
Abstract
In patients with diabetes mellitus, the incidence of cardiovascular disease is increased, and the outcome following cardiovascular events is worse. The antihyperglycemic drug metformin appears to limit cardiovascular death in patients with type 2 diabetes. Indeed, preclinical studies have demonstrated that metformin limits (myocardial) ischemia and reperfusion injury, independent from its glucose-lowering effect. This cardioprotection is mediated by activation of the Reperfusion Injury Salvage Kinase (RISK) pathway, activation of AMPK and by an increased formation of adenosine. In addition, metformin can modulate several cardiovascular risk factors and reduces the development of heart failure in murine models. Consequently, treatment with metformin might potentially improve cardiovascular outcome in patients at risk for myocardial ischemia, even if these patients do not have diabetes. In the current paper, we focus on the direct cardioprotective actions of metformin and the mechanisms that underlie these effects.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Coronary artery disease (CAD) is the leading cause of morbidity and mortality worldwide, accounting for nearly one third of all deaths [1]. In patients with diabetes mellitus, cardiovascular morbidity and mortality is even more pronounced. In part, this is explained by an accelerated development of atherosclerosis [2]. In addition, when suffering a myocardial infarction, the outcome is worse [3–6], with success rates of angioplasty and coronary artery bypass grafting also being lower [7–9]. Patients with diabetes also have a high prevalence of congestive heart failure secondary to diabetic changes in the myocardium. This unique clinical entity, which is characterized by fibrotic changes in the myocardium and functional alterations in diastolic relaxation and ventricular compliance, is referred to as ‘diabetic cardiomyopathy’ [10]. The pathological changes occurring in diabetic cardiomyopathy include increased reactive oxygen species (ROS) [11], abnormal handling of calcium and increased release of inflammatory mediators [12]. Although in part related to myocardial infarction, this type of heart failure in diabetics can occur independent from cardiovascular risk factors and ischemia [12]. The prevalence of patients with diabetes and heart failure is rapidly increasing, and these patients have a worse outcome than heart failure patients without diabetes [13•]. These observations, alongside the evidence that diabetic patients have a worse prognosis following myocardial infarction, suggest that the diabetic heart is more vulnerable to ischemic damage.
Therefore, novel strategies to prevent the development of atherosclerosis, and to increase the intrinsic tolerance of the myocardial cells to ischemia and reperfusion are urgently needed. In accordance with current guidelines, establishing early reperfusion is the primary therapy in patients with a myocardial infarction [14]. The beneficial effect of re-establishing coronary blood flow, however, is limited by a phenomenon called ‘reperfusion injury’ [14]. The process of reperfusion triggers various pathways, including oxidative stress, pH changes, calcium overload, an acute inflammatory response and metabolic changes, which cause tissue injury themselves and contribute to the final myocardial infarct size. Many of these factors promote the opening of the mitochondrial permeability transition pore (mPTP), a critical determinant of cell death in the setting of acute ischemia reperfusion (IR)-injury [1, 15]. To further optimize the results of coronary angioplasty and coronary artery bypass grafting, strategies to limit IR-injury are needed. In the past two decades, both pharmacological and non-pharmacological interventions have been developed that can limit or prevent acute myocardial IR-injury.
As a nonpharmacological cardioprotective strategy, ischemic preconditioning (IPC), has received much attention for its potent infarct size-limiting effect since it was first reported by Murry et al. in 1986 [16]. Murry demonstrated that short intermittent episodes of ischemia prior to a prolonged ischemic event potently limit the final infarct size. Over the years, a tremendous amount of effort has been done to mimic the cardioprotective effect of IPC with pharmacological agents [17, 18]. Interestingly, several large studies have suggested that the glucose lowering drug metformin might have such a cardioprotective effect, independent of its glucose lowering effect [13•]. In this article, we aim to summarize these cardioprotective effects of metformin and the underlying mechanisms.
The Effect of Metformin on Cardiovascular Outcome in Patients With Type 2 Diabetes
Metformin, a biguanide glucose-lowering agent, is the first-line oral treatment option for patients with type 2 diabetes mellitus [19]. Metformin lowers plasma glucose levels by reducing insulin resistance, particularly in the liver and skeletal muscle cells. It supresses hepatic gluconeogenesis, increases insulin sensitivity, enhances peripheral glucose utilization and has beneficial effects on lipid metabolism. The mechanisms underlying these effects have yet to be fully elucidated, although recent data have implicated AMP-activated protein kinase (AMPK) activation as an important mediator [20, 21]. One of the most significant breakthroughs in the understanding of the cellular mechanism of metformin was made in the early 2000s by two independent research groups reporting that metformin induces specific inhibition of complex 1 of the mitochondrial respiratory chain [22, 23]. It has been hypothesized that metformin activates AMPK by inhibiting complex 1 of the mitochondrial respiratory chain, resulting in a fall in the intracellular adenosine triphosphate (ATP) concentration and an increase in the adenosine monophosphate (AMP) to ATP ratio. In turn, AMPK activation leads to enhanced glucose uptake in skeletal muscle, stimulates oxidation of free fatty acids and inhibits glucose production by hepatocytes [24]. The AMPK hypothesis, however, has recently been challenged by Foretz et al. [25]. In their study, they demonstrated that administration of metformin in mice suppressed hepatic gluconeogenesis directly, by a decrease in hepatic energy state independent from AMPK activation.
Several large observational studies in patients with type 2 diabetes have reported that treatment with metformin limits cardiovascular morbidity and mortality independent from its glucose-lowering action [26•]. In the United Kingdom, Prospective Diabetes Study (UKPDS) treatment of diabetic patients with metformin was associated with lower mortality rates compared to treatment with sulphonylurea derivatives (SUDs) [27]. The study showed that patients with type 2 diabetes treated with metformin had a 36 % lower all-cause mortality and a 39 % lower risk of myocardial infarction compared with conventional treatment. This risk reduction was greater for metformin than for treatment with insulin or SUDs, despite similar glycaemic control. Also in patients with diabetes who have suffered from a myocardial infarction in the past, treatment with metformin is associated with a lower mortality rate than treatment with SUDs [28, 29].
The reduction in cardiovascular mortality by metformin was confirmed by a systematic review and a recently published Cochrane analysis [30, 31]. Selvin et al. concluded that treatment with metformin of overweight diabetic patients was associated with a decreased risk of cardiovascular mortality (pooled OR 0.74; 95 % CI, 0.62–0.89) compared with any other oral antidiabetic agent or placebo. The results for cardiovascular morbidity and all-cause mortality showed a similar trend, but this was not statistically significant. It needs to be mentioned, however, that this analysis was restricted to the overweight patients.
A more recent meta-analysis was performed by Lamanna et al., in which they studied 35 clinical trials including 18,472 participants treated with metformin or comparator [32]. In this meta-analysis, the overweight group and the non-overweight group were combined. In addition to patients with diabetes, they included studies on non-diabetic patients with HIV or polycystic ovary syndrome. In an overall analysis, metformin therapy, when compared to an active comparator, did not produce any significant effect on the incidence of cardiovascular events. Monotherapy with metformin, however, induced a trend towards improved overall mortality, which was abolished when metformin was administered in addition to sulphonylurea derivatives.
The contradicting results of the recent meta-analysis, and the striking beneficial effect of metformin in the UKPDS study, might be related to the fact that the UKPDS was performed in a much earlier time period where the treatment of other cardiovascular risk factors and the treatment of acute coronary events was inferior to the treatment in the present era. Therefore, in the current era, establishing a beneficial cardioprotective effect of metformin on top of its glucose-lowering effect is a greater challenge.
Laboratory studies, animal studies, and studies in healthy volunteers and patients with diabetes have provided evidence that metformin modulates several risk factors for atherosclerosis, increases the intrinsic tolerance of the myocardium to ischemia and reperfusion, and attenuates the subsequent development of heart failure. We will consecutively discuss these studies and focus on the direct cardioprotective effects of metformin.
Effect of Metformin on Other Cardiovascular Risk Factors
In addition to its glucose lowering effect, metformin therapy also has beneficial effects on other cardiovascular risk factors. Patients with diabetes exhibit an atherogenic lipid profile characterized by hypertriglyceridemia, decreased levels of high-density lipoprotein (HDL) cholesterol and elevated levels of small, dense atherogenic low-density lipoprotein (LDL) cholesterol particles [33, 34]. Free fatty acid levels are increased, and consequently, hepatic production of very-low-density lipoprotein (VLDL) is increased and clearance of VLDL particles is reduced [35, 36]. Metformin seems to improve the lipid profile by lowering triglyceride levels, total cholesterol and LDL cholesterol levels, while maintaining or increasing HDL levels [37–42]. By lowering glucose levels, metformin also seems to reduce oxidative stress and lipid oxidation [43, 44].
An increase in bodyweight is also associated with an increased risk for cardiovascular disease (CDV) and is deemed an independent risk factor for CVD by the Framingham Heart Study [45]. Metformin reduces bodyweight by enhancing carbohydrate utilization in the gastrointestinal tract, adverse gastrointestinal side effect, carbohydrate malabsorption, and/or through associated anorexia [37].
Another suggested beneficial effect of metformin is its ability to lower blood pressure. Even a small elevation in blood pressure significantly increases death from cardiovascular disease and risk for myocardial infarction, stroke and congestive heart failure in patients with diabetes [46]. Therefore, even a minimal reduction in blood pressure during treatment with metformin may contribute to a significant decrease in diabetes related morbidity and mortality. Several studies indicate an antihypertensive effect of metformin in animals and humans [38, 47–52]. Data regarding the effect of metformin on blood pressure however, are not entirely consistent. In a study by Nagi et al., metformin had no effect on blood pressure in patients with diabetes, and a recent study by He et al. showed the same results for non-diabetics [53, 54].
Platelet aggregation plays an important role in the pathophysiology of myocardial infarction. Metformin has been shown to reverse platelet hyperaggregation, but this effect is more pronounced in animal experimental models than in humans [55]. Gin et al. was able to demonstrate that in patients with diabetes mellitus treated with insulin, administration of metformin decreased the maximum platelet aggregation induced by adenosine diphosphate (ADP) in vitro [56]. Another study, however, failed to demonstrate any effect on platelet aggregation or platelet aggregation induced by ADP [53].
Another risk factor for cardiovascular arterial thrombotic events is an elevated fibrinogen level [57]. In patients with insulin resistance, fibrinolysis is reduced, which seems correlated to increased levels of plasminogen activator inhibitor (PAI-)1 [58]. Metformin appears to reduce PAI-1 levels in patients with and without diabetes, hereby reducing the risk of developing cloth formation [59–61].
Endothelial dysfunction, as characterized by an impairment in endothelium dependent relaxation and reduced nitric oxide (NO) bioavailability, is an early characteristic of atherogenesis. Studies have shown that treatment with metformin improves endothelium-dependent vasodilation by increased availability of NO [62].
All of the abovementioned beneficial effects of metformin on cardiovascular risk factors could potentially lead to slowing down the process of atherosclerosis, and therefore decrease the incidence of cardiovascular events.
Direct Cardiovascular Effects of Metformin
Effect of Metformin on Ischemia Reperfusion Injury
The administration of metformin, either before the ischemic stimulus or at the moment of coronary reperfusion, profoundly reduces infarct size in murine models of myocardial infarction. Already in 1988, Charlon et al. showed that oral administration of metformin could reduce infarct size in rats [63]. Solskov et al. also showed in a rat isolated perfused heart model that the administration of a single dose of metformin 24 hours before transient coronary artery occlusion reduced infarct size [64]. In subsequent studies in isolated rat hearts, coronary perfusion with metformin during the first 15 min of reperfusion reduced infarct size with approximately 40–50 % [65–67]. This was also confirmed in in-vivo studies in rats and mice [66, 68]. Recently, several intracellular mechanisms that mediate this cardioprotective effect have been discovered.
Reperfusion Injury Salvage Kinase (RISK) Pathway
Opening of the mitochondrial permeability transition pore (mPTP) during (early) reperfusion is a pivotal mechanism of ischemia-reperfusion (IR)-injury. This nonselective pore in the mitochondrial inner membrane is closed during ischemia. Immediately after reperfusion, the mPTP opens, which will induce ATP depletion and release of pro-apoptotic factors, including cytochrome C, ultimately leading to necrosis and apoptosis [69, 70]. It has been demonstrated that the administration of metformin at reperfusion leads to activation of several kinases of the Reperfusion Injury Salvage Kinase (RISK) pathway, including phosphatidylinositol-3-kinase (PI3K) and Akt, which in turn can prevent opening of the mPTP.
In an isolated rat heart model, Bhamra et al. found that the administration of metformin during the first 15 min of reperfusion reduced infarct size in nondiabetic Wistar rats and type 2 diabetic Goto-Kakizaki rats [65]. Metformin also induced a significant increase in Akt phosphorylation after reperfusion. Concomitant administration of LY294002, a PI3K inhibitor, prevented Akt phosphorylation and abolished the protective effect on infarct size. In isolated rat cardiomyocytes, incubation with metformin prevented mPTP opening by activation of the PI3K/Akt pathway. The authors concluded that the protective effect of metformin against myocardial IR-injury is PI3K dependent, and that the effect of metformin on Akt phosphorylation is similar in both nondiabetic and diabetic rats.
AMPK
In addition to the RISK pathway, the cardioprotective effect of metformin is also critically dependent on activation of AMPK. AMPK is a major regulator of energy balance in the cell. Environmental stress including exercise, starvation, inflammation and hypoxia increases AMPK activity. AMPK also fulfils a crucial role in preserving myocardial viability during myocardial infarction. By increasing ATP synthesis and lowering ATP utilization, AMPK functions to maintain normal cellular energy stores during ischemia [24]. Calvert et al. showed that the protective effect of metformin is AMPK dependent [68]. Immediately after the onset of myocardial ischemia, phosphorylation of AMPK occurs, and remains active for more than 24 hours following reperfusion in a murine model of coronary artery occlusion. Pretreatment with a very low dose of metformin increased the phosphorylation of AMPK in hearts not exposed to ischemia, as well as in hearts exposed to ischemia reperfusion. Metformin failed to provide protection in cardiac-specific AMPKα2 dominate-negative transgenic mice, demonstrating that AMPK activation is essential for metformin-induced protection. In an additional experiment, it was shown that metformin increased eNOS phosphorylation via activation of AMPK and that eNOS activation was also indispensible for its cardioprotective action [68]. Yellon’s group also studied the involvement of AMPK in metformin-induced cardioprotection [67]. In a rat isolated heart model, metformin was added for 15 min at the onset of reperfusion alone or with the AMPK inhibitor Compound C. Metformin significantly reduced infarct-size, which was completely abolished by adding Compound C simultaneously. Interestingly, delayed administration of Compound C after 5 min of reperfusion no longer blocked the protective effect of metformin. The authors concluded that the cardioprotective effect of metformin was mediated by AMPK activation very early during reperfusion.
Adenosine
Most recently, the endogenous nucleoside adenosine has been shown to be involved in metformin-induced cardioprotection. Metformin induces phosphorylation of AMPK, which is mediated, at least in part, by an increase in the cytosolic AMP concentration [71]. The concentration of free AMP in the cytosol is also a major determinant of the intracellular formation of adenosine. It is well known that adenosine receptor stimulation limits infarct size by activation of the RISK pathway [17, 72]. Therefore, Paiva et al. hypothesized that adenosine receptor stimulation contributes to the infarct size limiting effect of metformin [66]. In isolated perfused nondiabetic rat hearts, perfusion with metformin during the first 15 min of reperfusion limited infarct size. Both 8-p-sulfophenyltheophylline (a nonspecific adenosine receptor blocker) and nitrobenzylthioinosine (transmembranous adenosine transport inhibitor) completely prevented the protective effect, suggesting that intracellular formation of adenosine, and subsequent adenosine receptor stimulation mediates the protective effect of metformin.
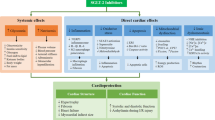
In conclusion, metformin potently reduces myocardial infarct size by activation of multiple, and probably parallel, intracellular pathways. These pathways include activation of the RISK pathway, activation of AMPK and eNOS and increased adenosine receptor stimulation, resulting in the prevention of MPTP opening at reperfusion. These signaling pathways are illustrated in Fig. 1.

Inhibition of complex 1 increases the AMP concentration. This activates AMPK and increases the formation of adenosine. AMPK activation increases tolerance against ischaemia and reperfusion and, by phosphorylation of eNOS, prevents MPTP opening at reperfusion. In addition, increased adenosine receptor stimulation activates the PI3K/Akt pathway, which contributes to eNOS activation (Reproduced with permission from El Messaoudi et al., Curr Opin Lipidol. 2011; 22:445–453)
Effect of Metformin on Cardiac Remodeling and Heart Failure
In patients suffering a myocardial infarction, the prognosis is determined not only by the final infarct size, but also by long-term changes that occur in the myocardium following an infarction. Following IR-injury, but also in patients suffering from volume overload (e.g. due to valvular disease), pressure overload (e.g. due to hypertension) and diabetic cardiomyopathy, a complex cascade of events occurs in the myocardial cells, including inflammation and fibrosis, ultimately leading to changes in size, shape and function of the heart, a process which has collectively been termed ‘remodeling’ [73, 74]. In time, this cardiac remodeling will lead to clinically overt heart failure in a significant portion of the patients. Several experimental animal studies showed that metformin, in addition to limiting IR-injury, can also limit the process of cardiac remodeling and the development of heart failure [75].
Gundewar et al. have shown in a series of experiments that metformin significantly improves left ventricular function and survival in a murine model of heart failure [76•]. The administration of a single dose of metformin at reperfusion following 60 min of coronary artery occlusion reduced infarct size, but did not prevent the development of severe cardiomyopathy. In contrast, when metformin was administered daily for a period of 4 weeks, the development of cardiac hypertrophy was limited and heart function was preserved. In a model of permanent coronary artery ligation, treatment with metformin did not reduce infarct size and did not affect ejection fraction, although this treatment increased overall survival.
In their rat model of permanent coronary artery ligation, Yin et al. have recently shown that the long term administration of metformin preserves left ventricular function [77]. In this study, animals were randomly allocated to the treatment with normal water or metformin-containing water (250 mg/kg/day). In the rats that received permanent coronary artery ligation, infarct size was significantly smaller after 12 weeks of metformin treatment compared to the control group. Moreover, metformin treatment resulted in less left ventricular dilatation and preservation of left ventricular ejection fraction compared with the control group. In addition, in a dog model of cardiac pacing-induced heart failure and in a mouse model of heart failure due to thoracic aortic constriction, the chronic administration of metformin attenuated the hemodynamic and structural changes that were characteristic of the development of heart failure [78, 79]. In contrast, metformin did not prevent heart failure development in a rat model of volume overload-induced heart failure [80].
Probably, several mechanisms contribute to this protective effect. Metformin is thought to improve myocardial mitochondrial respiration, and ATP synthesis by an underlying mechanism requiring the activation of AMPK and its downstream mediators, eNOS and peroxisome proliferator-activated receptor-γ coactivator-(PGC) 1α [76•]. Also, metformin seems to reduce collagen expression that occurs after coronary artery ligation [77]. Yin et al. also suggested that the beneficial effect of metformin on post-myocardial infarction remodeling could be attributed to a decrease in plasma insulin concentration, given the observation that hyperinsulinemia is associated with exacerbation of cardiac remodeling [77, 81].
Alternative Glucose-Lowering Drugs and Cardioprotection
Interestingly, metformin is not the only glucose-lowering agent that can modulate (myocardial) IR-injury. Sulphonylurea derivatives (SUDs) do not induce cardioprotection, but have been shown to prevent the cardioprotective effect of ischemic preconditioning and pharmacological preconditioning, most likely due to prevention of opening of the mitochondrial ATP-sensitive K+ channel [28, 29, 82–84]. This effect of SUDs might contribute to the findings that patients who are treated with a combination of SUD and metformin appeared to have a worse outcome on mortality, as compared with those who are treated with metformin alone [32]. A recent national cohort study of veterans initiating oral treatment for diabetes mellitus found that sulfonylurea use was associated with an increased hazard of acute myocardial infarction, stroke, or death compared with metformin use. The findings do not clarify whether the difference in cardiovascular disease risk is due to harm from sulfonylureas, benefit from metformin, or both [27]. Recent comparative effectiveness reviews and meta-analyses concluded that metformin was associated with a slightly lower risk for all-cause mortality compared with sulfonylureas, but results were inconsistent and imprecise [30, 85, 86]. This study provides further evidence of a risk difference in cardiovascular outcomes for sulfonylurea and metformin users. Insulin, on the other hand, profoundly protects the myocardium from IR-injury via activation of the RISK pathway [87]. Similarly, the novel antihyperglycemic glucagon like-peptide-1 ( GLP-1) analogues and the dipeptidylpeptidase-4 (DPP-4) inhibitors have also been shown to exert direct cardioprotective effects in murine models of myocardial infarction. Very recently, the GLP-1 analogue exenatide was tested in patients suffering an acute ST-segment elevation myocardial infarction [88]. It appeared that a six-hours intravenous administration of exenatide, commencing immediately before reperfusion, significantly reduced infarct size, measured with cardiac magnetic resonance. Since this is outside the scope of our review, we would like to refer the reader to contemporary reviews on these alternative glucose-lowering drugs [89–91].
Conclusion
Animal studies have provided consistent evidence that metformin can limit myocardial ischemia reperfusion injury and infarct size. Metformin therapy also seems to attenuate postinfarction cardiac remodeling. In addition, metformin can modulate several risk factors for atherosclerosis, independent of glucose control, although these findings are less consistent. The most likely underlying mechanisms of cardioprotection are activation of the RISK-pathway, activation of AMPK, and increased adenosine receptor stimulation. Currently, several randomized clinical trials are being performed to explore the cardioprotective effect of metformin in nondiabetic patients suffering a myocardial infarction [92] and in nondiabetic patients undergoing cardiac surgery (ClinicalTrials.gov registration number NCT01439723). Metformin is cheap, and its long-term safety has been well established. Therefore, a positive result from these clinical studies could result in a quick implementation of metformin in clinical management to prevent and treat cardiovascular events, also in patients without diabetes mellitus.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Miki T, Itoh T, Sunaga D, Miura T. Effects of diabetes on myocardial infarct size and cardioprotection by preconditioning and postconditioning. Cardiovasc Diabetol. 2012;11:67.
Nicholls SJ, Tuzcu EM, Kalidindi S, Wolski K, Moon KW, Sipahi I, et al. Effect of diabetes on progression of coronary atherosclerosis and arterial remodeling: a pooled analysis of 5 intravascular ultrasound trials. J Am Coll Cardiol. 2008;52:255–62.
Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998;339:229–34.
Malmberg K, Yusuf S, Gerstein HC, Brown J, Zhao F, Hunt D, et al. Impact of diabetes on long-term prognosis in patients with unstable angina and non-q-wave myocardial infarction: Results of the oasis (organization to assess strategies for ischemic syndromes) registry. Circulation. 2000;102:1014–9.
Norhammar A, Lindback J, Ryden L, Wallentin L, Stenestrand U. Improved but still high short- and long-term mortality rates after myocardial infarction in patients with diabetes mellitus: A time-trend report from the swedish register of information and knowledge about swedish heart intensive care admission. Heart. 2007;93:1577–83.
Jonas M, Reicher-Reiss H, Boyko V, Behar S, Grossman E. Hospital and 1-year outcome after acute myocardial infarction in patients with diabetes mellitus and hypertension. J Hum Hypertens. 2003;17:665–70.
Mathew V, Gersh BJ, Williams BA, Laskey WK, Willerson JT, Tilbury RT, et al. Outcomes in patients with diabetes mellitus undergoing percutaneous coronary intervention in the current era: a report from the prevention of restenosis with tranilast and its outcomes (presto) trial. Circulation. 2004;109:476–80.
Alserius T, Hammar N, Nordqvist T, Ivert T. Risk of death or acute myocardial infarction 10 years after coronary artery bypass surgery in relation to type of diabetes. Am Heart J. 2006;152:599–605.
Calafiore AM, Di Mauro M, Di Giammarco G, Contini M, Vitolla G, Iaco AL, et al. Effect of diabetes on early and late survival after isolated first coronary bypass surgery in multivessel disease. J Thorac Cardiovasc Surg. 2003;125:144–54.
Hayat SA, Patel B, Khattar RS, Malik RA. Diabetic cardiomyopathy: mechanisms, diagnosis and treatment. Clin Sci (Lond). 2004;107:539–57.
Mehta JL, Rasouli N, Sinha AK, Molavi B. Oxidative stress in diabetes: a mechanistic overview of its effects on atherogenesis and myocardial dysfunction. Int J Biochem Cell Biol. 2006;38:794–803.
Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115:3213–23.
• Aguilar D, Chan W, Bozkurt B, Ramasubbu K, Deswal A. Metformin use and mortality in ambulatory patients with diabetes and heart failure. Circ Heart Fail. 2011;4:53–8. In this cohort study in patients with diabetes and heart failure, metformin therapy was associated with a lower mortality rate.
Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–35.
Ansley DM, Wang B. Oxidative stress and myocardial injury in the diabetic heart. J Pathol. 2012.
Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–36.
Riksen NP, Smits P, Rongen GA. Ischaemic preconditioning: from molecular characterisation to clinical application—part i. Neth J Med. 2004;62:353–63.
Riksen NP, Smits P, Rongen GA. Ischaemic preconditioning: from molecular characterisation to clinical application–part ii. Neth J Med. 2004;62:409–23.
Qaseem A, Humphrey LL, Sweet DE, Starkey M, Shekelle P. Oral pharmacologic treatment of type 2 diabetes mellitus: a clinical practice guideline from the american college of physicians. Ann Intern Med. 2012;156:218–31.
Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond). 2012;122:253–70.
Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of amp-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–74.
El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex i. J Biol Chem. 2000;275:223–8.
Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348(Pt 3):607–14.
Boyle JG, Salt IP, McKay GA. Metformin action on amp-activated protein kinase: a translational research approach to understanding a potential new therapeutic target. Diabet Med. 2010;27:1097–106.
Foretz M, Hebrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the lkb1/ampk pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120:2355–69.
• Roussel R, Travert F, Pasquet B, Wilson PW, Smith Jr SC, Goto S, et al. Metformin use and mortality among patients with diabetes and atherothrombosis. Arch Intern Med. 2010;170:1892–9. In this large retrospective cohort study in patients with diabetes and established cardiovascular disease, the use of metformin was associated with a reduced mortality rate, also in patients with heart failure.
Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (ukpds 34). Uk prospective diabetes study (ukpds) group. Lancet. 1998;352:854–865
Schramm TK, Gislason GH, Vaag A, Rasmussen JN, Folke F, Hansen ML, et al. Mortality and cardiovascular risk associated with different insulin secretagogues compared with metformin in type 2 diabetes, with or without a previous myocardial infarction: a nationwide study. Eur Heart J. 2011;32:1900–8.
Jorgensen CH, Gislason GH, Andersson C, Ahlehoff O, Charlot M, Schramm TK, et al. Effects of oral glucose-lowering drugs on long term outcomes in patients with diabetes mellitus following myocardial infarction not treated with emergent percutaneous coronary intervention—a retrospective nationwide cohort study. Cardiovasc Diabetol. 2010;9:54.
Selvin E, Bolen S, Yeh HC, Wiley C, Wilson LM, Marinopoulos SS, et al. Cardiovascular outcomes in trials of oral diabetes medications: a systematic review. Arch Intern Med. 2008;168:2070–80.
Saenz A, Fernandez-Esteban I, Mataix A, Ausejo M, Roque M, Moher D. Metformin monotherapy for type 2 diabetes mellitus. Cochrane Database Syst Rev. 2005:CD002966.
Lamanna C, Monami M, Marchionni N, Mannucci E. Effect of metformin on cardiovascular events and mortality: a meta-analysis of randomized clinical trials. Diabetes Obes Metab. 2011;13:221–8.
Ginsberg HN. Review: efficacy and mechanisms of action of statins in the treatment of diabetic dyslipidemia. J Clin Endocrinol Metab. 2006;91:383–92.
Betteridge DJ. Long-term risk reduction: who needs treatment? Diabetes Res Clin Pract. 2005;68 Suppl 2:S15–22.
Karam JH. Type ii diabetes and syndrome x. Pathogenesis and glycemic management. Endocrinol Metab Clin North Am. 1992;21:329–50.
Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–607.
Bailey CJ, Turner RC. Metformin. N Engl J Med. 1996;334:574–9.
Giugliano D, De Rosa N, Di Maro G, Marfella R, Acampora R, Buoninconti R, et al. Metformin improves glucose, lipid metabolism, and reduces blood pressure in hypertensive, obese women. Diabetes Care. 1993;16:1387–90.
Hollenbeck CB, Johnston P, Varasteh BB, Chen YD, Reaven GM. Effects of metformin on glucose, insulin and lipid metabolism in patients with mild hypertriglyceridaemia and non-insulin dependent diabetes by glucose tolerance test criteria. Diabete Metab. 1991;17:483–9.
Jeppesen J, Zhou MY, Chen YD, Reaven GM. Effect of metformin on postprandial lipemia in patients with fairly to poorly controlled niddm. Diabetes Care. 1994;17:1093–9.
Reaven GM, Johnston P, Hollenbeck CB, Skowronski R, Zhang JC, Goldfine ID, et al. Combined metformin-sulfonylurea treatment of patients with noninsulin-dependent diabetes in fair to poor glycemic control. J Clin Endocrinol Metab. 1992;74:1020–6.
Robinson AC, Burke J, Robinson S, Johnston DG, Elkeles RS. The effects of metformin on glycemic control and serum lipids in insulin-treated niddm patients with suboptimal metabolic control. Diabetes Care. 1998;21:701–5.
Tessier D, Maheux P, Khalil A, Fulop T. Effects of gliclazide versus metformin on the clinical profile and lipid peroxidation markers in type 2 diabetes. Metabolism. 1999;48:897–903.
Ghatak SB, Dhamecha PS, Bhadada SV, Panchal SJ. Investigation of the potential effects of metformin on atherothrombotic risk factors in hyperlipidemic rats. Eur J Pharmacol. 2011;659:213–23.
Kannel WB, Dawber TR, Kagan A, Revotskie N, Stokes 3rd J. Factors of risk in the development of coronary heart disease—six year follow-up experience. The framingham study. Ann Intern Med. 1961;55:33–50.
Adler AI, Stratton IM, Neil HA, Yudkin JS, Matthews DR, Cull CA, et al. Association of systolic blood pressure with macrovascular and microvascular complications of type 2 diabetes (ukpds 36): prospective observational study. BMJ. 2000;321:412–9.
Petersen JS, DiBona GF. Acute sympathoinhibitory actions of metformin in spontaneously hypertensive rats. Hypertension. 1996;27:619–25.
Katakam PV, Ujhelyi MR, Hoenig M, Miller AW. Metformin improves vascular function in insulin-resistant rats. Hypertension. 2000;35:108–12.
Bhalla RC, Toth KF, Tan E, Bhatty RA, Mathias E, Sharma RV. Vascular effects of metformin. Possible mechanisms for its antihypertensive action in the spontaneously hypertensive rat. Am J Hypertens. 1996;9:570–6.
Verma S, Yao L, Dumont AS, McNeill JH. Metformin treatment corrects vascular insulin resistance in hypertension. J Hypertens. 2000;18:1445–50.
Landin K, Tengborn L, Smith U. Treating insulin resistance in hypertension with metformin reduces both blood pressure and metabolic risk factors. J Intern Med. 1991;229:181–7.
Landin-Wilhelmsen K. Metformin and blood pressure. J Clin Pharm Ther. 1992;17:75–9.
Nagi DK, Yudkin JS. Effects of metformin on insulin resistance, risk factors for cardiovascular disease, and plasminogen activator inhibitor in niddm subjects. A study of two ethnic groups. Diabetes Care. 1993;16:621–9.
He H, Zhao Z, Chen J, Ni Y, Zhong J, Yan Z, et al. Metformin-based treatment for obesity-related hypertension: a randomized, double-blind, placebo-controlled trial. J Hypertens. 2012;30:1430–9.
Tremoli E, Ghiselli G, Maderna P, Colli S, Sirtori CR. Metformin reduces platelet hypersensitivity in hypercholesterolemic rabbits. Atherosclerosis. 1982;41:53–60.
Gin H, Freyburger G, Boisseau M, Aubertin J. Study of the effect of metformin on platelet aggregation in insulin-dependent diabetics. Diabetes Res Clin Pract. 1989;6:61–7.
Kakafika AI, Liberopoulos EN, Mikhailidis DP. Fibrinogen: a predictor of vascular disease. Curr Pharm Des. 2007;13:1647–59.
Anfosso F, Chomiki N, Alessi MC, Vague P, Juhan-Vague I. Plasminogen activator inhibitor-1 synthesis in the human hepatoma cell line hep g2. Metformin inhibits the stimulating effect of insulin. J Clin Invest. 1993;91:2185–93.
Vague P, Juhan-Vague I, Alessi MC, Badier C, Valadier J. Metformin decreases the high plasminogen activator inhibition capacity, plasma insulin and triglyceride levels in non-diabetic obese subjects. Thromb Haemost. 1987;57:326–8.
Krysiak R, Okopien B. Haemostatic effects of metformin in simvastatin-treated volunteers with impaired fasting glucose. Basic Clin Pharmacol Toxicol. 2012.
McCoy RG, Irving BA, Soop M, Srinivasan M, Tatpati L, Chow L, et al. Effect of insulin sensitizer therapy on atherothrombotic and inflammatory profiles associated with insulin resistance. Mayo Clin Proc. 2012;87:561–70.
Agarwal N, Rice SP, Bolusani H, Luzio SD, Dunseath G, Ludgate M, et al. Metformin reduces arterial stiffness and improves endothelial function in young women with polycystic ovary syndrome: a randomized, placebo-controlled, crossover trial. J Clin Endocrinol Metab. 2010;95:722–30.
El Messaoudi S, Rongen GA, de Boer RA, Riksen NP. The cardioprotective effects of metformin. Curr Opin Lipidol. 2011;22:445–53.
Solskov L, Lofgren B, Kristiansen SB, Jessen N, Pold R, Nielsen TT, et al. Metformin induces cardioprotection against ischaemia/reperfusion injury in the rat heart 24 hours after administration. Basic Clin Pharmacol Toxicol. 2008;103:82–7.
Bhamra GS, Hausenloy DJ, Davidson SM, Carr RD, Paiva M, Wynne AM, et al. Metformin protects the ischemic heart by the akt-mediated inhibition of mitochondrial permeability transition pore opening. Basic Res Cardiol. 2008;103:274–84.
Paiva M, Riksen NP, Davidson SM, Hausenloy DJ, Monteiro P, Goncalves L, et al. Metformin prevents myocardial reperfusion injury by activating the adenosine receptor. J Cardiovasc Pharmacol. 2009;53:373–8.
Paiva MA, Goncalves LM, Providencia LA, Davidson SM, Yellon DM, Mocanu MM. Transitory activation of ampk at reperfusion protects the ischaemic-reperfused rat myocardium against infarction. Cardiovasc Drugs Ther. 2010;24:25–32.
Calvert JW, Gundewar S, Jha S, Greer JJ, Bestermann WH, Tian R, et al. Acute metformin therapy confers cardioprotection against myocardial infarction via ampk-enos-mediated signaling. Diabetes. 2008;57:696–705.
Hausenloy DJ, Yellon DM. The mitochondrial permeability transition pore: Its fundamental role in mediating cell death during ischaemia and reperfusion. J Mol Cell Cardiol. 2003;35:339–41.
Hausenloy DJ, Maddock HL, Baxter GF, Yellon DM. Inhibiting mitochondrial permeability transition pore opening: a new paradigm for myocardial preconditioning? Cardiovasc Res. 2002;55:534–43.
Zhang L, He H, Balschi JA. Metformin and phenformin activate amp-activated protein kinase in the heart by increasing cytosolic amp concentration. Am J Physiol Heart Circ Physiol. 2007;293:H457–66.
Mubagwa K, Flameng W. Adenosine, adenosine receptors and myocardial protection: an updated overview. Cardiovasc Res. 2001;52:25–39.
Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling—concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an international forum on cardiac remodeling. J Am Coll Cardiol. 2000;35:569–82.
Kirpichnikov D, McFarlane SI, Sowers JR. Metformin: an update. Ann Intern Med. 2002;137:25–33.
Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic ove26 mice. Diabetes. 2011;60:1770–8.
• Gundewar S, Calvert JW, Jha S, Toedt-Pingel I, Ji SY, Nunez D, et al. Activation of amp-activated protein kinase by metformin improves left ventricular function and survival in heart failure. Circ Res. 2009;104:403–11. In a mouse model of coronary artery occlusion, long term administration of metformin reduced infarct size and prevented adverse cardiac remodeling, via activation of AMPK.
Yin M, van der Horst IC, van Melle JP, Qian C, van Gilst WH, Sillje HH, et al. Metformin improves cardiac function in a nondiabetic rat model of post-mi heart failure. Am J Physiol Heart Circ Physiol. 2011;301:H459–68.
Sasaki H, Asanuma H, Fujita M, Takahama H, Wakeno M, Ito S, et al. Metformin prevents progression of heart failure in dogs: role of amp-activated protein kinase. Circulation. 2009;119:2568–77.
Xiao H, Ma X, Feng W, Fu Y, Lu Z, Xu M, et al. Metformin attenuates cardiac fibrosis by inhibiting the tgfbeta1-smad3 signalling pathway. Cardiovasc Res. 2010;87:504–13.
Benes J, Kazdova L, Drahota Z, Houstek J, Medrikova D, Kopecky J, et al. Effect of metformin therapy on cardiac function and survival in a volume-overload model of heart failure in rats. Clin Sci (Lond). 2011;121:29–41.
Shimizu I, Minamino T, Toko H, Okada S, Ikeda H, Yasuda N, et al. Excessive cardiac insulin signaling exacerbates systolic dysfunction induced by pressure overload in rodents. J Clin Invest. 2010;120:1506–14.
Meier JJ, Gallwitz B, Schmidt WE, Mugge A, Nauck MA. Is impairment of ischaemic preconditioning by sulfonylurea drugs clinically important? Heart. 2004;90:9–12.
Gross GJ, Auchampach JA. Blockade of atp-sensitive potassium channels prevents myocardial preconditioning in dogs. Circ Res. 1992;70:223–33.
Tavackoli S, Ashitkov T, Hu ZY, Motamedi M, Uretsky BF, Birnbaum Y. Simvastatin-induced myocardial protection against ischemia-reperfusion injury is mediated by activation of atp-sensitive k+ channels. Coron Artery Dis. 2004;15:53–8.
Bolen S, Feldman L, Vassy J, Wilson L, Yeh HC, Marinopoulos S, et al. Systematic review: comparative effectiveness and safety of oral medications for type 2 diabetes mellitus. Ann Intern Med. 2007;147:386–99.
Bennett WL, Maruthur NM, Singh S, Segal JB, Wilson LM, Chatterjee R, et al. Comparative effectiveness and safety of medications for type 2 diabetes: an update including new drugs and 2-drug combinations. Ann Intern Med. 2011;154:602–13.
Yu Q, Gao F, Ma XL. Insulin says no to cardiovascular disease. Cardiovasc Res. 2011;89:516–24.
Lonborg J, Vejlstrup N, Kelbaek H, Botker HE, Kim WY, Mathiasen AB, et al. Exenatide reduces reperfusion injury in patients with st-segment elevation myocardial infarction. Eur Heart J. 2012;33:1491–9.
Matheeussen V, Jungraithmayr W, De Meester I. Dipeptidyl peptidase 4 as a therapeutic target in ischemia/reperfusion injury. Pharmacol Ther. 2012;136:267–82.
Ye Y, Perez-Polo JR, Aguilar D, Birnbaum Y. The potential effects of anti-diabetic medications on myocardial ischemia-reperfusion injury. Basic Res Cardiol. 2011;106:925–52.
Riksen NP, Hausenloy D. Limitation of myocardial ischemia-reperfusion injury in clinical practice: new hopes and dissapointments. Curr Opin Lipidol. 2012;23:588–90
Lexis CP, van der Horst IC, Lipsic E, van der Harst P, van der Horst-Schrivers AN, Wolffenbuttel BH, et al. Metformin in non-diabetic patients presenting with st elevation myocardial infarction: rationale and design of the glycometabolic intervention as adjunct to primary percutaneous intervention in st elevation myocardial infarction (gips)-iii trial. Cardiovasc Drugs Ther. 2012;26:417–26.
Acknowledgements
This work was financially supported by a grant of the Dutch Heart Foundation (to NPR and GA) and the Netherlands Organisation for Health Research and Development (ZonMW; Clinical Fellowship to NPR).
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Coronary Heart Disease
Rights and permissions
About this article
Cite this article
El Messaoudi, S., Rongen, G.A. & Riksen, N.P. Metformin Therapy in Diabetes: The Role of Cardioprotection. Curr Atheroscler Rep 15, 314 (2013). https://doi.org/10.1007/s11883-013-0314-z
Published:
DOI: https://doi.org/10.1007/s11883-013-0314-z