen A-M, et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe. 2015;17(2):260–73. doi:10.1016/j.chom.2015.01.001.
de Goffau M, Fuentes S, van den Bogert B, Honkanen H, de Vos W, Welling G, et al. Aberrant gut microbiota composition at the onset of type 1 diabetes in young children. Diabetologia. 2014;57(8):1569–77.
Alkanani AK, Hara N, Gottlieb PA, Ir D, Robertson CE, Wagner BD, et al. Alterations in intestinal microbiota correlate with susceptibility to type 1 diabetes. Diabetes. 2015. doi:10.2337/db14-1847.
Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334(6052):105–8.
De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A. 2010;107(33):14691–6.
Holmes E, Li J-á, Marchesi J-á, Nicholson J-á. Gut microbiota composition and activity in relation to host metabolic phenotype and disease risk. Cell Metab. 2012;16(5):559–64.
Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly Y, et al. The microbial metabolites, short-chain fatty acids, regulate colonic treg cell homeostasis. Science. 2013;341(6145):569–73.
Cole DK, Bulek AM, Dolton G, Schauenberg AJ, Szomolay B, Rittase W, et al. Hotspot autoimmune T cell receptor binding underlies pathogen and insulin peptide cross-reactivity. J Clin Invest. 2016;126(6):2191–204. doi:10.1172/JCI85679.
Costa FRC, Françozo MCS, de Oliveira GG, Ignacio A, Castoldi A, Zamboni DS, et al. Gut microbiota translocation to the pancreatic lymph nodes triggers NOD2 activation and contributes to T1D onset. J Exp Med. 2016;213(7):1223–39. doi:10.1084/jem.20150744.
Acknowledgments
Our studies were supported by grants from the JDRF.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
James C. Needell and Danny Zipris declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
Our studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in our studies. The studies involving animals were in accordance with the ethical standards of the University of Colorado Denver.
Additional information
This article is part of the Topical Collection on Pathogenesis of Type 1 Diabetes
Rights and permissions
About this article

Cite this article
Needell, J.C., Zipris, D. The Role of the Intestinal Microbiome in Type 1 Diabetes Pathogenesis. Curr Diab Rep 16, 89 (2016). https://doi.org/10.1007/s11892-016-0781-z
Published:
DOI: https://doi.org/10.1007/s11892-016-0781-z