
Abstract
Purpose of Review
The aim of this paper is to summarize the current treatment landscape in metastatic colorectal cancer, as well as those on the horizon in the third-line and beyond settings.
Recent Findings
Herein, recent data regarding TAS-102, regorafenib, and novel anti-angiogenic agents are described. Data on chemotherapy re-challenge and EGFR re-challenge is reviewed. A summary of data on the use of BRAF-targeted therapies, HER-2-targeted therapies, rare fusions (NTRK, RET), MET amplification, and KRAS G12C is included, as well as a brief review on the current role of immune checkpoint inhibitors in metastatic colorectal cancer.
Summary
Multiple new agents are on the horizon. There is increasing relevance of next generation sequencing to look for rare targets, and potentially to assess tumor mutational burden. ctDNA appears to be a valuable asset which may guide the use of therapies in the re-challenge setting.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer is the third most common cancer diagnosed in the USA and the second most common cause of cancer-related deaths in men and women combined [1]. Approximately 20% of patients present with metastatic disease, and 20–50% of patients with early stage disease go on to develop distant metastases [2]. Following progression after two lines of treatment with classic chemotherapies (a fluoropyrimidine, oxaliplatin, irinotecan, and as applicable, anti-VEGF/R and/or anti-EGFR antibodies), expected survival with best supportive care alone is a dismal 4–6 months [3]. Both regorafenib and trifluridine/tipiracil are now approved in this space. Over the past decade, a myriad of novel therapies have been introduced, in part related to advances in molecular profiling. Growing data also supports the re-introduction of prior therapy, namely EGFR antibodies, in select situations. This has both expanded treatment options and complicated treatment decisions for patients with relapsed and refractory metastatic colorectal cancer (mCRC) [4]. Therapies that have been recently approved and/or are showing significant promise include BRAF inhibitors, anti-HER2 therapies, NTRK inhibitors, and direct KRAS G12C inhibitors [5]. In addition, angiogenesis remains an important target in mCRC, and fruquitinib represents a new generation of highly selective tyrosine kinase inhibitors (TKIs) specific for VEGFR-1, VEGFR-2, and VEGFR-3 [5].
Target Agnostic Therapies for mCRC
Trifluridine/Tipiracil (TAS-102)
Trifluridine/tipiracil is a novel oral agent composed of trifluridine, a thymidine nucleoside analog, and tipiracil, a thymidine phosphorylase inhibitor. Once the active anti-neoplastic agent, trifluridine, is taken up by tumor cells, it is incorporated into replicating strands of DNA. This interrupts DNA synthesis and halts cell proliferation. Tipiracil inhibits the degradation of trifluridine and, in doing so, increases its bioavailability [6•].
RECOURSE was the pivotal global phase III trial of TAS-102 vs. placebo (randomized in a 2:1 ratio) in 800 patients who had tumor progression after two lines of standard chemotherapy, including a fluoropyrimidine, oxaliplatin, irinotecan, bevacizumab, and (if KRAS wild-type) cetuximab or panitumumab. While responses were uncommon (1.1%), median OS improved to 7.1 months with TAS-102 from 5.3 months with placebo (HR 0.68, P < 0.001). Hematologic toxicity was the most significant adverse effect. The incidence of grade ≥ 3 neutropenia was 38% in patients treated with TAS-102 [7]. However, despite the high rate of neutropenia, just 4% of patients developed febrile neutropenia [7, 8]. Gastrointestinal toxicities were also greater in the active treatment arm, though generally grades 1–2: nausea 48% vs 24%, vomiting 28% vs 14%, and diarrhea 32% vs 12%.
The subsequent Phase III TERRA study evaluated the efficacy of TAS-102 in the refractory setting among 406 patients from 30 sites in China, Korea, and Thailand. Patients were randomly assigned in a 2:1 ratio to TAS-102 or placebo. In this population, median OS was again improved: 7.8 months with TAS-102 compared to 7.1 months with placebo (HR: 0.79, P < 0.035). PFS was similarly improved as seen in RECOURSE (HR 0.43, P < 0.001) [9].
Regorafenib
Regorafenib is an oral small molecule TKI. Its targets include angiogenic (VEGFR-1, VEGFR-2, and VEGFR-3), oncogenic (KIT, RET, RAF, FGFR), and stromal (PDGFR-β and FGFR) tyrosine kinases [10]. These diverse sets of kinases help regulate tumor angiogenesis, metastasis, oncogenesis, and tumor immunity. Regorafenib, thereby, has a multifaceted mechanism of action [11].
The phase III placebo-controlled CORRECT study enrolled 760 mCRC patients who had progressed after treatment with 5-fluorouracil (5-FU), irinotecan, oxaliplatin, bevacizumab, and, in patients with KRAS wild-type tumors, an EGFR-inhibitor. The study demonstrated an improvement in OS with a median OS of 6.4 months in the regorafenib arm compared to 5.0 months in the placebo arm. The objective response rates were very low in both groups (1% vs 0.4%); however, the disease control rate was significantly greater in the regorafenib group (41% vs 15%, P < 0.0001), as was PFS (HR 0.49, P < 0.0001) [12]. The CORRECT trial was replicated in an Asian population within CONCUR. In this trial, regorafenib achieved a median OS of 8.8 months compared to 6.3 months in the placebo group (HR 0.55, P = 0.00016), a clinically significant difference [13].
As it is observed with multi-kinase inhibitors, regorafenib has a high incidence of dermatologic toxicity, with a rate of hand-foot skin reactions (HFSR) of 47%, 17% being grade 3. Importantly, the incidence of this toxicity is highest with the first cycle and subsequently lessens. Within CORRECT, fatigue (47% vs 28%), diarrhea (34% vs 8%), and anorexia (30% vs 15%) occurred at greater rates in the investigational arm than with placebo, though the majority of these AEs were grades 1 and 2. Hypertension was also frequent, occurring in 30% of subjects [12]. Upon approval, real-world use saw preserved efficacy, which was accompanied by significant rates of dose interruptions (31%) and dose reductions (42.5%). Patients with ECOG PS 2, who would not have been eligible for the refractory studies, unsurprisingly had worse survival outcomes [14].
To mitigate toxicities associated with regorafenib, alternate dosing strategies have been studied, similar to the previous experience with the related drug, sorafenib [15]. The ReDOS trial compared standard dose regorafenib (160 mg/day for 21 days on a 28-day cycle) to a dose escalation strategy (80 mg/day initially, with weekly escalation in 40-mg increments, as tolerated). Grade 3 and greater adverse events were reduced with the dose-escalation strategy, and with this approach, a greater number of patients were able to initiate the third cycle of treatment (43% vs. 26%). In addition, mOS was 9.8 months in the dose-escalation group vs 6.0 months in the standard-dose group (HR = 0.72, P = 0.12), and mPFS was 2.8 months vs 2.0 months (HR = 0.84, P = 0.38) [16•]. Within cycle 2, the mean percentage of planned dose administered was 93% in the dose-escalation group vs 73% in the standard group. Thus, this dose escalation strategy proved to be efficacious, representing the preferred approach in this setting.
Sequencing and Selection of Regorafenib and TAS-102
There are no head-to-head trials to guide selection of regorafenib versus TAS-102. The observational REGOTAS study demonstrated a similar median OS among 550 patients in Japan who received either regorafenib or TAS-102 in the refractory setting. Propensity score adjusted analysis demonstrated a HR of 0.96 (95% CI 0.78–1.18) [17]. A smaller retrospective study of 146 patients showed no significant difference in either PFS or OS [18]. Additional analyses have not been able to identify predictors of efficacy, looking at RAS mutational status and clinical parameters such as age [9, 19]. On the other hand, some inferences can be made from existing data regarding sequencing. In the RECOURSE trial, 17% of patients in the TAS-102 and 20% of patients in the placebo group had been previously exposed to regorafenib. The hazard ratio for OS was equivalent (0.69) in patients who had previously been treated with regorafenib and in those who had not, suggesting preserved efficacy after regorafenib [20].
At this time, selection should largely be driven by the differing toxicity profiles of these agents. TAS-102 is associated primarily with hematologic toxicity, while regorafenib is associated with HFSR, fatigue, and hypertension; both elicit gastrointestinal side effects [7, 12, 18]. For a patient with an ECOG PS of 2, TAS-102 may be preferable. Regardless, the major current interest lies not in prospective studies to optimize the sequencing, but rather in testing approaches to integrate these within novel regimens, which might provide greater yields. These efforts include trials of TAS-102 with oxaliplatin, with bevacizumab, and also with irinotecan and bevacizumab (NCT04109924) [21,22,23]. Key completed studies in the 3rd-line space are detailed in Table 1.
Re-challenge Strategies in the Refractory Setting
Re-challenge with Oxaliplatin
FOLFOX (5-FU, leucovorin, and oxaliplatin) represents a standard chemotherapy regimen in the advanced setting, with neurotoxicity being a nearly unavoidable cumulative toxicity, such that a majority of patients stop therapy for reasons other than progressive disease [24]. This has led to interest in both maintenance stop-and-go studies, as well as re-use of oxaliplatin in the refractory setting, to maximize effect. The ORION study evaluated re-challenge with capecitabine and oxaliplatin in patients who had previously received oxaliplatin. Out of the 46 patients enrolled, 45.5% has discontinued oxaliplatin due to progression, while the remainder had discontinued treatment for other reasons. They found a median time-to-treatment failure (TTF) of 3.4 months, and a median OS of > 9.2 months [25]. REOX, a retrospective study of 83 patients who underwent re-exposure to an oxaliplatin-containing regimen (mFOLFOX in 84.3% of patients), demonstrated a median TTF of 6.04 months and an OS of 10.04 months. Disease control was observed in 56.6% of patients [26]. Collectively, this data suggests the potential for a modest benefit from oxaliplatin re-introduction, though response rates are widely variable, reflecting the heterogeneity. For the most part, both prospective and more recent studies suggest fairly low rates of response. Recently, a phase Ib trial was conducted combining TAS-102 with oxaliplatin in a refractory population. This study achieved a median PFS of 2.7 months (95% CI, 2.4–4.8 months) and a median OS of 6.8 months (95% CI, 5.7–10 months), with only 1 response (2.4%) [22]. A follow-up phase II trial of TAS-102 and oxaliplatin (with bevacizumab as appropriate) in patients with prior exposure to 5FU, oxaliplatin, irinotecan, and appropriate biologics is currently underway (NCT04294264).
Re-challenge with Anti-EGFR Therapy
Anti-EGFR monoclonal Abs (MAbs) such as cetuximab and panitumumab significantly improve outcomes in patients with BRAF and RAS wild-type tumors, particularly in left-sided tumors, and are approved for use as part of a 1st-line regimen with chemotherapy, as well as in the refractory setting [27]. As use in earlier lines of therapy has increased, anti-EGFR re-challenge has been investigated, though with widely variable efficacy outcomes. A retrospective single institution analysis of 68 patients who were re-exposed to anti-EGFR therapy described significant activity of this strategy, though efficacy was greater in patients who had discontinued prior anti-EGFR therapy for reasons other than progression (75% of the population) vs those that had previously discontinued therapy due to progressive disease (25%): objective response 52% vs 18%, median OS 33.4 vs 7.5 months, and median PFS 8.4 vs 3.3 months, respectively [28]. In contrast, in one study where panitumumab was utilized following cetuximab progression, no objective responses were observed, with a median PFS of 1.7 months and OS of 5.2 months [29].
Exposing neoplastic cells to targeted therapies inherently drives clonal evolution. Alterations of KRAS, NRAS, MET, ERBB2, FLT3, EGFR, and MAP2K1 are clearly linked to resistance. The proliferation of technologies which measure circulating tumor DNA (ctDNA) in blood specimen has permitted real-time observation of such mutations and amplifications emerging during therapy, followed by a decline in levels upon withdrawal of EGFR antibodies [30]. Data suggests an exponential decay in relevant mutant allele frequency, with a cumulative half-life of 4.4 months [31••]. Consistent with this, a retrospective review examining anti-EGFR-re-challenge among patients who previously achieved at least stable disease demonstrated an association between longer anti-EGFR-free interval and ORR. Overall activity was encouraging, with an ORR of 19.8%, PFS of 3.8 months, and OS of 10.2 months [32]. The CRICKET trial evaluated irinotecan and cetuximab re-challenge in 28 patients who had received 1st-line cetuximab and irinotecan containing chemotherapy, with tumors that were previously confirmed to be RAS and BRAF wt. At least 6 months of treatment on 1st-line therapy and 4-month lapse between enrollment and prior anti-EGFR treatment were required. Six (21%) responses were observed with a disease control rate of 54%. Analysis of ctDNA demonstrated 12 (48%) of the 25 evaluable patients to have RAS mutations at the time of re-challenge. Of note, none of the patients who achieved confirmed response had a detected plasma RAS mutation, and PFS was significantly longer among those with RAS wt ctDNA (4 vs 1.9 months, HR 0.44, P = 0.03) [33••]. A post hoc analysis of the CAVE trial, utilizing cetuximab re-challenge plus the PD-L1 inhibitor, avelumab, subsequently replicated these outcomes. Of the 67 (87%) patients with evaluable baseline plasma samples, 19 (28%) harbored ctDNA RAS or BRAF mutations at enrollment. Here, median PFS (4.1 vs 3 months, P = 0.004) and OS (17.3 vs 10.4, P = 0.02) were significantly improved among patients with RAS/BRAF wt ctDNA vs those with detected mutations [34••].
While the optimal time lapse from prior related-therapy to re-challenge remains to be established, the potential to utilize ctDNA for selection has garnered excitement. Initial results of the CHRONOS trial were recently presented. This trial enrolled patients with tissue-based RAS/BRAF wt mCRC, who had a history of prior response to anti-EGFR therapy, and required an absence of RAS, BRAF, or EGFR extracellular domain (ECD) mutations by ctDNA. Patients received panitumumab monotherapy. Remarkably, 8 (30%) achieved a response with 17 patients (63%) achieving stable disease or better at 4 months. Median PFS was 16.4 weeks [35]. Liquid biopsy at progression demonstrated the acquisition of resistance conferring alterations in the vast majority of analyzed patients. Prospective randomized trials are underway to further assess this strategy. The PULSE trial will randomize patients without ctDNA resistance alterations to panitumumab re-challenge vs standard of care (TAS-102 or regorafenib) (NCT03992456). The PARARE study will assess optimal sequencing of re-challenge in the refractory setting within the context of a RAS/BRAF wt ctDNA assay (NCT04787341). These studies will be critical to our understanding of optimal patient management. Table 2 highlights key data sets involving therapeutic re-challenge in mCRC.
Emerging Therapies
Anti-VEGF Strategies
Tumor-driven angiogenesis is a well-established target in colorectal cancer. Bevacizumab combined with chemotherapy is a cornerstone of first- and second-line therapy in mCRC, and several studies have demonstrated the benefit of continuation of bevacizumab after progression on first-line therapy [24, 36,37,38,39]. Regorafenib in the refractory setting confers modest additional benefits, as previously discussed. Thus, angiogenic inhibition provides benefit into the 3rd line of therapy and, perhaps, even beyond.
Fruquintinib is a highly selective, oral small molecular inhibitor of VEGFR-1, VEGFR-2, and VEGFR-3, which exhibited high potency in early investigation [40, 41]. The FRESCO trial assessed fruquintinib in a population of 416 patients in China with tumor progression following at least 2 lines of chemotherapy. Patients were randomized in a 2:1 ratio to receive either fruquintinib or placebo. Prior treatment with VEGFR inhibitors was not permitted. OS was improved at 9.3 vs 6.57 months, with fruquintinib vs placebo (HR 0.65, P < 0.001). Median PFS was improved with fruquintinib at 3.71 vs 1.84 months (HR 0.26, P < 0.001). The ORR (5% vs 0%) and DCR (62% vs 12%) were also improved. The most frequent adverse events with fruquintinib were hand-foot-skin reaction, proteinuria, and thrombocytopenia [42••]. Hypertension was seen in more than half of fruquintinib-treated patients, with 21% experiencing ≥ grade 3 hypertension. However, consistent with the more selective profile, fatigue was more similar to placebo (12% vs 7%, with just 1% ≥ grade 3). Further studies are needed to assess the efficacy of fruquintinib outside of a Chinese population, especially given that VEGF inhibition is less commonly incorporated into first- and second-line treatment in China. Only 26% of patients had received a prior VEGF-targeted therapy in FRESCO [42••]. A global phase III study, FRESCO-2, is currently underway and is enrolling patients in Europe, Japan, and the USA (NCT04322539). This study will enroll a more refractory population that has received either TAS-102 and/or regorafenib in addition to the classic chemotherapy regimens.
The C-TASK FORCE study combined TAS-102 with bevacizumab in 25 patients who were refractory to two lines of therapy that included fluoropyrimidine, irinotecan, oxaliplatin, anti-VEGF therapy, and anti-EGFR therapy. The trial demonstrated activity with a centrally assessed PFS of 42.9% at 16 weeks, similar to the outcomes seen with TAS-102 and nintedanib, as well as capecitabine and nintedanib [43,44,45]. Given these encouraging results, a phase II was launched, randomizing 93 patients with refractory mCRC to TAS-102 monotherapy or TAS-102 + bevacizumab. Median PFS was improved at 4.6 vs 2.6 months (HR 0.45, P = 0.0015) [46••]. This led to the development of an ongoing phase III study to definitively evaluate the value of adding bevacizumab to TAS-102, SUNLIGHT (NCT04737187).
HER2-Targeted Therapy
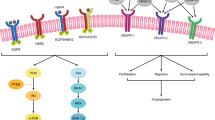
The ERBB protein family consists of 4 receptor tyrosine kinases (RTKs): ERBB1-4 or HER1-4. These RTKs drive multiple downstream signaling pathway, notably the RAS/RAF/MEK/ERK and the PI3K/AKT/mTOR pathways. ERBB2 (human epidermal growth factor receptor 2, HER-2) is a transmembrane glycoprotein receptor, which does not bind extracellular ligands, but readily partners with other ERBB receptors in heterodimers. In the setting of HER-2 amplification, ligand-independent activation occurs through the formation of homodimers [47]. HER-2 is a well-established driver of oncogenesis, successfully targeted pharmacologically in breast and gastric cancer. Two to five percent of mCRC tumors overexpress HER2, though criteria for calling positivity by immunohistochemistry (IHC) slightly differ from other tumor types [48, 49]. RAS/BRAF wild-type and left-sided tumors are enriched for HER-2 amplification, particularly rectosigmoid tumors [50]. Preclinical and clinical data has consistently demonstrated HER2 to be associated with resistance to anti-EGFR therapies, further emphasizing its biologic significance in mCRC [51,52,53,54]. Preclinical models demonstrated significant in vivo activity with dual EGFR/HER2 inhibition, as compared to monotherapy [53]. Given the predictive capacity and potential targeted options (discussed below), the most recent NCCN guidelines advocate routine evaluation for HER-2 amplification [55].
Several non-randomized phase II studies have examined the efficacy of multipronged anti-HER2 therapy. The HERACLES-A study enrolled patients with HER2-positive, KRAS wild-type refractory mCRC, treating them with trastuzumab and lapatinib. The ORR was 30% with a median PFS of 21 weeks and OS of 46 weeks [56]. The MyPathway trial treated HER2-positive, refractory mCRC patients with trastuzumab and pertuzumab, achieving an ORR of 32% and a median PFS of 2.9 months. In the subgroup of patients with KRAS wild-type mCRC, ORR was improved to 40% and median PFS was improved to 5.3 months [57]. Similar results were replicated in the TRIUMPH trial, utilizing the same regimen in patients who were found to be HER2 positive based on tissue analysis or on circulating tumor DNA [58]. Finally, the MOUNTAINEER trial is an ongoing phase II trial testing the combination of tucatinib and trastuzumab in patients with HER2-positive, RAS wild-type refractory mCRC. Initial results revealed an ORR of 55%, median PFS of 6.2 months, and a median OS of 17.3 months [59].
Etiology of non-response to these therapies remains unclear for many patients. Within MyPathway, of the 13 (23%) KRAS-mutated patients, a response was only seen in 1 (8%), with median PFS of 1.4 months, suggesting a lack of benefit. PIK3CA-mutated patients and those with right-sided tumors similarly had worse outcomes, though only 8 (17%) PIK3CA mutations were identified and 2 had concurrent KRAS mutations. In HERACLES, patients were selected specifically for KRAS wt status. Here, the majority of responses were seen in patients with HER2 scores of 3 + on IHC. An exploratory analysis identified a discriminatory HER2 gene copy number (CN) of 9.45, whereby no patients with a CN below 9.45 responded; PFS was 29 weeks vs 16 weeks for patients above vs below that threshold, respectively [56]. Subsequent analysis indicated that ctDNA correctly identified 97% of the pre-treatment samples as HER2 amplified, with an adjusted copy number (aCN) correlating well with the tissue HER2 CN. Furthermore, the plasma HER2 aCN predicted the benefit in a manner similar to that of the tissue analysis [60••].
Trastuzumab deruxtecan-nxki (T-DXd) is a novel antibody–drug conjugate (ADC) composed of the anti-HER2 antibody, trastuzumab, linked to a topoisomerase I inhibitor, deruxtecan. It has shown promising results in HER2-positive metastatic gastric adenocarcinoma trials and was approved for this indication by the FDA in January 2021 [61]. DESTINY-CRC01 is a phase II, multicenter trial evaluating the efficacy of T-DXd in HER2-positive mCRC patients with RAS/BRAF wild-type tumors who have previously received 2 or more lines of treatment (including prior anti-HER2 therapy in 20.5% of patients). Results showed an ORR of 45.3% and a DCR of 83% (95% CI, 70.2–91.9%). In the subgroup of patients with high HER-2 expression (3 + by immunohistochemistry), the ORR improved to 57.5% [62].
This body of data supports the use of dual HER2-directed therapy as well as HER-2 ADCs in HER2-positive mCRC, particularly for those with RAS wild-type disease. Randomized data is lacking, though the ongoing SWOG1613 is an important trial which will compare trastuzumab and pertuzumab to cetuximab and irinotecan in HER2 + tumors as 2nd/3rd-line therapy (NCT03365882).
BRAF V600E-Targeted Therapy
BRAFV600E- mutated mCRC occurs at a frequency of 6–8% and represents a distinct disease subtype, established to have poor prognosis [63]. BRAFV600E mutations constitutively activate the mitogen-activated protein kinase (MAPK) pathway, acting as major drivers of oncogenesis. The MAPK pathway is comprised of a tiered phosphorylation cascade which involves a range of kinases including RAS, RAF, MEK, and ERK [64]. While monotherapy BRAF or dual BRAF and MEK inhibition exhibit significant activity in BRAF V600E-mutated melanoma, the activity of BRAF inhibitors alone is very limited in colorectal cancer [65]. Resistance mechanisms converge to produce the adaptive reactivation of MAPK signaling, frequently through EGFR-mediated mechanisms [66, 67]. Dual EGFR and BRAF inhibition, with or without concomitant MEK inhibition, demonstrated promise in vitro, as well as in early clinical studies [66, 68,69,70].
The BEACON trial was designed to evaluate the efficacy of doublet therapy with encorafenib and cetuximab vs. triplet therapy with encorafenib, binimetinib, and cetuximab vs standard-of-care cetuximab and irinotecan-based chemotherapy. The trial found that both experimental regimens outperformed the standard of care comparator, based upon superior OS (9 months vs 8.4 months vs 5.4 months) and ORR (26% vs 20% vs 2%), for the triplet, doublet, and standard of care, respectively [71••]. The trial was not powered to directly compare the triplet regimen with the doublet regimen, though preclinical data and preliminary studies suggested that the triplet might outperform the doublet. However, OS results showed a median OS of 9.3 months with either the triplet or doublet regimen, suggesting the lack of a large additional benefit with triplet therapy [72]. Based upon this data, encorafenib and cetuximab are currently approved for pre-treated BRAF V600E-mutated colorectal cancer. Current efforts are focused on integrating these agents with chemotherapeutics, moving them into earlier lines of therapy, and evaluating the impact of combination with checkpoint inhibitors (NCT03693170, NCT04607421, NCT04017650).
Promising New Agents
Multiple novel targets in CRC treatment are on the horizon, including NTRK fusions, RET fusions, MET amplification, and KRAS G12C. Fusions involving NTRK1, 2, and 3 occur in 0.5–2% of colorectal cancers [73]. The TRK-inhibitors, larotrectinib and entrectenib, gained tumor-agnostic approval in 2018 and 2019, respectively, based upon pooled results of single-arm studies, demonstrating response rates of 75% and 79% [74•, 75,76,77,78]. As well as being frequent, the responses are highly durable, making this an attractive target to identify. DNA-based sequencing or IHC assays will pick up the majority of these rare alterations, though use of platforms including RNA-seq may be desirable to maximize sensitivity [79]. Whether other rare fusions can be successfully targeted in colorectal cancer, such as ALK, ROS1, and RET, remains to be established. Data presented at AACR 2021 suggests that the targeting of RET fusions is viable across cancers; the RET inhibitor, selpercatinib, achieved 47% ORR overall, including partial responses (PR) in 4/9 colorectal cancers [80]. Responses to ALK inhibitors in the setting of EML4-ALK fusions have also been reported anecdotally, though rigorous trial data does not support routine use at the moment [81, 82]. Additional data and follow-up are needed.
MET amplification is estimated to occur in 4% of metastatic colorectal cancers and is associated with both de novo and acquired resistance to EGFR targeted therapies [83,84,85]. In phase I studies of two novel MET inhibitors, tepotinib and capmatinib, responses were seen across tumor types, including in colorectal cancer [86, 87]. A phase Ib study of capmatinib and cetuximab did not yield responses, though 31% (4/13) of patients experienced tumor regression, ranging from 29 to 44% [88]. Studies are ongoing to test additional MET inhibitors (NCT03592641) as well as the strategy of dual EGFR and MET inhibition in MET-amplified CRC (NCT04515394).
Finally, inhibitors of KRAS G12C have rapidly emerged, with the first, sotorasib, approved for use in non-small cell lung cancer (NSCLC) in early 2021. KRAS G12C is found in approximately 3% of mCRC cases [89]. In initial studies of sotorasib (AMG510), responses were seen in just 7% (3/42) colorectal cancer patents, though 67% achieved stable disease, lasting a median duration of 5.4 months [90••]. Initial data with a different KRAS G12C inhibitor, MRTX849, appeared comparable, if not slightly improved, with 17% (3/18) of tumors responding and 94% (17/18) with disease control [91]. Similar to what was previously seen with BRAF-mutated CRC, pre-clinical data suggests that dual EGFR and KRAS G12C inhibition might produce greater activity [92•]. With this as well as data on other combinations, including SHP2 inhibition, multi-arm phase I studies are underway to optimize the targeting of KRAS G12C (NCT04185883, NCT03785249, NCT04330664).
Immunotherapy
Microsatellite instability is a biomarker of response to immune checkpoint inhibition. About 3–4% of stage IV CRCs have deficient mismatch repair (dMMR) resulting in the microsatellite instability high (MSI-H) phenotype. Loss of mismatch repair results in the accumulation of multiple mutations, particularly frameshift alterations, facilitating the development of multiple potential tumor neoantigens [93]. The Phase 2 KEYNOTE-016 trial demonstrated a 40% response rate to Pembrolizumab in dMMR CRC vs. 0% in pMMR CRC [94]. Follow-up investigation, KEYNOTE-164 and Checkmate-142, confirmed the highly durable activity of PD-1 blockade in this tumor type, which in 2017 led to FDA approval of pembrolizumab, and later nivolumab (with or without low-dose ipilimumab), for use in refractory MSI-H CRC [95•, 96].
Keynote-158 demonstrated the activity of pembrolizumab across multiple tumor types and was subsequently analyzed on the basis of tumor mutational burden (TMB). From the > 1000 patients, 13% (102) were noted to have a high TMB from archived tissue testing (≥ 10 mutations/Mb). ORR was greater in the TMB-high group (29%) vs the TMB-low group (6%), with 57% of responses lasting > 12 months [97]. Pembrolizumab was approved in 2020 for TMB-high tumors on the basis of this data, though has sparked considerable controversy; colorectal cancer not represented in this study [98]. The TAPUR study evaluated monotherapy with pembrolizumab in a cohort of heavily pre-treated CRC patients with high TMB, defined as ≥ 9 Mut/Mb, and noted an ORR of 11% and DCR of 28%. Here, median OS and PFS were 51.9 and 9.3 weeks respectively [99]. Preliminary data from MyPathway, utilizing atezolizumab in TMB-high tumors (≥ 10 mutations/Mb), suggested that a greater TMB cut-off of 16 mutations/Mb had far improved discrimination for response. The ORR was 38% (16/42) vs 2% (1/48) in patients enrolled with TMBs above vs below the cut-off of 16. This included responses in 7 (70%) of 10 patients with colorectal cancer, 3 of whom were confirmed to be MSS. Thus, TMB appears to be a valid biomarker for PD-1/L1 inhibition, though 10 is likely not the optimal cut-off in mCRC.
Pathogenic mutations in POLE, which encodes the catalytic subunit of DNA polymerase ε, are thought to occur in approximately 1% of colorectal cancers [100]. These mutations result in an excess of replication errors, producing TMBs far greater than non-mutated MSS or MSI-H tumors, upwards of 100 muts/Mb. Several case reports suggested activity of immune checkpoint inhibitors in this patient population [101,102,103,104]. A multinational study through the AsCé immunotherapy program reported on a cohort of 16 patients with POLE mutations who were treated with nivolumab, including 7 CRC patients. Thirty-eight percent (6) achieved a response, including 71% (5/7) with mCRC [105]. It is important to note than not all observed POLE mutations are pathogenic, and the benefit was restricted to those patients with established pathogenic mutations.
Conclusion
Advances in the treatment of colorectal cancer have led to improved survival in patients with metastatic disease [106]. With patients surviving beyond both first- and second-line therapy, researchers have turned their attention to expanding treatment options for third-line therapy and beyond. While regorafenib and TAS-102 are currently the primary considerations for these patients, the standard of care in this setting remains undefined. The treatment landscape in this arena will continue to evolve as data from clinical trials continues to accumulate. With the advent of targeted therapies, expanded molecular profiling will likely become increasingly necessary, as it is an important factor in selecting the right patient for particular targets.
References
Papers of particular interest, published recently, have been highlighted as:• Of importance ••Of major importance
Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC, et al. Colorectal cancer statistics, 2020. CA Cancer J Clin. 2020;70(3):145–64.
Field K, Lipton L. Metastatic colorectal cancer-past, progress and future. World J Gastroenterol. 2007;13(28):3806–15.
Jonker DJ, O’Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357(20):2040–8.
NCC N. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®). Colon Cancer Version 2.2021. 2021.
Xie YH, Chen YX, Fang JY. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct Target Ther. 2020;5(1):22.
Kish T, Uppal P. Trifluridine/tipiracil (lonsurf) for the treatment of metastatic colorectal cancer. P T. 2016;41(5):314–25.
Mayer RJ, Van Cutsem E, Falcone A, Yoshino T, Garcia-Carbonero R, Mizunuma N, et al. Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N Engl J Med. 2015;372(20):1909–19.
Yoshino T, Mizunuma N, Yamazaki K, Nishina T, Komatsu Y, Baba H, et al. TAS-102 monotherapy for pretreated metastatic colorectal cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2012;13(10):993–1001.
Xu J, Kim TW, Shen L, Sriuranpong V, Pan H, Xu R, et al. Results of a randomized, double-blind, placebo-controlled, phase iii trial of trifluridine/tipiracil (TAS-102) monotherapy in Asian patients with previously treated metastatic colorectal cancer: the TERRA study. J Clin Oncol. 2018;36(4):350–8.
Ettrich T.J. ST. Regorafenib. Small Molecules in Oncology Recent Results in Cancer Research. 211. August 2, 2018 ed: Springer, Cham; 2018.
Grothey A, Prager G, Yoshino T. The mechanism of action of regorafenib in colorectal cancer: a guide for the community physician. Clin Adv Hematol Oncol. 2019;17(Suppl 12(8)):1–19.
Grothey A, Van Cutsem E, Sobrero A, Siena S, Falcone A, Ychou M, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):303–12.
Li J, Qin S, Xu R, Yau TC, Ma B, Pan H, et al. Regorafenib plus best supportive care versus placebo plus best supportive care in Asian patients with previously treated metastatic colorectal cancer (CONCUR): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2015;16(6):619–29.
Adenis A, de la Fouchardiere C, Paule B, Burtin P, Tougeron D, Wallet J, et al. Survival, safety, and prognostic factors for outcome with Regorafenib in patients with metastatic colorectal cancer refractory to standard therapies: results from a multicenter study (REBECCA) nested within a compassionate use program. BMC Cancer. 2016;16:412.
Reiss KA, Yu S, Mamtani R, Mehta R, D’Addeo K, Wileyto EP, et al. Starting dose of sorafenib for the treatment of hepatocellular carcinoma: a retrospective, multi-institutional study. J Clin Oncol. 2017;35(31):3575–81.
Bekaii-Saab TS, Ou FS, Ahn DH, Boland PM, Ciombor KK, Heying EN, et al. 2018 Regorafenib dose-optimisation in patients with refractory metastatic colorectal cancer (ReDOS): a randomised, multicentre, open-label, phase 2 study. Lancet Oncol. 2019;20(8):1070–82. Novel study demonstrating effiacy of modified dosing schema with regorafenib.
Moriwaki T, Fukuoka S, Taniguchi H, Takashima A, Kumekawa Y, Kajiwara T, et al. Propensity score analysis of regorafenib versus trifluridine/tipiracil in patients with metastatic colorectal cancer refractory to standard chemotherapy (REGOTAS): a Japanese Society for Cancer of the Colon and Rectum Multicenter Observational Study. Oncologist. 2018;23(1):7–15.
Masuishi T, Taniguchi H, Hamauchi S, Komori A, Kito Y, Narita Y, et al. Regorafenib versus trifluridine/tipiracil for refractory metastatic colorectal cancer: a retrospective comparison. Clin Colorectal Cancer. 2017;16(2):e15–22.
Van Cutsem E, Mayer RJ, Laurent S, Winkler R, Gravalos C, Benavides M, et al. The subgroups of the phase III RECOURSE trial of trifluridine/tipiracil (TAS-102) versus placebo with best supportive care in patients with metastatic colorectal cancer. Eur J Cancer. 2018;90:63–72.
Vogel A, Hofheinz RD, Kubicka S, Arnold D. Treatment decisions in metastatic colorectal cancer - beyond first and second line combination therapies. Cancer Treat Rev. 2017;59:54–60.
Satake H, Kato T, Oba K, Kotaka M, Kagawa Y, Yasui H, et al. Phase Ib/II study of biweekly TAS-102 in combination with bevacizumab for patients with metastatic colorectal cancer refractory to standard therapies (BiTS study). Oncologist. 2020;25(12):e1855–63.
Cecchini M, Kortmansky JS, Cui C, Wei W, Thumar JR, Uboha NV, et al. A phase 1b expansion study of TAS-102 with oxaliplatin for refractory metastatic colorectal cancer. Cancer. 2020.
Varghese AM, Cardin DB, Hersch J, Benson AB, Hochster HS, Makris L, et al. Phase I study of trifluridine/tipiracil plus irinotecan and bevacizumab in advanced gastrointestinal tumors. Clin Cancer Res. 2020;26(7):1555–62.
Saltz LB, Clarke S, Diaz-Rubio E, Scheithauer W, Figer A, Wong R, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol. 2008;26(12):2013–9.
Matsuda C, Honda M, Tanaka C, Fukunaga M, Ishibashi K, Munemoto Y, et al. Multicenter randomized phase II clinical trial of oxaliplatin reintroduction as a third- or later-line therapy for metastatic colorectal cancer-biweekly versus standard triweekly XELOX (The ORION Study). Int J Clin Oncol. 2016;21(3):566–72.
Costa T, Nunez J, Felismino T, Boente L, Mello C. REOX: evaluation of the efficacy of retreatment with an oxaliplatin-containing regimen in metastatic colorectal cancer: a retrospective single-center study. Clin Colorectal Cancer. 2017;16(4):316–23.
Yin J, Cohen R, Jin Z, Liu H, Pederson L, Adams R, et al. Prognostic and predictive impact of primary tumor sidedness for previously untreated advanced colorectal cancer. J Natl Cancer Inst. 2021.
Karani A, Felismino TC, Diniz L, Macedo MP, Silva VSE, Mello CA. Is there a role for rechallenge and reintroduction of anti-EGFR plus chemotherapy in later lines of therapy for metastatic colorectal carcinoma? A retrospective analysis Ecancermedicalscience. 2020;14:1069.
Wadlow RC, Hezel AF, Abrams TA, Blaszkowsky LS, Fuchs CS, Kulke MH, et al. Panitumumab in patients with KRAS wild-type colorectal cancer after progression on cetuximab. Oncologist. 2012;17(1):14.
Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21(7):827.
Parseghian CM, Loree JM, Morris VK, Liu X, Clifton KK, Napolitano S, et al. Anti-EGFR-resistant clones decay exponentially after progression: implications for anti-EGFR re-challenge. Ann Oncol. 2019;30(2):243–9. Important analysis providing insight on decay of resistant clones following EGFR targeted therapy.
Rossini D, Germani MM, Pagani F, Pellino A, Dell’Aquila E, Bensi M, et al. Retreatment with anti-EGFR antibodies in metastatic colorectal cancer patients: a multi-institutional analysis. Clin Colorectal Cancer. 2020;19(3):191-9 e6.
Cremolini C, Rossini D, Dell’Aquila E, Lonardi S, Conca E, Del Re M, et al. Rechallenge for patients with RAS and BRAF wild-type metastatic colorectal cancer with acquired resistance to first-line cetuximab and irinotecan: a phase 2 single-arm clinical trial. JAMA Oncol. 2019;5(3):343–50. Key study demonstrating efficacy of re-challenge and potential value of ctDNA.
Martinelli E, Martini G, Famiglietti V, Troiani T, Napolitano S, Pietrantonio F, et al. Cetuximab rechallenge plus avelumab in pretreated patients with RAS wild-type metastatic colorectal cancer: the phase 2 single-arm clinical CAVE trial. JAMA Oncol. 2021. Recent study reinforcing value of ctDNA in predicting efficacy of EGFR re-challenge.
Sartore-Bianchi A PF, Lonardi S, Mussolin B, Rua F, Fenocchio E, et al. Phase II study of anti-EGFR rechallenge therapy with panitumumab driven by circulating tumor DNA molecular selection in metastatic colorectal cancer: the CHRONOS trial. Journal of Clinical Oncology. 2021;39, no. 15_suppl 3506.
Hochster HS, Hart LL, Ramanathan RK, Childs BH, Hainsworth JD, Cohn AL, et al. Safety and efficacy of oxaliplatin and fluoropyrimidine regimens with or without bevacizumab as first-line treatment of metastatic colorectal cancer: results of the TREE Study. J Clin Oncol. 2008;26(21):3523–9.
Bennouna J, Sastre J, Arnold D, Osterlund P, Greil R, Van Cutsem E, et al. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): a randomised phase 3 trial. Lancet Oncol. 2013;14(1):29–37.
Bennouna J, Hiret S, Bertaut A, Bouche O, Deplanque G, Borel C, et al. Continuation of bevacizumab vs cetuximab plus chemotherapy after first progression in KRAS wild-type metastatic colorectal cancer: the UNICANCER PRODIGE18 randomized clinical trial. JAMA Oncol. 2019;5(1):83–90.
Masi G, Salvatore L, Boni L, Loupakis F, Cremolini C, Fornaro L, et al. Continuation or reintroduction of bevacizumab beyond progression to first-line therapy in metastatic colorectal cancer: final results of the randomized BEBYP trial. Ann Oncol. 2015;26(4):724–30.
Sun Q, Zhou J, Zhang Z, Guo M, Liang J, Zhou F, et al. Discovery of fruquintinib, a potent and highly selective small molecule inhibitor of VEGFR 1, 2, 3 tyrosine kinases for cancer therapy. Cancer Biol Ther. 2014;15(12):1635–45.
Cao J, Zhang J, Peng W, Chen Z, Fan S, Su W, et al. A phase I study of safety and pharmacokinetics of fruquintinib, a novel selective inhibitor of vascular endothelial growth factor receptor-1, -2, and -3 tyrosine kinases in Chinese patients with advanced solid tumors. Cancer Chemother Pharmacol. 2016;78(2):259–69.
Li J, Qin S, Xu RH, Shen L, Xu J, Bai Y, et al. Effect of fruquintinib vs placebo on overall survival in patients with previously treated metastatic colorectal cancer: the FRESCO Randomized clinical trial. JAMA. 2018;319(24):2486–96. Pivotal phase III study demonstrating efficacy of fruquintinib in Asian population.
Kuboki Y, Nishina T, Shinozaki E, Yamazaki K, Shitara K, Okamoto W, et al. TAS-102 plus bevacizumab for patients with metastatic colorectal cancer refractory to standard therapies (C-TASK FORCE): an investigator-initiated, open-label, single-arm, multicentre, phase 1/2 study. Lancet Oncol. 2017;18(9):1172–81.
Yamazaki K KY, Shinozaki E, Hara H, Komatsu Y, Nishina T, et al. A multicentre phase I/II study of TAS-102 with nintedanib in patients with metastatic colorectal cancer refractory to standard therapies (N-task force: EPOC1410). Ann Oncol. 2017;28, suppl_5.
Boland PM FM, Lim D, Attwood K, Tan W, Mastri M, et al. A phase I/II study of nintedanib and capecitabine in refractory metastatic colorectal cancer. J Clin Oncol. 2018;36, no. 15_suppl.
Pfeiffer P, Yilmaz M, Moller S, Zitnjak D, Krogh M, Petersen LN, et al. TAS-102 with or without bevacizumab in patients with chemorefractory metastatic colorectal cancer: an investigator-initiated open-label randomised phase 2 trial. Lancet Oncol. 2020;21(3):412–20. Randomized study demonstrating efficacy of adding anti-angiogenic therapy to fluoropyrimidine in 3rd-line setting.
Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell. 2014;25(3):282–303.
Ross JS, Fakih M, Ali SM, Elvin JA, Schrock AB, Suh J, et al. Targeting HER2 in colorectal cancer: the landscape of amplification and short variant mutations in ERBB2 and ERBB3. Cancer. 2018;124(7):1358–73.
Nowak JA. HER2 in colorectal carcinoma: are we there yet? Surg Pathol Clin. 2020;13(3):485–502.
Salem ME, Weinberg BA, Xiu J, El-Deiry WS, Hwang JJ, Gatalica Z, et al. Comparative molecular analyses of left-sided colon, right-sided colon, and rectal cancers. Oncotarget. 2017;8(49):86356–68.
Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J, et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med. 2011;3(99):99ra86.
Jeong JH, Kim J, Hong YS, Kim D, Kim JE, Kim SY, et al. HER2 amplification and cetuximab efficacy in patients with metastatic colorectal cancer harboring wild-type RAS and BRAF. Clin Colorectal Cancer. 2017;16(3):e147–52.
Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011;1(6):508–23.
Sartore-Bianchi A, Amatu A, Porcu L, Ghezzi S, Lonardi S, Leone F, et al. HER2 positivity predicts unresponsiveness to EGFR-targeted treatment in metastatic colorectal cancer. Oncologist. 2019;24(10):1395–402.
Benson AB, Venook AP, Al-Hawary MM, Arain MA, Chen YJ, Ciombor KK, et al. Colon cancer, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2021;19(3):329–59.
Sartore-Bianchi A, Trusolino L, Martino C, Bencardino K, Lonardi S, Bergamo F, et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016;17(6):738–46.
Meric-Bernstam F, Hurwitz H, Raghav KPS, McWilliams RR, Fakih M, VanderWalde A, et al. Pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer (MyPathway): an updated report from a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 2019;20(4):518–30.
Nakamura Y, Okamoto W, Kato T, Hasegawa H, Kato K, Iwasa S, et al. 526PDTRIUMPH: primary efficacy of a phase II trial of trastuzumab (T) and pertuzumab (P) in patients (pts) with metastatic colorectal cancer (mCRC) with HER2 (ERBB2) amplification (amp) in tumour tissue or circulating tumour DNA (ctDNA): A GOZILA sub-study. Annals of Oncology. 2019;30.
Strickler JH ZT, Ou FS, Cercek A, Wu C, Sanchez FA, et al. Trastuzumab and tucatinib for the treatment of HER2 amplified metastatic colorectal cancer (mCRC): initial results from the MOUNTAINEER trial. Ann Oncol. 2019;30, suppl_5.
Siravegna G, Sartore-Bianchi A, Nagy RJ, Raghav K, Odegaard JI, Lanman RB, et al. Plasma HER2 (ERBB2) copy number predicts response to HER2-targeted therapy in metastatic colorectal cancer. Clin Cancer Res. 2019;25(10):3046–53. Novel ctDNA analysis demonstrating predictive capacity for HER-2-targeted therapy.
Shitara K, Bang YJ, Iwasa S, Sugimoto N, Ryu MH, Sakai D, et al. Trastuzumab deruxtecan in previously treated HER2-positive gastric cancer. N Engl J Med. 2020;382(25):2419–30.
Siena S, Bartolomeo MD, Raghav KPS, Masuishi T, Loupakis F, Kawakami H, et al. A phase II, multicenter, open-label study of trastuzumab deruxtecan (T-DXd; DS-8201) in patients (pts) with HER2-expressing metastatic colorectal cancer (mCRC): DESTINY-CRC01. Journal of Clinical Oncology. 2020;38(15_suppl):4000.
Clarke CN, Kopetz ES. BRAF mutant colorectal cancer as a distinct subset of colorectal cancer: clinical characteristics, clinical behavior, and response to targeted therapies. J Gastrointest Oncol. 2015;6(6):660–7.
Paton EL, Turner JA, Schlaepfer IR. Overcoming resistance to therapies targeting the MAPK pathway in BRAF-mutated tumours. J Oncol. 2020;2020:1079827.
Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015;373(8):726–36.
Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2(3):227–35.
Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483(7387):100–3.
Corcoran RB, Atreya CE, Falchook GS, Kwak EL, Ryan DP, Bendell JC, et al. Combined BRAF and MEK inhibition with dabrafenib and trametinib in BRAF V600-mutant colorectal cancer. J Clin Oncol. 2015;33(34):4023–31.
Hong DS, Morris VK, El Osta B, Sorokin AV, Janku F, Fu S, et al. Phase IB study of vemurafenib in combination with irinotecan and cetuximab in patients with metastatic colorectal cancer with BRAFV600E mutation. Cancer Discov. 2016;6(12):1352–65.
Tabernero J, Geel RV, Guren TK, Yaeger RD, Spreafico A, Faris JE, et al. Phase results: encorafenib (ENCO) and cetuximab (CETUX) with or without alpelisib (ALP) in patients with advanced BRAF mutant colorectal cancer (BRAFm CRC). Journal of Clinical Oncology. 2016;34(15_suppl):3544.
Kopetz S, Grothey A, Yaeger R, Van Cutsem E, Desai J, Yoshino T, et al. Encorafenib, binimetinib, and cetuximab in BRAF V600E-mutated colorectal cancer. N Engl J Med. 2019;381(17):1632–43. First-phase III trial demonstrating efficacy of multiagent BRAF targeted approach in mCRC.
Kopetz S, Grothey A, Cutsem EV, Yaeger R, Wasan HS, Yoshino T, et al. Encorafenib plus cetuximab with or without binimetinib for BRAF V600E metastatic colorectal cancer Updated survival results from a randomized. three-arm, phase III study versus choice of either irinotecan or FOLFIRI plus cetuximab (BEACCON CRC). Journal of Clinical Oncology. 2020;38(15_suppl):4001.
Ricciuti B, Genova C, Crino L, Libra M, Leonardi GC. Antitumor activity of larotrectinib in tumors harboring NTRK gene fusions: a short review on the current evidence. Onco Targets Ther. 2019;12:3171–9.
Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med. 2018;378(8):731–9. Key trial of larotrectinib demonstrating pan-cancer efficacy.
Pagani F, Randon G, Guarini V, Raimondi A, Prisciandaro M, Lobefaro R, et al. The landscape of actionable gene fusions in colorectal cancer. Int J Mol Sci. 2019;20(21).
Drilon A, Siena S, Ou SI, Patel M, Ahn MJ, Lee J, et al. Safety and antitumor activity of the multitargeted Pan-TRK, ROS1, and ALK inhibitor entrectinib: combined results from two phase I trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 2017;7(4):400–9.
Braud FGD, Niger M, Damian S, Bardazza B, Martinetti A, Pelosi G, et al. Alka-372-001: first-in-human, phase I study of entrectinib - an oral pan-trk, ROS1, and ALK inhibitor - in patients with advanced solid tumors with relevant molecular alterations. J Clin Oncol. 2015;33(15):2517.
Patel MR, Bauer TM, Liu SV, Drilon AE, Wheler JJ, Shaw AT, et al. STARTRK-1: phase 1/2a study of entrectinib, an oral Pan-Trk, ROS1, and ALK inhibitor, in patients with advanced solid tumors with relevant molecular alterations. J Clin Oncol. 2015;33(15):2596.
Solomon JP, Hechtman JF. Detection of NTRK fusions: merits and limitations of current diagnostic platforms. Cancer Res. 2019;79(13):3163–8.
Subbiah V KB, Bauer T, McCoach C, Falchook G, Takeda M, et al. Abstract CT011: efficacy and safety of selpercatinib in RET fusion-positive cancers other than lung or thyroid cancers. Cancer Res July 1 2021 (81) (13 Supplement) CT011. 2021.
He X, Jiao XD, Liu K, Qin BD, Wu Y, Ling Y, et al. Clinical responses to crizotinib, alectinib, and lorlatinib in a metastatic colorectal carcinoma patient with ALK gene rearrangement: a case report. Jco Precis Oncol. 2021;5.
Hsiao SY, He HL, Weng TS, Lin CY, Chao CM, Huang WT, et al. Colorectal cancer with EML4-ALK fusion gene response to alectinib: a case report and review of the literature. Case Rep Oncol. 2021;14(1):232–8.
Zhang M, Li G, Sun X, Ni S, Tan C, Xu M, et al. MET amplification, expression, and exon 14 mutations in colorectal adenocarcinoma. Hum Pathol. 2018;77:108–15.
Kwak Y, Yun S, Nam SK, Seo AN, Lee KS, Shin E, et al. Comparative analysis of the EGFR, HER2, c-MYC, and MET variations in colorectal cancer determined by three different measures: gene copy number gain, amplification status and the 2013 ASCO/CAP guideline criterion for HER2 testing of breast cancer. J Transl Med. 2017;15(1):167.
Parseghian CM, Napolitano S, Loree JM, Kopetz S. Mechanisms of innate and acquired resistance to anti-EGFR therapy: a review of current knowledge with a focus on rechallenge therapies. Clin Cancer Res. 2019;25(23):6899–908.
Bang YJ, Su WC, Schuler M, Nam DH, Lim WT, Bauer TM, et al. Phase 1 study of capmatinib in MET-positive solid tumor patients: dose escalation and expansion of selected cohorts. Cancer Sci. 2020;111(2):536–47.
Falchook GS, Kurzrock R, Amin HM, Xiong W, Fu S, Piha-Paul SA, et al. First-in-man phase I trial of the selective MET inhibitor tepotinib in patients with advanced solid tumors. Clin Cancer Res. 2020;26(6):1237–46.
Delord JP, Argiles G, Fayette J, Wirth L, Kasper S, Siena S, et al. A phase 1b study of the MET inhibitor capmatinib combined with cetuximab in patients with MET-positive colorectal cancer who had progressed following anti-EGFR monoclonal antibody treatment. Invest New Drugs. 2020;38(6):1774–83.
Araujo LH, Souza BM, Leite LR, Parma SAF, Lopes NP, Malta FSV, et al. Molecular profile of KRAS G12C-mutant colorectal and non-small-cell lung cancer. BMC Cancer. 2021;21(1):193.
Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS(G12C) inhibition with sotorasib in advanced solid tumors. N Engl J Med. 2020;383(13):1207–17. Initial phase I trial of KRAS G12C inhibitor demonstrating variable efficacy across tumor types.
Johnson ML OS, Barve M, Rybki II, Papadopoulos KP, Leal TA, et al. KRYSTAL-1: activity and safety of adagrasib (MRTX849) in patients with colorectal cancer (CRC) and other solid tumors harboring a KRAS G12C mutation. Eur J Cancer 138S2 (2020) S1–S62. 2020.
Amodio V, Yaeger R, Arcella P, Cancelliere C, Lamba S, Lorenzato A, et al. EGFR blockade reverts resistance to KRAS(G12C) inhibition in colorectal cancer. Cancer Discov. 2020;10(8):1129–39. Critical pre-clinical data paving the way for combinatorial approaches in KRAS G12C-mutated mCRC.
Overman MJ, Ernstoff MS, Morse MA. Where we stand with immunotherapy in colorectal cancer: deficient mismatch repair, proficient mismatch repair, and toxicity management. Am Soc Clin Oncol Educ Book. 2018;38:239–47.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–20.
Le DT, Kim TW, Van Cutsem E, Geva R, Jager D, Hara H, et al. Phase II open-label study of pembrolizumab in treatment-refractory, microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer: KEYNOTE-164. J Clin Oncol. 2020;38(1):11–9. Important follow-up study evaluating the efficacy of pembrolizumab in pretreated MSI-H mCRC.
Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18(9):1182–91.
Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020;21(10):1353–65.
Marcus L, Fashoyin-Aje LA, Donoghue M, Yuan M, Rodriguez L, Gallagher PS, et al. FDA approval summary: pembrolizumab for the treatment of tumor mutational burden-high solid tumors. Clin Cancer Res. 2021.
Meiri E, Garrett-Mayer E, Halabi S, Mangat PK, Shrestha S, Ahn ER, et al. Pembrolizumab (P) in patients (Pts) with colorectal cancer (CRC) with high tumor mutational burden (HTMB): results from the Targeted Agent and Profiling Utilization Registry (TAPUR) study. J Clin Oncol. 2020;38(4_suppl):133.
Domingo E, Freeman-Mills L, Rayner E, Glaire M, Briggs S, Vermeulen L, et al. Somatic POLE proofreading domain mutation, immune response, and prognosis in colorectal cancer: a retrospective, pooled biomarker study. Lancet Gastroenterol Hepatol. 2016;1(3):207–16.
Silberman R, Steiner DF, Lo AA, Gomez A, Zehnder JL, Chu G, et al. Complete and prolonged response to immune checkpoint blockade in POLE-mutated colorectal cancer. JCO Precis Oncol. 2019;3:1–5.
Chen J, Lou H. Complete response to pembrolizumab in advanced colon cancer harboring somatic POLE F367S mutation with microsatellite stability status: a case study. Onco Targets Ther. 2021;14:1791–6.
Gong J, Wang C, Lee PP, Chu P, Fakih M. Response to PD-1 blockade in microsatellite stable metastatic colorectal cancer harboring a POLE mutation. J Natl Compr Canc Netw. 2017;15(2):142–7.
Huyghe N, Baldin P, Van den Eynde M. Immunotherapy with immune checkpoint inhibitors in colorectal cancer: what is the future beyond deficient mismatch-repair tumours? Gastroenterol Rep (Oxf). 2020;8(1):11–24.
Rousseau BJCBI, Pasmant E, et al. High activity of nivolumab in patients with pathogenic exonucleasic domain POLE (edPOLE) mutated mismatch repair proficient (MMRp) advanced tumours. Annal Oncol. 2020;31(supp1_4):S462–504.
Kopetz S, Chang GJ, Overman MJ, Eng C, Sargent DJ, Larson DW, et al. Improved survival in metastatic colorectal cancer is associated with adoption of hepatic resection and improved chemotherapy. J Clin Oncol. 2009;27(22):3677–83.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Conflict of Interest
Sadaf Qureshi declares no potential conflicts of interest. Lyudmyla Berim has received grants from Natera and CARIS. Patrick M Boland has received grants from Taiho, Processa, Abbvie, Macrogenics, and Athenex. Dr. Boland has also received grants and non-financial support from Merck, personal fees from Bayer and Celgene, and grants and personal fees from Ipsen.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Systemic Therapies in Colorectal Cancer
Rights and permissions
About this article

Cite this article
Qureshi, S., Berim, L. & Boland, P.M. Current Treatment Landscape for Third- or Later-Line Therapy in Metastatic Colorectal Cancer. Curr Colorectal Cancer Rep 17, 131–141 (2021). https://doi.org/10.1007/s11888-021-00469-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11888-021-00469-4