
Abstract
Large vessel vasculitis (LVV) covers a spectrum of primary vasculitides predominantly affecting the aorta and its major branches. The two main subtypes are giant cell arteritis (GCA) and Takayasu arteritis (TA). Less commonly LVV occurs in various other diseases. Clinical manifestations result from vascular stenosis, occlusion, and dilation, sometimes complicated by aneurysm rupture or dissection. Occasionally LVV is discovered unexpectedly on pathological examination of a resected aortic aneurysm. Clinical evaluation is often unreliable in determining disease activity. Moreover, the diagnostic tools are imperfect. Acute phase reactants can be normal at presentation and available imaging modalities are more reliable in delineating vascular anatomy than in providing reliable information on degree of vascular inflammation. Glucocorticoids are the mainstay of therapy of LVV. Patients may develop predictable adverse effects from long-term glucocorticoid use. Several steroid-sparing agents have also shown some promise and are currently in use. Endovascular revascularization procedures and open surgical treatment for aneurysms and dissections are sometimes necessary, but results are not always favorable and relapses are common. This article, the first in a series of two, will be devoted to GCA and isolated (idiopathic) aortitis, while TA will be covered in detail in the next article.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Large vessel vasculitis (LVV) covers a spectrum of primary vasculitides predominantly of the aorta and its major branches. The two main subclasses of LVV are giant cell arteritis (GCA) and Takayasu arteritis (TA). These vasculitides differ in the age of onset with GCA rarely occurring before the age of 50 years and TA rarely after 40 years. It has been postulated that TA and GCA may represent different phenotypes within the continuum of the same disease [1]. These diseases and their treatments can cause significant morbidity and have the potential to cause premature mortality. LVV, especially in GCA, is not uncommon and may be under-recognized. Clinical manifestations can be protean - vascular stenosis and occlusion, leading to tissue ischemia, and vascular dilation (aneurysm formation) sometimes complicated by aneurysm rupture or dissection. At the outset, constitutional symptoms (fever, malaise, weight loss, night sweats, polyarthralgia or arthritis) can be more prominent than localizing features. So, if not suspected and appropriately investigated, diagnosis can be difficult and often missed. Vascular pathology can be erroneously attributed to atherosclerotic disease, and an incorrect management approach can lead to unfavorable outcomes. Sometimes LVV is discovered unexpectedly on histopathological examination of a resected aortic aneurysm or during repair of an aortic dissection. In this situation, if the disease is found to be limited to the aorta, it is classified as an isolated (idiopathic) aortitis. Less commonly, LVV has been associated with a number of other diseases (please refer to Table 1 in the next article on TA).
The importance of a careful and focused history and a thorough physical examination cannot be over-emphasized, particularly that of the vascular system – assessment of pulse and blood pressure in all four extremities, and listening for bruits and for an aortic regurgitation murmur. However, clinical evaluation is often unreliable in determining disease activity, and disease progression may occur silently in about 50 % of cases. Moreover progressive vascular narrowing or vascular occlusion does not necessarily reflect active inflammation, but could largely be a result of progressive fibrosis. Similarly, aneurysm enlargement does not automatically indicate activity, but could be a result of hemodynamic perturbations, especially when hypertension is uncontrolled.
The diagnostic tools of LVV are imperfect and have their limitations. Acute phase reactants can be normal at presentation in about a third of patients, when disease is active, and also do not always accurately reflect remission or relapse. It is often difficult to differentiate activity (vascular inflammation, which is potentially reversible with immunosuppressive medications) from damage (which is cumulative and potentially irreversible). Available imaging modalities are better and more reliable in delineating vascular anatomy (luminal narrowing, occlusion, ectasia or dissection) [2] and demonstrating wall thickening and calcification, than in providing reliable information on degree of vascular inflammation, and thus guide treatment recommendations. We definitely need better surrogate markers and imaging techniques to demonstrate ongoing vascular inflammation that will accurately indicate activity and response to therapy, and also provide long term prognostic information.
Glucocorticoids have remained the mainstay of therapy of LVV. Patients may have a chronic course and need glucocorticoids for several years and be faced with their predictable adverse effects. Depending on the etiology of LVV, several steroid-sparing agents (conventional disease modifying agents and biologic response modifiers) that have shown some efficacy are currently in use. Some other agents are undergoing clinical trials. Surgical treatment is sometimes necessary, but results are not always favorable, especially if LVV is active peri-operatively. Close vigilance during and post-treatment is also needed as relapses are not uncommon.
This article, the first in a series of two, will be devoted to GCA and isolated (idiopathic) aortitis, while TA will be discussed in detail in the next article.
Giant Cell Arteritis
Giant cell arteritis (GCA) is a chronic granulomatous arteritis of large- and medium-sized vessels. The mean age of onset is around 72 years, and the disease does not occur below the age of 50 [3]. The overall prevalence is about one in 500 individuals [4]. The American College of Rheumatology (ACR) classification criteria for GCA were developed in 1990 [4]. It is typically a systemic illness and vascular involvement may be extensive. However, it most frequently involves the medium-sized cranial branches of the arteries originating from the aortic arch. The most dreaded complication of this disease, visual loss, is a consequence of the cranial arteritis associated with GCA.
The diagnosis of GCA should be considered in a patient over the age of 50 who develops: (a) new headaches, (b) jaw claudication, (c) acute onset of visual disturbances, (d) symptoms of polymyalgia rheumatica (PMR), (e) unexplained fever or anemia, (f) elevated acute phase reactants, and (g) temporal artery abnormality on examination.
In addition to involvement of the cranial arteries, GCA can also sometimes present with involvement of other vascular territories, such as the great vessels. In this situation the temporal arteries are often spared (about 40 %) [5] and ACR criteria [4] may be unhelpful. These patients may present with upper extremity claudication because of involvement of subclavian and axillary arteries.
Clinical Features
In GCA, there are four main sets of clinical manifestations which might present in isolation or in various combinations:
-
(a)
Cranial (temporal) arteritis: This is characterized by temporal headache, amaurosis fugax, jaw and/or tongue claudication, scalp tenderness and/or hyperesthesia. The affected temporal artery is often thickened, tortuous, tender, nodular, or have diminished pulsation. The main concern of cranial GCA is “arteritic” anterior ischemic optic neuropathy (AION) as a result of inflammation and occlusion of blood vessels supplying the optic nerve (posterior ciliary arteries and/or ophthalmic artery), leading to sudden, nearly complete and often permanent unilateral loss of vision (Fig. 1). If left untreated, the other eye is also likely to be affected within weeks. Rarely, patients can present with scalp necrosis [6, 7].
Fig. 1 
Severe AION in GCA: choroidal hypoperfusion resulted in rapidly progressive vision loss in the right eye, despite being on intravenous methylprednisolone: (a) Pallor of the optic disk, disk edema, and retinal cherry red spot from central retinal artery occlusion; (b) Fluorescein angiogram showing severe choroidal ischemia
-
(b)
PMR is characterized by predominantly proximal muscle pain and stiffness, significant morning stiffness, and much less commonly a peripheral inflammatory arthritis. Approximately 40-50 % of patients with GCA have associated PMR, and about 15 % of patients with PMR develop GCA [8].
-
(c)
Constitutional symptoms are common in both GCA and PMR: fever, malaise, fatigue, weight loss, anorexia, night sweats.
-
(d)
Extracranial disease manifesting as LVV: It has become evident in the last few decades that large artery involvement is a common but under-recognized manifestation of GCA, and tends to occur in up to a third of patients with this disease. This could be in the form of (a) arterial stenoses, occlusions or ectasias affecting subclavian and axillary arteries, carotid and vertebro-basilar arteries, and also sometimes the iliac arteries and their distal branches, and (b) aortitis, which is often asymptomatic, manifesting as ectasias and aneurysm formation in the thoracic and less commonly the abdominal aorta. This can sometimes be complicated by aortic rupture or dissection.
Large vessel involvement associated with GCA (LV-GCA) will be reviewed in detail in this article:

Arterial Stenoses and Occlusion
This complication is not uncommon; the prevalence is variable depending on the vascular imaging modality used for evaluation. Fluorodeoxyglucose (18F)-positron emission tomography (FDG-PET) scans show that >80 % of patients with GCA have large vessel disease [9–11]. Even in about a third of patients with isolated PMR, there is inflammatory uptake in the subclavian arteries. On Doppler ultrasound scan of axillary arteries about a third of the patients with cranial GCA will have upper extremity involvement [12, 13, 14•]. Bilateral disease is quite common. On computerized tomographic angiography (CTA), a significant proportion (42 %) of patients with GCA will have subclavian involvement. Patients are often asymptomatic (only 4-6 % will have upper extremity claudication), but the condition may be under-recognized. Arterial involvement in the arms either seems to develop concurrent with or 1–2 years after the diagnosis of GCA [14•, 15–17]. Compared to patients with cranial disease, on average, those with LV-GCA tend to be about 6 years younger. Also, only about 40 % of patients with LV-GCA will have cranial symptoms; in particular, vision loss is distinctly uncommon. On the other hand, LV-GCA tends to present with vascular symptoms and/or findings, such as absent upper extremity pulses, large artery bruits, and claudication [5, 14•, 15, 17]. Less commonly, similar symptoms can also occur in the lower extremities. Studies with Doppler ultrasound or FDG-PET scans show that up to 37 % of LV-GCA patients can have inflammatory activity of the proximal leg arteries, such as the superficial femoral, iliac and popliteal arteries, and involvement is often bilateral. Clinical manifestations are usually uncommon because of development of exuberant collaterals, but in one series, symptoms were present in up to 20 % of patients [9, 18–20]. Patients with LV-GCA who develop rapidly progressive bilateral leg claudication can have marked improvement of imaging findings and resolution of claudication after successful immunosuppressive therapy. Hence, the outcome of medical therapy can be much more rewarding as opposed to those who have atherosclerotic peripheral arterial disease, where medical treatment options are very limited. Interestingly, in one series, a substantial number of patients (84 % women) with GCA involving the lower extremities presented with leg claudication, and the diagnosis only became apparent a few months later. In these patients cranial symptoms were absent in about 42 %. Diagnosis of GCA depends on vascular imaging studies, often on cross sectional imaging demonstrating vessel wall thickening (Figs. 2 and 3) to differentiate it from atherosclerosis. Though many patients improve with medical therapy, in contrast to those with arm disease, a significant proportion (15-30 %) may develop critical leg ischemia requiring surgery and rarely amputation [20, 21].

Idiopathic aortitis: (a) Contrast-enhanced CT, axial image at the level of the mid-ascending aorta demonstrating circumferential wall thickening measuring up to 9 mm (white arrows); (b) Contrast-enhanced CT, axial image at the level of the mid-aortic arch demonstrating mild circumferential wall thickening measuring up to 3 mm (white arrows). In addition there is a focal, saccular aneurysm at the lateral aspect of the mid-arch (black asterisk); (c) 3D-reconstruction of a contrast-enhanced CT demonstrating the mild enlargement of the ascending aorta and focal, saccular aneurysm at the lateral aspect of the mid-arch. Note that the circumferential wall thickening cannot be seen on this reconstruction as it is a “luminogram” demonstrating only the lumen of the vessels; (d) Contrast-enhanced CT, in an oblique axial maximum intensity projection image at the level of the aortic root demonstrating the circumferential wall thickening, again measuring up to 9 mm (between the black arrows). The proximal left and right coronary arteries are also included and are seen to be surrounded by the wall thickening: the left main coronary artery (short black arrow) is not narrowed by the wall thickening, while the proximal right coronary artery (RCA, short white arrow) has moderate to severe stenosis

Giant cell arteritis: (a) 3D-reconstruction of a contrast-enhanced MRA demonstrating normal caliber suprarenal abdominal aorta, and mild uniform narrowing of the infrarenal abdominal aorta; (b) Coronal source image from a contrast-enhanced MRA of the abdominal aorta demonstrating uniform wall thickening of the infrarenal abdominal aorta (the black aortic wall highlighted by the white arrows); (c) Axial steady-state free-precision image obtained at the level of the superior mesenteric artery (SMA) origin. Note the circumferential wall thickening (white arrows) that involves the SMA origin, but does not cause significant narrowing of either the SMA or mesenteric segment of the abdominal aorta
Aortitis
The aorta is also frequently involved in GCA. Aortitis leads to an increased risk of aortic aneurysms. The thoracic aorta is preferentially affected. As it is mostly asymptomatic, periodic vascular imaging studies are required for its detection. Aneurysms may cause premature mortality due to dissection or rupture. Hence screening is generally recommended, though the optimal timing is controversial. A small number of patients (15 %) have aortic damage already at diagnosis of GCA. However, in the majority, aortic aneurysms tend to be a late complication, occurring about 3–5 years from diagnosis, when the disease seems to be clinically quiet and the inflammatory markers are normal [14•, 19, 22]. Recent epidemiology data also show that there is a progressive increase in the incidence of aortic aneurysms even beyond 5 years of diagnosis. Hence patients need to be followed more consistently over time, because their risk of aortic aneurysm formation continues to increase [16].
In some cases of cranial GCA maintained on daily low dose prednisone, a widened mediastinum may be discovered on a chest X-ray, and further imaging may reveal a dilated aorta. PET scans may show FDG uptake in the aorta (Fig. 4). Similarly, magnetic resonance angiogram (MRA) may show edema, thickening, and contrast enhancement of the aortic wall (Fig. 3). Whether PET scan (Fig. 4) [23] or CTA (Fig. 2) [14•] is used, about 50 % of patients with new onset GCA have involvement of the aorta. In an elegant study, it was shown that if aortitis was detectable on a PET scan at the onset of GCA, the same patients showed evidence of progressive aortic dilation when evaluated 4 years later [23].

PET-CT images of a 67-year-old female with large vessel vasculitis (aortitis): before (a, b, c, and d) and after (e, f, g, h) treatment with prednisone and methotrexate for 6 months: (a-d) Wall thickening was demonstrated in the ascending aorta and aortic arch with increased FDG uptake [peak standardized uptake value (SUV) = 5.07], consistent with active inflammation. Tracer uptake in the rest of the body regions imaged was unremarkable. Physiologic tracer uptake was noted in heart, liver, and renal collecting system; (e-h) Significant reduction in degree of FDG uptake (peak SUV = 3.55) associated with the wall of the ascending aorta and aortic arch, when compared to the previous study (previous peak SUV = 5.07). These findings are consistent with a positive response of the hypermetabolic inflammation of the aortic wall to therapy; the hypermetabolic changes have not totally resolved however. Physiologic FDG tracer accumulation was noted in portions of the myocardium, several loops of bowel and urinary tract
Epidemiology data suggest that patients with GCA have about 17-fold and 2.5-fold higher risk of thoracic and abdominal aortic aneurysms respectively, compared to the general population [24]. A recent study from a large UK database estimated a twofold increased risk of aortic aneurysms [25]. Another epidemiologic study indicated that up to 12 % of patients with GCA, when followed for 10 years, developed an aortic aneurysm [16]. Nevertheless, it is apparent that retrospective studies may underestimate the true risk of aneurysms in GCA patients. In a prospective study, 54 patients with GCA who were in clinical remission and had normal acute phase reactants were followed for 5 years after their diagnosis. Systematic screening with imaging revealed that 22 % had evidence of aortic damage [26]. When the same prospective cohort was systematically followed further for a median period of 10 years [27•], 33 % of those patients showed evidence of aortic structural damage.
There are no consistent clinical predictors across studies that allow clinicians to identify the patients at risk of aortic dilation and aneurysm formation. At the bedside, presence of an aortic regurgitation murmur can be a helpful clue, because as the ascending aorta dilates, aortic regurgitation develops in some of these patients.
Unfortunately there are no data to guide clinicians about how these patients should be screened [28, 29]. There are expert recommendations suggesting that a chest X-ray may be used [29], but this is notoriously insensitive. An echocardiogram can help us image the ascending aorta, but not the descending thoracic aorta. The American College of Cardiology and Society of Vascular Medicine recommend a baseline CTA or MRA to look for evidence of aortic damage at diagnosis of GCA [30]. However, there are no recommendations about the optimum interval for repeating these imaging studies. It would seem reasonable that after about 3 years from diagnosis we need to periodically re-image these patients. In a recent meta-analysis from the UK, it was stated that in order to detect a previously unknown aneurysm, we would need to screen as few as five to ten GCA patients. The number needed to screen being so small, this should definitely be implemented in our clinical practice [31•].
Pathology
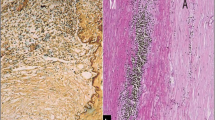
Histopathology of aortic GCA will demonstrate variable degrees of inflammation as a function of the chronicity of the disease. The typical pathologic findings in aortic GCA in the elderly are confined to the media and include areas of medial necrosis that are surrounded by mononuclear inflammatory cells and giant cells (Fig. 5b and e). Medial necrosis refers to areas with loss of smooth muscle cells and collapse of the elastic lamellae often bordered by proliferating vasa vasorum, with perivascular inflammation. Connective tissue stains such as Movat or elastic van Gieson are necessary to demonstrate the fragmentation and loss of elastic lamellae in the media. The intima shows reactive hyperplasia that can become fibrotic with or without superimposed atherosclerosis over time. There is variable thickening and inflammation of the adventitia but these are often milder in degree compared to that seen in TA. In chronic lesions, the media may appear thinned out due to the destruction of the elastic lamellae and inflammatory cells may be sparse and giant cells may be absent.

Histopathology of large vessel vasculitis: The panel shows (a) photomicrographs of a normal aorta, (b) giant cell arteritis, and (c) Takayasu arteritis, with corresponding sections stained with Movat pentachrome (d, e, and f). The images are all taken at the same low magnification: (a) The intima and adventitial layers of a normal aorta are very thin; (d) The media shows multiple orderly layers of elastic lamellae stained black with Movat; (b) A case of giant cell aortitis shows medial necrosis surrounded by mononuclear inflammatory cells and multinucleated giant cells (inset); (e) The elastic lamellae are disrupted in areas of inflammation but remain visible in the area of medial necrosis; (c) A case of Takayasu arteritis shows inflammation in the media and adventitia with predominantly mononuclear cells and occasional multinucleated giant cells (inset); (f) The media shows disruption and collapse of the elastic layer. Note that the intima in both aortitis cases is thicker than the media due to reactive hyperplasia and fibrosis. (e and f) The intimal hyperplasia appears blue green on Movat due to increased ground substance. The adventitia shows fibrous thickening which becomes progressively more prominent in Takayasu arteritis. (e and f) The dense adventitial fibrosis is delineated by the yellow staining of collagen on Movat
Diagnosis
In addition to a careful history and physical examination as described above, diagnosis of GCA should be based on laboratory studies, temporal artery biopsy, and vascular imaging studies.
Laboratory Studies
In most patients, the erythrocyte sedimentation rate (ESR) is significantly elevated [32–34], but sometimes it is only modestly elevated or even normal, even before treatment is initiated. In a meta-analysis, out of 941 cases, only 4 % had a normal ESR [32]. A normal ESR significantly reduces the probability of a positive temporal artery biopsy [32]. In another series 5.4 % had an ESR <40 mm/hour, and were less likely to experience systemic symptoms such as malaise, fever, or weight loss, but their risk of visual loss was not significantly different compared to those with a higher ESR [35]. Serum C-reactive protein (CRP) levels also tend to rise in GCA, and is a more reliable marker of disease activity when the ESR elevation is attributable to other concomitant conditions such as a paraproteinemia [36]. Even though acute phase reactants are helpful in supporting clinical decisions, treatment recommendations should still be based primarily on clinical assessment.
Serum interleukin-6 concentrations tend to correlate with disease activity [34] and may be a better predictor of a relapse than the ESR [37]. However, the assay is expensive, is not routinely available, and the turnaround time is typically long, thus limiting its cost-effectiveness and its clinical usefulness in the acute setting where quick therapeutic decisions need to be made.
The presence of constitutional symptoms and significantly elevated inflammatory markers may be associated with a reduced risk of developing cranial ischemic events [38, 39]. Although this suggested association is thought-provoking, the usefulness of this information in any given patient is debatable. This possible discrepancy can be partly explained by the fact that earlier diagnosis and earlier institution of therapy in those with constitutional symptoms associated with systemic inflammation and elevated inflammatory markers reduce the risk of cranial ischemic events.
Temporal Artery Biopsy
The distinctive histopathology of the superficial temporal artery confirms the diagnosis of cranial (temporal) GCA [40, 41]. This simple procedure has no absolute contraindication and complications are uncommon. Temporal artery biopsy should be performed soon when cranial GCA is suspected, ideally prior to starting glucocorticoids. However, sometimes this is not practical, as treatment should be started promptly in those with threatened vision [28, 42], before the opportunity of getting a biopsy. In that situation, the biopsy should be obtained within a few days of starting treatment [43]. Studies have suggested that a biopsy can still be useful weeks or sometimes even months following treatment initiation [44–47]. Thus, if not done earlier, it is recommended to get the biopsy even up to 4 weeks after initiation of therapy [48]. Unilateral biopsy is often sufficient [42, 49]; however, it may be negative in up to 25 % of patients [50–57]. The side of the biopsy is usually based on lateralizing features (pain, visual disturbances, temporal artery nodularity or tenderness) when they are present [58, 59]. When lateralizing features are absent, bilateral biopsies have been suggested to increase the yield [50–53]. Due to segmental involvement sufficient specimen length is also important [51]. Vasculitis experts suggest a minimum in vivo biopsy length of 2 cm to optimize the yield. If the temporal arteries are not abnormal on examination but the facial or occipital arteries are, then these arteries should be biopsied instead. This may also be deemed necessary if bilateral temporal artery biopsies are negative and the diagnosis of cranial-GCA is still being considered. The yield of a temporal artery biopsy in extracranial GCA is rather low [5].
Isolated (Idiopathic) Aortitis
In a patient with biopsy-proven cranial GCA, subsequent involvement of the aorta or occlusions or ectasias of its first order branches can be assumed to be due to LV-GCA, and obtaining tissue for histopathology would be considered unnecessary. It is neither practical nor essential to get a histopathologic diagnosis in most cases of extra-cranial LV-GCA. However, in a patient who is otherwise asymptomatic, with no indication of a systemic illness at the time of surgery, histopathological examination of a resected ascending aortic aneurysm sometimes shows evidence of active aortitis. This clinical scenario of isolated (idiopathic) aortitis is not infrequent, and has been evaluated in a number of retrospective cohort studies. The histopathology is often indistinguishable from that seen in LV-GCA (Fig. 5b and e). Laminar medial necrosis is often observed (Fig. 5b and e), which may precede progressive aortic dilation and aneurysm formation. It is not clear whether this entity is part of the clinical spectrum of GCA or it is a distinct disorder. If the latter, potentially these patients may have had vasculitis in only a single vascular territory that had been surgically excised and conceivably that could be considered curative and therefore subsequent immunosuppressive therapy would not be deemed necessary [60]. However, careful vascular imaging in cohort studies have demonstrated that often the disease is more extensive than was originally appreciated, and that in some cases inflammation had extended beyond the ascending aorta. Hence, it may be necessary to treat at least a subset of ‘isolated aortitis’ patients with corticosteroids. Unfortunately, the evidence based recommendations about the appropriate management of this condition are sparse.
Imaging [61, 62]
There are no published comparative studies to suggest that one vascular imaging modality is better than another for evaluation of any subtype of LVV. Each modality has its own specific advantages and disadvantages as described in Table 1. Moreover, it is not clear what the significance of the abnormal findings on vascular imaging is, as histopathologic correlation is often not available. Cost and accessibility must be weighed against the concerns of radiation and contrast toxicity. The pros and cons of the different imaging modalities for LVV are discussed below:
-
Catheter directed angiography (CDA) has been the historic gold standard for visualization of vascular lesions (Fig. 6) and has the additional advantages that: (a) the intra-arterial transducer can obtain an accurate central blood pressure measurement at the level of interest, and (b) it can also permit endovascular procedures when indicated. However, CDA has a number of drawbacks: it only images the lumen and does not provide information about the vessel wall; there are risks associated with radiation and intravenous contrast exposure; and, as an invasive procedure there is risk of infection, hemorrhage, and vascular injury. For all these reasons, CDA has largely been supplanted by non-invasive imaging modalities such as CT angiography (CTA) (Fig. 2), MR angiography (MRA) (Fig. 3), Doppler ultrasound (Fig. 7), and PET / PET-CT scans (Fig. 4).
Fig. 6 
Large vessel vasculitis: (a) MRA aortic arch: 3D-reconstruction of a contrast-enhanced magnetic resonance angiogram of the thoracic aorta and proximal arch branch vessels demonstrates a widely patent innominate artery, severe stenosis of the left common carotid artery origin (white arrow), and total occlusion of the left subclavian artery origin (white arrowhead). The left subclavian artery fills via retrograde flow from the left vertebral artery; (b) MRA aortic arch (same patient, 2 years later): 3D-reconstruction of a contrast-enhanced magnetic resonance angiogram of the thoracic aorta and proximal arch branch vessels again demonstrates a widely patent innominate artery, and total occlusion of the left subclavian artery origin (white arrowhead). However, there has also been interval occlusion of the left common carotid artery (the expected origin is denoted by white arrow, and the dashed line indicates the expected course of the left common carotid artery); (c) Digital subtraction angiography (DSA) of the aortic arch demonstrating a widely patent innominate artery (white arrow); (d) DSA of the aortic arch, moments later, demonstrating faint retrograde filling of the left vertebral artery (between arrowheads), and left subclavian artery (arrows); (e) DSA of the aortic arch (without contrast) demonstrating successful positioning of a stent in the proximal left subclavian artery (arrow); (f) DSA of the aortic arch demonstrating widely patent left subclavian and left vertebral arteries following stent placement
Fig. 7 
Giant cell arteritis: Ultrasound Doppler scans in an elderly man with headache and upper extremity pulse deficits. (a) Irregularity in the right temporal artery; (b) Halo around the left axillary artery
-
MRA [2] not just images the lumen (demonstrating stenoses, occlusions and aneurysms) but also the vessel wall identifying wall thickening and enhancement (Fig. 3). Also, MRA is non-invasive and there is no radiation exposure. One of the shortcomings however is interpretation of the significance of the increased signal intensity that suggests inflammation, and wall thickening, particularly in follow-up scans in the face of treatment. Cost and availability are other concerns. Gadolinium toxicity in susceptible patients, claustrophobia, and difficulty with pacemaker and implantable devices – all constitute drawbacks to this approach.
-
CTA has the advantage of being more readily available and it is well tolerated. It can also identify structural abnormalities, thickening and enhancement of the vessel wall (Fig. 2), as well as calcification (which can indicate concomitant atheromatous disease). However, it also has drawbacks such as risk of radiation exposure and contrast toxicity. Similar to MRA, there is controversy over the relevance of persistent thickening or enhancement.
-
Doppler ultrasound has the advantages of being well tolerated, and there is no radiation or contrast exposure. It can also identify structural abnormalities as well as wall edema (Fig. 7). However it does have the disadvantages that its utility is dependent on the skill of the operator, and it may not be readily available. For LV-GCA, the critical drawback of Doppler ultrasound is the inability to visualize the thoracic aorta.
-
PET [63, 64] scans can provide excellent images based on the vascular FDG uptake (Fig. 4), it is non-invasive and can also provide a whole body assessment. However, a concomitant CT scan is required (PET-CT) to study the lumen (Fig. 4), and there is some uncertainty with regard to interpretation of low grade uptake and differentiation from atherosclerosis. Moreover, it does involve isotope and radiation exposure, and there are concerns with cost and availability as well.


There is no consensus about how often imaging should be repeated. Factors determining the frequency of monitoring may include availability of the different modalities, patient factors, and cost issues. It is certainly reasonable that if a relapse is suspected based on clinical findings and acute phase response, then imaging studies should be performed. Some experts recommend that it should also be performed at least annually, even when clinical and inflammatory parameters suggest inactive disease: (a) to rule out asymptomatic disease progression, and (b) to evaluate for worsening due to mechanical factors, even if there is no evidence of disease activity. Certain patients may require more frequent monitoring based on their disease pattern.
Once arterial narrowing or occlusion occurs, it is exceedingly uncommon for therapy to reverse the damage. Therefore, it is unusual to see improvement in luminal changes in subsequent imaging studies. Thus, in clinical practice, repeated imaging does not help as much in detecting reversal as in detecting new lesions. This applies to PET scan as well, as studies show that follow-up imaging with PET scan does not have a very good predictive value.
There are some potential pitfalls of imaging for LVV. Worsening claudication can occur with progression of pre-existing stenosis, and this may occur due to both fibrosis and to atherosclerotic disease progression, independent of active disease, particularly in the setting of other co-morbidities for vascular disease such as smoking, hypertension, dyslipidemia, and diabetes mellitus. Moreover, progressive aneurysmal enlargement may occur purely due to mechanical factors, often as a consequence of uncontrolled hypertension. So, if the only suggestive evidence of ongoing vascular inflammation is enlargement of a preexisting aneurysm, it would be inadvisable to escalate immunosuppressive therapy on that basis alone. Instead, aggressive management of hypertension and careful monitoring for a potential relapse would be recommended. There are uncertainties about the significance of the changes that occur in wall thickening and enhancement despite treatment. Therefore, only the development of a new lesion in a previously unaffected vascular territory should be regarded as unequivocal evidence of active disease. Unfortunately, this finding will only detect active disease after it has caused the damage. So the information cannot be used pre-emptively to prevent a flare by enhancing immunosuppressive therapy.
In patients with established LVV, serial screening is advised. If there is evidence of a new vascular lesion in one vascular territory, this should prompt a thorough evaluation for new lesions in other territories, which may often be asymptomatic, by performing imaging of the aorta and all first order branches. Other dedicated imaging, e.g., of the cerebral or coronary (Fig. 2d) vessels may be required based on clinical manifestations or past involvement in those areas.
Management
Medical Treatment
Since the discovery of cortisone in 1950, glucocorticoids have remained the mainstay of therapy for GCA. Though these agents have never been studied in any randomized controlled trial in GCA, their efficacy in this disease has never been disputed, to the extent that if a therapeutic trial of glucocorticoids does not provide complete symptomatic relief within 24–48 hours, the diagnosis of GCA is in question. Since the potential visual consequences of untreated GCA are grave, it would be ethically unacceptable at this time to conduct a placebo-controlled study using glucocorticoids. Hence, we have to settle with evidence of efficacy from studies in which glucocorticoids provided symptomatic relief and reduced the risk of vascular complications compared to historical GCA controls from the pre-glucocorticoid era [65–68].
It has been found that daily glucocorticoid dosing is more effective than alternate day dosing, but the results are comparable with single or divided daily dosing [69]. If visual loss is not an immediate threat, an initial daily dose of 40 to 60 mg of prednisone (or its equivalent) is recommended [69]. However, with an unequivocal diagnosis of GCA, when potentially reversible symptoms persist or worsen, even higher doses of glucocorticoids may be necessary to provide complete symptomatic relief. One study suggested that intravenous pulse methylprednisolone (15 mg/kg/day for 3 doses) at the start of therapy may be more efficacious in the long run than daily oral dosing [70], by achieving and maintaining more prolonged remission with ≤5 mg of prednisone daily, and by allowing adequate control with a lower median cumulative dose of prednisone, side effects being comparable to the oral regimen. However, the results of this small trial need to be further validated in larger studies before this regimen can be recommended as standard of care.
Considering the dismal prognosis of arteritic AION in GCA [71], patients presenting with visual loss should receive intravenous pulse methylprednisolone 1000 mg daily for 3 days before starting high dose oral prednisone (1 mg/kg/day). The same regimen should also be used in patients presenting with amaurosis fugax, diplopia, or other significant visual symptoms, in an attempt to prevent permanent blindness, even if this recommendation has not been validated in randomized trials. High dose oral prednisone should be continued for 2–4 weeks. Dramatic improvement of GCA symptoms is expected within 1–2 days of glucocorticoid initiation and acute phase reactants are expected to improve. In the absence of this predicted pattern of response, the diagnosis of GCA should be questioned.
As glucocorticoid use in these elderly patients is anticipated to continue for at least 1–2 years, it is important to adopt prophylactic measures against osteoporosis. Bone density measurement is recommended when therapy is initiated. Calcium, vitamin D supplements, and bisphosphonates are also necessary. Patients should be warned about other common side effects of long term glucocorticoid use such as weight gain, glucose intolerance, hypertension, and opportunistic infections. Treating physicians should also be vigilant in monitoring for these side effects.
Glucocorticoid dose reduction should begin no sooner than 2–4 weeks to avoid relapse from premature tapering. Usually prednisone can be reduced from 60 to 50 mg/day in 2 weeks and to 40 mg/day in 4 weeks. Thereafter, the dose can be reduced by approximately 10 % of the daily dose every 1–2 weeks. Once the dose reaches 10 mg/day, subsequent rate of reduction should be slower, at the rate of 1 mg/month. During tapering, flares are not uncommon and should be watched for. Those who relapse often need prolonged prednisone therapy and are more at risk of adverse effects. Recognizing disease relapse during prednisone taper is not easy; acute phase reactants, which are often used to guide therapy, are imperfect markers and do not always correlate precisely with clinical disease activity. Other potential biomarkers such as interleukin-6 [34, 37] and soluble intercellular adhesion molecule-1 [72] have been found to help assess disease activity, but are expensive and not widely available.
The disease course in those with LV-GCA has been compared to cranial-GCA. Disparate results have been reported. One study indicates that cranial-GCA and LV-GCA tend to have the same disease course [73], whereas another study [17] suggests that those with LV-GCA get more relapses, and require a higher cumulative glucocorticoid dose. Also, it takes the LV-GCA patients longer to discontinue prednisone altogether - about 4.5 years on average, as opposed to about 2 years in cranial-GCA patients. Though the data are retrospective, it certainly raises concern that patients with LV-GCA have more refractory disease compared to those with cranial-GCA.
A relapse should be suspected when the original symptoms return, when symptoms suggestive of PMR occur, or when there is a significant rise of the acute phase reactants. Patients should be advised to seek medical attention urgently if they suspect a relapse. In a patient with a history of GCA, discovery of an enlarging aortic aneurysm associated with elevated inflammatory markers should prompt the clinician to restart (or increase the dose of) glucocorticoids. The efficacy of other immunosuppressive agents, though often used in this setting, has not been established.
In the absence of contraindications, low-dose aspirin (81 mg/day) is also recommended in GCA, especially in patients who have already lost vision in one eye. It is believed that platelet inhibition may reduce risk of thrombosis of the diseased posterior ciliary and ophthalmic arteries. Therefore, aspirin is particularly recommended in patients with thrombocytosis [74]. Nevertheless, the evidence is equivocal, as some studies demonstrate a significant reduction in incidence of visual loss and cerebral ischemic events with anti-platelet therapy [75, 76], while others do not demonstrate any such protective effect [77, 78].
Resistant Disease and Glucocorticoid-Sparing Agents
When patients need a prolonged course of high dose glucocorticoids for adequate disease control, and hence develop significant side effects, a steroid sparing agent needs to be considered. In such circumstances, methotrexate has been tried, though controversies abound, as the results are mixed in different studies [79–81]. A meta-analysis involving these trials (161 patients) suggest that concomitant use of methotrexate allows significant reduction in the overall glucocorticoid dose in the subsequent 48 weeks [82], indicating that methotrexate may be a moderately effective steroid-sparing agent in GCA.
Interleukin-6 is believed to play an important role in the pathogenesis of GCA. Several case studies have suggested that tocilizumab, an interleukin-6 inhibitor, may be effective in patients with GCA, thus allowing glucocorticoid dose reduction to a more tolerable level [83•, 84–87].
Some small, uncontrolled trials have suggested that both oral [88] and intravenous pulse [89] cyclophosphamide may be a useful steroid-sparing agent in GCA [88–90]. However, relapses can still occur and adverse effects such as infections and bone marrow suppression limit long term use.
Leflunomide also seems to be an effective steroid-sparing agent when response to the conventional glucocorticoid regimen is inadequate [91•, 92]. However, randomized controlled trials are necessary to confirm the usefulness of leflunomide in this setting.
Though it makes theoretical sense to use a tumor necrosis factor-α (TNF)-inhibitor, several small randomized trials have proven the ineffectiveness of infliximab [93], etanercept [94], and adalimumab [95] in GCA.
Surgical Treatment
Most symptoms of LV-GCA improve with medical therapy alone. Revascularization procedures (e.g., angioplasty, stent placement, or bypass surgery) are seldom required because of the exuberant collateral circulation that develops over time (Fig. 6). These collaterals are often adequate in maintaining tissue perfusion, in spite of frequent absence of peripheral pulses and development of symptomatic limb claudication. Only on rare situations (e.g., in subclavian steal syndrome) should revascularization be considered (Fig. 6). Though successful revascularization has been reported in a few cases of GCA [96–100], restenosis is common.
Aortic valve replacement should be performed when indicated [101]. Surgical resection of an aortic aneurysm or repair of a dissection may be life-saving [101]. However, unlike aneurysms in the general population, the critical size of inflammatory aneurysms that will lead to an increased risk of rupture or dissection and hence necessitate surgery is unknown.
Prognosis
Once visual loss occurs in cranial-GCA, the prognosis of arteritic AION is grave. In a study of 84 consecutive patients with visual loss (114 eyes), only 4 % showed any signs of meaningful improvement [71].
Studies indicate that in LV-GCA the overall survival as well as those that develop large-artery stenoses are similar to that in the general population [16]. However, survival is significantly reduced among those who develop aortic aneurysms and/or dissection (standardized mortality ratio: 2.63) [16]. Therefore, periodic pre-emptive screening with an appropriate vascular imaging study is justified.
Conclusions
As we learnt from this article, LVV primarily affects the aorta and its major branches, the two major subtypes being GCA and TA. Less frequently, LVV has also been associated with various other diseases. As discussed, GCA and the side effects of immunosuppressive therapy can cause significant morbidity and premature mortality. Clinical manifestations of LV-GCA result from vascular stenoses, occlusions, and vascular dilation that can lead to aneurysmal rupture or dissection. Though obtaining a careful history and performing a thorough physical examination are critically important, clinical evaluation is often imprecise in making a diagnosis and determining disease activity. Disease progression may occur silently. Also, the diagnostic modalities of LV-GCA are often inadequate. Acute phase reactants can be normal and sometimes do not accurately indicate remission or relapse. Available imaging modalities are also imperfect, and are more reliable in defining vascular anatomy than in assessing disease activity and guiding treatment recommendations. Moreover, progressive vascular narrowing can simply be a result of progressive fibrosis, and aneurysm enlargement can be a consequence of hemodynamic changes, often aggravated by uncontrolled hypertension. Glucocorticoids remain the cornerstone of therapy, and despite all prophylactic measures, chronic glucocorticoid use leads to considerable adverse effects. Though randomized controlled trial data are often lacking, several steroid-sparing agents are currently in use for management of GCA, based mostly on efficacy data from small uncontrolled studies. Surgical treatment, which should preferably be avoided in LV-GCA, is sometimes necessary. Results of surgery are not favorable if the disease is active perioperatively. When inflammatory disease is felt to be in remission, the immediate results of surgery for revascularization may be acceptable, but recurrences are still common on the long run. It is clear that we need better surrogate markers and better imaging techniques to detect ongoing vascular inflammation that will accurately reflect response to therapy, and also provide long term prognostic information. A more in-depth understanding of the molecular mechanisms involved in the pathogenesis of GCA should help provide new targets for therapy.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Maksimowicz-McKinnon K, Clark TM, Hoffman GS. Takayasu arteritis and giant cell arteritis: a spectrum within the same disease? Medicine (Baltimore). 2009;88(4):221–6.
Tso E, Flamm SD, White RD, Schvartzman PR, Mascha E, Hoffman GS. Takayasu arteritis: utility and limitations of magnetic resonance imaging in diagnosis and treatment. Arthritis Rheum. 2002;46(6):1634–42.
Klein RG, Hunder GG, Stanson AW, Sheps SG. Large artery involvement in giant cell (temporal) arteritis. Ann Intern Med. 1975;83(6):806–12.
Hunder GG, Bloch DA, Michel BA, Stevens MB, Arend WP, Calabrese LH, et al. The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum. 1990;33(8):1122–8.
Brack A, Martinez-Taboada V, Stanson A, Goronzy JJ, Weyand CM. Disease pattern in cranial and large-vessel giant cell arteritis. Arthritis Rheum. 1999;42(2):311–7.
Abdullah AN, Keczkes K, Wyatt EH. Skin necrosis in giant cell (temporal) arteritis: report of three cases. Br J Dermatol. 1989;120(6):843–6.
Campbell FA, Clark C, Holmes S. Scalp necrosis in temporal arteritis. Clin Exp Dermatol. 2003;28(5):488–90.
Gonzalez-Gay MA, Barros S, Lopez-Diaz MJ, Garcia-Porrua C, Sanchez-Andrade A, Llorca J. Giant cell arteritis: disease patterns of clinical presentation in a series of 240 patients. Medicine (Baltimore). 2005;84(5):269–76.
Blockmans D, de Ceuninck L, Vanderschueren S, Knockaert D, Mortelmans L, Bobbaers H. Repetitive 18F-fluorodeoxyglucose positron emission tomography in giant cell arteritis: a prospective study of 35 patients. Arthritis Rheum. 2006;55(1):131–7.
Blockmans D, de Ceuninck L, Vanderschueren S, Knockaert D, Mortelmans L, Bobbaers H. Repetitive 18-fluorodeoxyglucose positron emission tomography in isolated polymyalgia rheumatica: a prospective study in 35 patients. Rheumatology (Oxford). 2007;46(4):672–7.
Ostberg G. Morphological changes in the large arteries in polymyalgia arteritica. Acta Med Scand Suppl. 1972;533:135–59.
Schmidt WA, Seifert A, Gromnica-Ihle E, Krause A, Natusch A. Ultrasound of proximal upper extremity arteries to increase the diagnostic yield in large-vessel giant cell arteritis. Rheumatology (Oxford). 2008;47(1):96–101.
Ghinoi A, Pipitone N, Nicolini A, Boiardi L, Silingardi M, Germano G, et al. Large-vessel involvement in recent-onset giant cell arteritis: a case–control colour-Doppler sonography study. Rheumatology (Oxford). 2012;51(4):730–4.
Prieto-Gonzalez S, Arguis P, Garcia-Martinez A, Espigol-Frigole G, Tavera-Bahillo I, Butjosa M, et al. Large vessel involvement in biopsy-proven giant cell arteritis: prospective study in 40 newly diagnosed patients using CT angiography. Ann Rheum Dis. 2012;71(7):1170–6. This prospective study detected CTA evidence of LVV in two-thirds of patients newly diagnosed with GCA. Aortic dilation was already present in 15% patients.
Schmidt WA. Takayasu and temporal arteritis. Front Neurol Neurosci. 2006;21:96–104.
Kermani TA, Warrington KJ, Crowson CS, Ytterberg SR, Hunder GG, Gabriel SE, et al. Large-vessel involvement in giant cell arteritis: a population-based cohort study of the incidence-trends and prognosis. Ann Rheum Dis. 2013;72(12):1989–94.
Muratore F, Kermani T, Crowson C, Green A, Matteson E, Warrington K. Large vessel giant cell arteritis: a cohort study. Arthritis Rheum. 64[10 (Abstract Supplement)], S994. 10-1-2012 [Abstract].
Aschwanden M, Kesten F, Stern M, Thalhammer C, Walker UA, Tyndall A, et al. Vascular involvement in patients with giant cell arteritis determined by duplex sonography of 2x11 arterial regions. Ann Rheum Dis. 2010;69(7):1356–9.
Nuenninghoff DM, Hunder GG, Christianson TJ, McClelland RL, Matteson EL. Incidence and predictors of large-artery complication (aortic aneurysm, aortic dissection, and/or large-artery stenosis) in patients with giant cell arteritis: a population-based study over 50 years. Arthritis Rheum. 2003;48(12):3522–31.
Czihal M, Tato F, Rademacher A, Kuhlencordt P, Schulze-Koops H, Hoffmann U. Involvement of the femoropopliteal arteries in giant cell arteritis: clinical and color duplex sonography. J Rheumatol. 2012;39(2):314–21.
Kermani TA, Matteson EL, Hunder GG, Warrington KJ. Symptomatic lower extremity vasculitis in giant cell arteritis: a case series. J Rheumatol. 2009;36(10):2277–83.
Gonzalez-Gay MA, Garcia-Porrua C, Pineiro A, Pego-Reigosa R, Llorca J, Hunder GG. Aortic aneurysm and dissection in patients with biopsy-proven giant cell arteritis from northwestern Spain: a population-based study. Medicine (Baltimore). 2004;83(6):335–41.
Blockmans D, Coudyzer W, Vanderschueren S, Stroobants S, Loeckx D, Heye S, et al. Relationship between fluorodeoxyglucose uptake in the large vessels and late aortic diameter in giant cell arteritis. Rheumatology (Oxford). 2008;47(8):1179–84.
Evans JM, O'Fallon WM, Hunder GG. Increased incidence of aortic aneurysm and dissection in giant cell (temporal) arteritis. A population-based study. Ann Intern Med. 1995;122(7):502–7.
Robson JC, Kiran A, Maskell J, Hutchings A, Arden N, Dasgupta B, et al. The relative risk of aortic aneurysm in patients with giant cell arteritis compared with the general population of the UK. Ann Rheum Dis 2013.
Garcia-Martinez A, Hernandez-Rodriguez J, Arguis P, Paredes P, Segarra M, Lozano E, et al. Development of aortic aneurysm/dilatation during the followup of patients with giant cell arteritis: a cross-sectional screening of fifty-four prospectively followed patients. Arthritis Rheum. 2008;59(3):422–30.
Garcia-Martinez A, Arguis P, Prieto-Gonzalez S, Espigol-Frigole G, Alba MA, Butjosa M, et al. Prospective long term follow-up of a cohort of patients with giant cell arteritis screened for aortic structural damage (aneurysm or dilatation). Ann Rheum Dis. 2013. doi:10.1136/annrheumdis-2013-203322. In this prospective study, 33% of GCA patients, who were apparently in clinical remission, had evidence of aortic structural damage over a median follow-up period of 10 years.
Salvarani C, Cantini F, Hunder GG. Polymyalgia rheumatica and giant-cell arteritis. Lancet. 2008;372(9634):234–45.
Bongartz T, Matteson EL. Large-vessel involvement in giant cell arteritis. Curr Opin Rheumatol. 2006;18(1):10–7.
Hiratzka LF, Bakris GL, Beckman JA, Bersin RM, Carr VF, Casey Jr DE, et al. ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the diagnosis and management of patients with thoracic aortic disease. A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology,American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons and Society for Vascular Medicine. J Am Coll Cardiol 2010. 2010;55(14):e27–e129.
Mackie SL, Hensor EM, Morgan AW, Pease CT. Should I send my patient with previous giant cell arteritis for imaging of the thoracic aorta? A systematic literature review and meta-analysis. Ann Rheum Dis. 2012. doi:10.1136/annrheumdis-2012-202145. This recent meta-analysis reported that only 5 to 10 GCA patients need to be screened to detect a previously unknown aneurysm, reinforcing the need for routine screening for aneurysms in GCA patients.
Smetana GW, Shmerling RH. Does this patient have temporal arteritis? JAMA. 2002;287(1):92–101.
Liozon E, Jauberteau-Marchan MO, Ly K, Loustaud-Ratti V, Soria P, Vidal E. Giant cell arteritis with a low erythrocyte sedimentation rate: comments on the article by Salvarani and Hunder. Arthritis Rheum. 2002;47(6):692–3.
Roche NE, Fulbright JW, Wagner AD, Hunder GG, Goronzy JJ, Weyand CM. Correlation of interleukin-6 production and disease activity in polymyalgia rheumatica and giant cell arteritis. Arthritis Rheum. 1993;36(9):1286–94.
Salvarani C, Hunder GG. Giant cell arteritis with low erythrocyte sedimentation rate: frequency of occurence in a population-based study. Arthritis Rheum. 2001;45(2):140–5.
Hazleman B. Laboratory investigations useful in the evaluation of polymyalgia rheumatica (PMR) and giant cell arteritis (GCA). Clin Exp Rheumatol. 2000;18(4 Suppl 20):S29–31.
Weyand CM, Fulbright JW, Hunder GG, Evans JM, Goronzy JJ. Treatment of giant cell arteritis: interleukin-6 as a biologic marker of disease activity. Arthritis Rheum. 2000;43(5):1041–8.
Gonzalez-Gay MA, Lopez-Diaz MJ, Barros S, Garcia-Porrua C, Sanchez-Andrade A, Paz-Carreira J, et al. Giant cell arteritis: laboratory tests at the time of diagnosis in a series of 240 patients. Medicine (Baltimore). 2005;84(5):277–90.
Cid MC, Font C, Oristrell J, de la Sierra A, Coll-Vinent B, Lopez-Soto A, et al. Association between strong inflammatory response and low risk of developing visual loss and other cranial ischemic complications in giant cell (temporal) arteritis. Arthritis Rheum. 1998;41(1):26–32.
Meisner RJ, Labropoulos N, Gasparis AP, Tassiopoulos AK. How to diagnose giant cell arteritis. Int Angiol. 2011;30(1):58–63.
Kermani TA, Schmidt J, Crowson CS, Ytterberg SR, Hunder GG, Matteson EL, et al. Utility of erythrocyte sedimentation rate and C-reactive protein for the diagnosis of giant cell arteritis. Semin Arthritis Rheum. 2012;41(6):866–71.
Dasgupta B. Concise guidance: diagnosis and management of giant cell arteritis. Clin Med. 2010;10(4):381–6.
Younge BR, Cook Jr BE, Bartley GB, Hodge DO, Hunder GG. Initiation of glucocorticoid therapy: before or after temporal artery biopsy? Mayo Clin Proc. 2004;79(4):483–91.
Achkar AA, Lie JT, Hunder GG, O'Fallon WM, Gabriel SE. How does previous corticosteroid treatment affect the biopsy findings in giant cell (temporal) arteritis? Ann Intern Med. 1994;120(12):987–92.
Lee YC, Padera RF, Noss EH, Fossel AH, Bienfang D, Liang MH, et al. Clinical course and management of a consecutive series of patients with "healed temporal arteritis". J Rheumatol. 2012;39(2):295–302.
Allison MC, Gallagher PJ. Temporal artery biopsy and corticosteroid treatment. Ann Rheum Dis. 1984;43(3):416–7.
Evans JM, Batts KP, Hunder GG. Persistent giant cell arteritis despite corticosteroid treatment. Mayo Clin Proc. 1994;69(11):1060–1.
Narvaez J, Bernad B, Roig-Vilaseca D, Garcia-Gomez C, Gomez-Vaquero C, Juanola X, et al. Influence of previous corticosteroid therapy on temporal artery biopsy yield in giant cell arteritis. Semin Arthritis Rheum. 2007;37(1):13–9.
Mari B, Monteagudo M, Bustamante E, Perez J, Casanovas A, Jordana R, et al. Analysis of temporal artery biopsies in an 18-year period at a community hospital. Eur J Intern Med. 2009;20(5):533–6.
Roth AM, Milsow L, Keltner JL. The ultimate diagnoses of patients undergoing temporal artery biopsies. Arch Ophthalmol. 1984;102(6):901–3.
Klein RG, Campbell RJ, Hunder GG, Carney JA. Skip lesions in temporal arteritis. Mayo Clin Proc. 1976;51(8):504–10.
Sorensen S, Lorenzen I. Giant-cell arteritis, temporal arteritis and polymyalgia rheumatica. A retrospective study of 63 patients. Acta Med Scand. 1977;201(3):207–13.
Hall S, Hunder GG. Is temporal artery biopsy prudent? Mayo Clin Proc. 1984;59(11):793–6.
Boyev LR, Miller NR, Green WR. Efficacy of unilateral versus bilateral temporal artery biopsies for the diagnosis of giant cell arteritis. Am J Ophthalmol. 1999;128(2):211–5.
Pless M, Rizzo III JF, Lamkin JC, Lessell S. Concordance of bilateral temporal artery biopsy in giant cell arteritis. J Neuroophthalmol. 2000;20(3):216–8.
Breuer GS, Nesher G, Nesher R. Rate of discordant findings in bilateral temporal artery biopsy to diagnose giant cell arteritis. J Rheumatol. 2009;36(4):794–6.
Hall S, Persellin S, Lie JT, O'Brien PC, Kurland LT, Hunder GG. The therapeutic impact of temporal artery biopsy. Lancet. 1983;2(8361):1217–20.
Kent III RB, Thomas L. Temporal artery biopsy. Am Surg. 1990;56(1):16–21.
Gonzalez-Gay MA, Garcia-Porrua C, Llorca J, Gonzalez-Louzao C, Rodriguez-Ledo P. Biopsy-negative giant cell arteritis: clinical spectrum and predictive factors for positive temporal artery biopsy. Semin Arthritis Rheum. 2001;30(4):249–56.
Rojo-Leyva F, Ratliff NB, Cosgrove III DM, Hoffman GS. Study of 52 patients with idiopathic aortitis from a cohort of 1,204 surgical cases. Arthritis Rheum. 2000;43(4):901–7.
Kermani TA, Warrington KJ. Recent advances in diagnostic strategies for giant cell arteritis. Curr Neurol Neurosci Rep. 2012;12(2):138–44.
Blockmans D, Bley T, Schmidt W. Imaging for large-vessel vasculitis. Curr Opin Rheumatol. 2009;21(1):19–28.
Salvarani C. Large vessel vasculitis. Clin Exp Rheumatol. 2003;21(6 Suppl 32):S133–4.
Blockmans D. The use of (18F)fluoro-deoxyglucose positron emission tomography in the assessment of large vessel vasculitis. Clin Exp Rheumatol. 2003;21(6 Suppl 32):S15–22.
Salvarani C, Macchioni PL, Tartoni PL, Rossi F, Baricchi R, Castri C, et al. Polymyalgia rheumatica and giant cell arteritis: a 5-year epidemiologic and clinical study in Reggio Emilia, Italy. Clin Exp Rheumatol. 1987;5(3):205–15.
Delecoeuillerie G, Joly P, de Cohen LA, Paolaggi JB. Polymyalgia rheumatica and temporal arteritis: a retrospective analysis of prognostic features and different corticosteroid regimens (11 year survey of 210 patients). Ann Rheum Dis. 1988;47(9):733–9.
Lundberg I, Hedfors E. Restricted dose and duration of corticosteroid treatment in patients with polymyalgia rheumatica and temporal arteritis. J Rheumatol. 1990;17(10):1340–5.
Kyle V, Hazleman BL. Treatment of polymyalgia rheumatica and giant cell arteritis. II. Relation between steroid dose and steroid associated side effects. Ann Rheum Dis. 1989;48(8):662–6.
Hunder GG, Sheps SG, Allen GL, Joyce JW. Daily and alternate-day corticosteroid regimens in treatment of giant cell arteritis: comparison in a prospective study. Ann Intern Med. 1975;82(5):613–8.
Mazlumzadeh M, Hunder GG, Easley KA, Calamia KT, Matteson EL, Griffing WL, et al. Treatment of giant cell arteritis using induction therapy with high-dose glucocorticoids: a double-blind, placebo-controlled, randomized prospective clinical trial. Arthritis Rheum. 2006;54(10):3310–8.
Hayreh SS, Zimmerman B, Kardon RH. Visual improvement with corticosteroid therapy in giant cell arteritis. Report of a large study and review of literature. Acta Ophthalmol Scand. 2002;80(4):355–67.
Macchioni P, Boiardi L, Meliconi R, Salvarani C, Grazia UM, Rossi F, et al. Elevated soluble intercellular adhesion molecule 1 in the serum of patients with polymyalgia rheumatica: influence of steroid treatment. J Rheumatol. 1994;21(10):1860–4.
Schmidt WA, Moll A, Seifert A, Schicke B, Gromnica-Ihle E, Krause A. Prognosis of large-vessel giant cell arteritis. Rheumatology (Oxford). 2008;47(9):1406–8.
Liozon E, Herrmann F, Ly K, Robert PY, Loustaud V, Soria P, et al. Risk factors for visual loss in giant cell (temporal) arteritis: a prospective study of 174 patients. Am J Med. 2001;111(3):211–7.
Nesher G, Berkun Y, Mates M, Baras M, Rubinow A, Sonnenblick M. Low-dose aspirin and prevention of cranial ischemic complications in giant cell arteritis. Arthritis Rheum. 2004;50(4):1332–7.
Lee MS, Smith SD, Galor A, Hoffman GS. Antiplatelet and anticoagulant therapy in patients with giant cell arteritis. Arthritis Rheum. 2006;54(10):3306–9.
Salvarani C, Della BC, Cimino L, Macchioni P, Formisano D, Bajocchi G, et al. Risk factors for severe cranial ischaemic events in an Italian population-based cohort of patients with giant cell arteritis. Rheumatology (Oxford). 2009;48(3):250–3.
Berger CT, Wolbers M, Meyer P, Daikeler T, Hess C. High incidence of severe ischaemic complications in patients with giant cell arteritis irrespective of platelet count and size, and platelet inhibition. Rheumatology (Oxford). 2009;48(3):258–61.
Hoffman GS, Cid MC, Hellmann DB, Guillevin L, Stone JH, Schousboe J, et al. A multicenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum. 2002;46(5):1309–18.
Jover JA, Hernandez-Garcia C, Morado IC, Vargas E, Banares A, Fernandez-Gutierrez B. Combined treatment of giant-cell arteritis with methotrexate and prednisone. a randomized, double-blind, placebo-controlled trial. Ann Intern Med. 2001;134(2):106–14.
Spiera RF, Mitnick HJ, Kupersmith M, Richmond M, Spiera H, Peterson MG, et al. A prospective, double-blind, randomized, placebo controlled trial of methotrexate in the treatment of giant cell arteritis (GCA). Clin Exp Rheumatol. 2001;19(5):495–501.
Mahr AD, Jover JA, Spiera RF, Hernandez-Garcia C, Fernandez-Gutierrez B, Lavalley MP, et al. Adjunctive methotrexate for treatment of giant cell arteritis: an individual patient data meta-analysis. Arthritis Rheum. 2007;56(8):2789–97.
Unizony S, Arias-Urdaneta L, Miloslavsky E, Arvikar S, Khosroshahi A, Keroack B, et al. Tocilizumab for the treatment of large-vessel vasculitis (giant cell arteritis, Takayasu arteritis) and polymyalgia rheumatica. Arthritis Care Res. 2012;64(11):1720–9. Largest published case series of tocilizumab therapy in large-vessel vasculitis.
Seitz M, Reichenbach S, Bonel HM, Adler S, Wermelinger F, Villiger PM. Rapid induction of remission in large vessel vasculitis by IL-6 blockade. A case series. Swiss Med Wkly. 2011;141:w13156.
Beyer C, Axmann R, Sahinbegovic E, Distler JH, Manger B, Schett G, et al. Anti-interleukin 6 receptor therapy as rescue treatment for giant cell arteritis. Ann Rheum Dis. 2011;70(10):1874–5.
Salvarani C, Magnani L, Catanoso M, Pipitone N, Versari A, Dardani L, et al. Tocilizumab: a novel therapy for patients with large-vessel vasculitis. Rheumatology (Oxford). 2012;51(1):151–6.
Sciascia S, Rossi D, Roccatello D. Interleukin 6 blockade as steroid-sparing treatment for 2 patients with giant cell arteritis. J Rheumatol. 2011;38(9):2080–1.
Quartuccio L, Maset M, De MG, Pontarini E, Fabris M, Mansutti E, et al. Role of oral cyclophosphamide in the treatment of giant cell arteritis. Rheumatology (Oxford). 2012;51(9):1677–86.
de Boysson H, Boutemy J, Creveuil C, Ollivier Y, Letellier P, Pagnoux C, et al. Is there a place for cyclophosphamide in the treatment of giant-cell arteritis? A case series and systematic review. Semin Arthritis Rheum. 2013;43(1):105–12.
Henes JC, Mueller M, Pfannenberg C, Kanz L, Koetter I. Cyclophosphamide for large vessel vasculitis: assessment of response by PET/CT. Clin Exp Rheumatol. 2011;29(1 Suppl 64):S43–8.
Adizie T, Christidis D, Dharmapaliah C, Borg F, Dasgupta B. Efficacy and tolerability of leflunomide in difficult-to-treat polymyalgia rheumatica and giant cell arteritis: a case series. Int J Clin Pract. 2012;66(9):906–9. In this case series, leflunomide effectively controlled 22 out of 23 GCA patients who had inadequate response to glucocorticoids.
Diamantopoulos AP, Hetland H, Myklebust G. Leflunomide as a corticosteroid-sparing agent in giant cell arteritis and polymyalgia rheumatica: a case series. Biomed Res Int. 2013;2013:120638.
Hoffman GS, Cid MC, Rendt-Zagar KE, Merkel PA, Weyand CM, Stone JH, et al. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med. 2007;146(9):621–30.
Martinez-Taboada VM, Rodriguez-Valverde V, Carreno L, Lopez-Longo J, Figueroa M, Belzunegui J, et al. A double-blind placebo controlled trial of etanercept in patients with giant cell arteritis and corticosteroid side effects. Ann Rheum Dis. 2008;67(5):625–30.
Seror R, Baron G, Hachulla E, Debandt M, Larroche C, Puechal X, et al. Adalimumab for steroid sparing in patients with giant-cell arteritis: results of a multicentre randomised controlled trial. Ann Rheum Dis 2013.
Do DD, Jeanneret C, Mahler F. Images in vascular medicine. Giant cell arteritis of axillary artery. Vasc Med. 1996;1(4):293–4.
Monte R, Gonzalez-Gay MA, Garcia-Porrua C, Lopez-Alvarez MJ, Pulpeiro JR. Successful response to angioplasty in a patient with upper limb ischaemia secondary to giant cell arteritis. Br J Rheumatol. 1998;37(3):344.
Dellaripa PF, Eisenhauer AC. Bilateral percutaneous balloon angioplasty of the axillary arteries in a patient with giant cell arteritis and upper extremity ischemic symptoms not responsive to corticosteroids. J Rheumatol. 1998;25(7):1429–33.
Amann-Vesti BR, Koppensteiner R, Rainoni L, Pfamatter T, Schneider E. Immediate and long-term outcome of upper extremity balloon angioplasty in giant cell arteritis. J Endovasc Ther. 2003;10(2):371–5.
Both M, Aries PM, Muller-Hulsbeck S, Jahnke T, Schafer PJ, Gross WL, et al. Balloon angioplasty of arteries of the upper extremities in patients with extracranial giant-cell arteritis. Ann Rheum Dis. 2006;65(9):1124–30.
Evans JM, Bowles CA, Bjornsson J, Mullany CJ, Hunder GG. Thoracic aortic aneurysm and rupture in giant cell arteritis. A descriptive study of 41 cases. Arthritis Rheum. 1994;37(10):1539–47.
Acknowledgments
The authors are grateful to Dr. Heather L. Gornik, MD, Medical Director, Non-invasive Vascular Laboratory, Department of Vascular Medicine, Heart and Vascular Institute, Cleveland Clinic, for providing the ultrasound Doppler images (Fig. 7).
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Soumya Chatterjee declares that he has no conflict of interest.
Scott D. Flamm has been a consultant for Bayer Healthcare on the Cardiac Imaging Advisory Board.
Carmela D. Tan declares that she has no conflict of interest.
E. Rene Rodriguez declares that he has no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Peripheral Vascular Disease
Rights and permissions
About this article

Cite this article
Chatterjee, S., Flamm, S.D., Tan, C.D. et al. Clinical Diagnosis and Management of Large Vessel Vasculitis: Giant Cell Arteritis. Curr Cardiol Rep 16, 498 (2014). https://doi.org/10.1007/s11886-014-0498-z
Published:
DOI: https://doi.org/10.1007/s11886-014-0498-z
Keywords
- Large vessel vasculitis
- Giant cell arteritis (GCA)
- Cranial (temporal) arteritis
- Anterior ischemic optic neuropathy
- Amaurosis fugax
- Claudication
- Stenosis
- Occlusion
- Ectasia
- Aneurysm
- Dissection
- Isolated idiopathic aortitis
- Acute phase reactant
- ESR
- CRP
- Interleukin-6
- Computerized tomographic angiogram (CTA)
- Magnetic resonance angiogram (MRA)
- Fluorodeoxyglucose(18F)-positron emission tomography (FDG-PET)
- Vascular Doppler ultrasound
- Glucocorticoid
- Prednisone
- Methylprednisolone
- Methotrexate
- Azathioprine
- Leflunomide
- Cyclophosphamide
- Tocilizumab
- Rituximab
- TNF inhibitor
- Angioplasty
- Stent
- Bypass