
Abstract
Purpose of Review
Commonly categorized as a rare disease, alpha-1 antitrypsin deficiency (AATD) is neither rare, when compared to many other genetic disorders, nor an actual disease, but rather a predisposition toward a wide variety of diseases. It is one of the most common genetic disorders which can lead to a spectrum of clinical manifestations, ranging from no symptoms to progressively debilitating systemic disease, most commonly affecting the lung and liver. It is therefore imperative for clinicians to recognize and be familiar with the spectrum of presentations, methods of diagnosis, and clinical management of AATD. It is also imperative for scientists to recognize the potential for progress in the management of this disorder.
Recent Findings
This review focuses on the current state of knowledge of AATD, including the wide range of presentations, diagnosis, and clinical management. In addition to the clinical implications of severe AATD, we discuss the relevance of heterozygous state with mild or moderate AATD in the development of both lung and liver disease. While our understanding of the multiple roles of alpha-1 antitrypsin (AAT) is on the rise, with appreciation of its immunomodulatory, anti-infective, and anti-inflammatory properties, this knowledge has yet to impact our ability to predict outcomes. We discuss nuances of augmentation therapy and review novel therapeutic approaches currently under investigation.
Summary
With the expanding knowledge about the complexities of AAT function and its clinical relevance, and with the increasing ability to diagnose early and intervene on AATD, it should be our goal to change the perception of AATD as a correctable inherited disorder rather than a fatal disease.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Alpha-1 antitrypsin deficiency (AATD) is a hereditary genetic disorder that can lead to lung and liver disease due to decreased circulating levels of alpha-1 antitrypsin (AAT) and accumulation of AAT within the liver, respectively. Since its identification in the 1960s by Laurell and Eriksson [1], there have been many advances in the diagnosis and medical management of AATD. Despite a heightened awareness of this clinical entity, it still remains an infrequently diagnosed disorder. Its clinical presentation is highly variable and often mimics other more common diseases making it difficult to diagnose. However, its prompt diagnosis is imperative to the initiation of disease-modifying therapy, if indicated.
Genetics of Alpha-1 Antitrypsin Deficiency
AATD is one of the most common genetic disorders caused by a mutation in the SERPINA1 gene located in the long arm of chromosome 14 [2]. While the most common M allele predisposes for normal production, function, and levels of the AAT protein, more than 150 mutations within this gene have been identified [2]. The most prevalent deficiency alleles include the Z and S alleles, both arising from single amino acid substitutions. These alleles are associated with production of altered protein which consequently leads to suboptimal systemic levels or function of AAT. Homozygosity for the Z allele, Pi*ZZ, is the most frequent genotype leading to severe AAT deficiency which results from abnormal protein misfolding and accumulation within hepatocytes and subsequent impedance of AAT secretion into the circulation. Aggregation of abnormal protein within the liver may lead to inflammation, cellular stress, and as a consequence, cirrhosis. Decreased levels of AAT lead to increased risk for emphysema.
Understanding that AATD is a consequence of multiple identified abnormal alleles, we can classify the abnormal phenotypes as (1) AAT deficient, (2) AAT dysfunctional (AAT has decreased or no ability to block neutrophil elastase or has altered function such as PiPittsburgh where AAT becomes a potent inhibitor of thrombin and factor XI which can result in a bleeding disorder), or (3) AAT absent (no production of AAT, so called null variants) [2, 3]. In terms of the impact that abnormal gene presence has on the development of liver disease, some alleles lead to significant protein polymerization and accumulation which leads to liver damage (i.e., Z, Mmalton, Siiyama), while most others have a low or no risk of development of liver disease (i.e., S, I, and others) [4].
As AATD is characterized by an autosomal co-dominant inheritance pattern, a presenting phenotype, reflected by total AAT level and function, results from cumulative transcription of both inherited alleles. The presence of one abnormal gene will affect the total production of AAT at varying degrees, depending on the type of allele present. Thus, presence of one Z allele in an individual with a normal M allele will roughly result in 60% of blood AAT levels, and presence of a “null” allele in combination with a normal M allele will lead to roughly 50% of normally anticipated AAT levels [5]. While heterozygosity is not traditionally considered to increase risk of disease development, recent studies point out that both liver and lung disease may be more prevalent in this population than previously appreciated, especially in the presence of other risk factors [6••, 7•]. As such, one of the priorities of AAT research in coming years will be to better understand and possibly treat heterozygous individuals with mild or moderate AAT deficiency.
Alpha-1 Antitrypsin Levels
Although AAT is expressed in multiple cell types such as mononuclear phagocytes and neutrophils [8], the majority of systemically measured AAT in the blood is produced by hepatocytes. Although the liver is the major of AAT, other organs, such as the lung, may contribute to the balance between proteases and anti-proteases through the local production of AAT, but this contribution is considered to be of limited overall significance as AAT mostly reaches the lung by diffusion from the circulation. The normal range of AAT in the serum is 20 to 53 μM; concentration of AAT within the epithelial lining fluid of the lower respiratory tract is suggested to be about 10% of plasma levels [9].
Quantitative measurements of AAT are obtained using rocket immunoelectrophoresis, radial immunodiffusion or nephelometry, with the protective threshold cutoff set at 80 mg/dL for radial immunodiffusion and 11 umol/L or 50 mg/dL for nephelometry [10, 11]. Since radial immunodiffusion overestimates the actual concentrations, most centers today use nephelometry with normal values ranging between 80 and 220 mg/dL. The conversion between nephelometry-measured milligrams per deciliter and micromolar values requires division of the measured value by a factor of 5.2 [12].
Historically, levels > 11 μM are considered to be protective of lung damage. This threshold value was derived from median levels detected in PiSZ individuals who are considered to demonstrate moderate AATD with levels thought to be sufficient to protect the lungs from emphysema progression [13].
Measurement of AAT levels is relevant, yet some caution in interpretation is required. As an acute-phase reactant, plasma AAT levels increase rapidly [14••, 15, 16, 17] in response to inflammation or infection. The expected normal activity of AAT is not only dependent on the appropriate absolute increase in concentration of ATT, but also on the adequate maintenance of protease-antiprotease balance in normal and diseased states. Published evidence suggests that about 25% of individuals with the Pi*MZ phenotype demonstrate signs of inflammation at the time of AAT level measurement; if AAT level alone were to be used as diagnostic testing for AATD, then these patients could be potentially misclassified as “normal” [18]. Therefore, measurement of AAT alone for the diagnosis of AATD may be suboptimal for several reasons: baseline AAT levels vary between people, especially in cases of heterozygosity; serum AAT levels have been used as a surrogate to estimate the risks for the development of lung disease but not for liver disease; and AAT levels do not reflect total cumulative risk for development of lung or liver disease as other risk factors that may impact disease development, even with less than severely reduced levels, are not taken into consideration when risk stratification is made based on AAT levels only.
Alpha-1 Antitrypsin Function
Alpha-1 antitrypsin (also: Protease Inhibitor 1, or PI1) is a protein with many known functions. It is a member of the SERPIN family of proteases, a family of proteins that have a conserved mechanism of conformational change to inhibit various target enzymes [19]. Largely synthesized within hepatocytes and excreted into circulation by the liver, one of its main functions is to protect the lung from potential damage caused by the unbalanced effect of enzyme neutrophil elastase (ELANE). In the presence of severe AAT deficiency, proteolytic effects of ELANE can lead to the development of emphysema due to an imbalance between the concentration of proteases and antiproteases [20]. In addition to ELANE, AAT inhibits cathepsin and proteinase 3 while recent studies highlight its activity on other classes of proteases, such as metalloproteases and cysteine-aspartic proteases, suggesting a wider role than simply targeting one specific protease [14].
AAT is now known to possess immunomodulatory, anti-infective, and anti-inflammatory properties. As an acute-phase reactant, the levels of AAT can increase significantly in response to inflammation or infection [18]. Tissue AAT levels may also increase as a result of local synthesis by inflammatory cells, monocytes, and alveolar macrophages in response to inflammatory cytokines. AAT plays an important role in modulating immunity, inflammation, proteostasis, apoptosis, and possibly cellular senescence programs [21]. AAT may be able to inhibit LPS-induced cytokine production (TNF-a and IL-1b), while enhancing the production of IL-10 from monocyte models [22]. AAT can downregulate other proinflammatory cytokines such as IL-6, IL-8, or IL-1β, while promoting anti-inflammatory mediators such as IL-10, IL-1βRa, or TGF-β [23]. It can also inhibit the production of matrix metalloproteinase-12 and can regulate the expression of CD14 and the associated signal transducer Toll-like receptor 4, which can downregulate proinflammatory signals [24]. It interacts with adaptive immunity affecting the proliferation of Th1, Th2, and Th17 cells while enhancing the production of T-regulatory cells [24]. AAT can also directly control antigen presentation, by preventing the maturation of dendritic cells [24], and has demonstrated to reduce cell death and apoptosis, specifically that induced by caspase-3 [25]. These multiple functions of AAT are plausible due to its conformational polymorphism which can lead to modification of its physical, structural, biochemical, and functional properties after interacting with other molecules participating in inflammatory responses [22].
On a clinical level, data indicate its possible therapeutic role in the management of graft versus host disease [26], acute myocardial infarction, where it has been shown to decrease myocardial leukocyte infiltration [27], diabetes, where it reduces beta-cell injury [28], inflammatory bowel disease [29], arthritis [30]. It may be able to reduce renal ischemia-reperfusion injury by reducing TNF-alpha expression [31]. While the wide spectrum of functions of AAT and its role in the pathogenesis of multiple clinical disorders is becoming apparent with ongoing translational research, the overall significance of functions and therapeutic potential of AAT are still largely unknown.
AATD Is Not So Rare
AATD is one of the most common inherited disorders among those of European ancestry [32]. The Pi*ZZ genotype has the highest prevalence in coastal communities of Europe, with a prevalence as high as 1:1500–1:2000 [33]. It has been estimated that around 100,000 individuals in the USA have severe AAT deficiency [34]. While the prevalence of AAT deficiency varies between geographical regions, it is estimated that more than 3 million people worldwide may suffer from severe AAT deficiency [35]. More interestingly, when the focus on severe disease is shifted to all degrees of AAT deficiency, it is estimated that more than 100 million worldwide suffer from this disorder [35].
One of the major problems related to the management of AAT deficiency is the “iceberg phenomenon” which characterizes the relationship between the relatively small percentage of diagnosed individuals among all deficient individuals. Affected patients often go undiagnosed or misdiagnosed for many years and are evaluated by multiple practitioners before the correct diagnosis is made. Improving the diagnostic approach to AAT may not only lead to better outcomes but may also offer a better understanding about the natural course and interaction with environmental factors of an AAT-deficient individual. Increased awareness of the true prevalence of AATD also helps researchers and pharmaceutical companies increase efforts to produce novel therapies.
AAT Is Not Necessarily a Disease
While not a rare disorder, AAT deficiency is not a disease per se, but rather a predisposition to a wide spectrum of diseases. Who will develop disease is not easily predictable as AATD symptoms and manifestations are likely a result of genetic, epigenetic, and environmental factors. Individual rates of lung function decline in AATD differ from person to person, and an individualized approach is recommended [36]. Smoking is a key risk factor for the development of lung disease in patients with AATD. Disease progression and survival are both significantly worse in smokers than never-smokers [11]. Importantly, even among those with severe deficiency, many can also reach a normal life expectancy without any clinical manifestations of either lung or liver disease [37]. Earlier studies looking into the natural course of lung and liver disease among AATD-deficient individuals showing high morbidity and mortality are, to certain degree, affected by selection bias; many studied individuals were identified because of advanced liver and lung disease, which may have selected for sicker patients. More recent studies evaluating mortality among AATD-affected individuals paint a slightly different picture indicating that never-smoking Pi*ZZ individuals identified through screening efforts can have better outcomes, possibly due to conscious avoidance of risk factors [37]. With increasing access to commercial genome sequencing, the number of individuals diagnosed with AAT deficiency will continue to increase and many of those will have no evidence of apparent disease [38]. This could, with life-style modifications and appropriate early interventions, allow us to change the common perception of an individual with this genetic disorder as chronically sick and disabled, to a picture of a healthy person with AATD. Unfortunately, the ability to predict clinical course and identify those at higher risk for poorer outcomes among AATD individuals, even with adequate risk management, still remains inadequate.
When AATD Is a Disease: Morbidity and Mortality in Alpha-1 Antitrypsin Deficiency
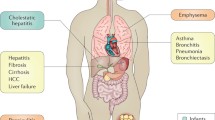
Currently, AATD is considered to be one of the most common fatal genetic disorders among adults; respiratory failure accounts for as much as 39% of deaths in never-smoking AATD patients [39]. In ever-smokers, respiratory failure can account for up to 60% of deaths [40]. Among patients with severe AATD, severe airflow obstruction, defined as FEV1 < 35% predicted, is associated with increased mortality [41]. Besides the classic presentation of lower lobe predominant emphysema, a patient with lung disease frequently presents with any of the common COPD phenotypes characterized by different patterns of emphysema, chronic bronchitis, or bronchiectasis. It can also present as asthma, vasculitis (granulomatosis with polyangiitis, GPA), or lung cancer. Other diseases related to AAT deficiency are liver fibrosis, cirrhosis, hepatocellular carcinoma, necrotizing panniculitis, urticaria, angioedema, psoriasis [11], connective tissue diseases such as GPA [42] glomerulonephritis [43], inflammatory bowel disease [44], and fertility abnormalities [45].
Liver disease, including hepatocellular carcinoma, cirrhosis, and neonatal jaundice, is the second most frequent clinical manifestation and the second leading cause of death among those with AATD [37]. About 10% of adults affected by AATD develop cirrhosis, and out of those affected, about 14% require liver transplantation [46]. It is unknown why some patients with AATD develop clinically significant liver disease while others do not. The bimodal distribution of liver disease adds a further layer of complexity. A significant portion of patients receiving liver transplant for AAT deficiency were actually heterozygous for the mutation (mostly Pi*MZ), but found to have another “second hit,” such as alcohol consumption or hepatitis, that could have caused or contributed to more rapid progression to end-stage liver disease [47].
In addition to commonly affected organ systems, AATD can undoubtedly be a progressively debilitating disease that can result in depression, anxiety, disability, and healthcare utilization [48]. In contrast to a classic, smoking-related COPD, where patients tend to manifest in the later stages of life, patients affected by AATD present as early as in between the third and fourth decade of life with symptoms that can drastically affect quality of life. Analysis of Medicare data from 2011 to 2013 demonstrated that 40.1% of AATD patients received disability benefits as compared to only 20.3% of patients with COPD [49], highlighting the significant disability experienced by AATD-affected individuals.
Diagnosis of Alpha-1 Antitrypsin Deficiency
Since the symptoms of AATD range from negligible to very severe, similar to common chronic diseases such as COPD, asthma, and even cirrhosis, it is difficult to establish a diagnosis of AATD based on symptoms alone. Cough, dyspnea, wheezing, reduced exercise tolerance, or muscle tissue loss are the most common presenting symptoms, but at the same time, some of the least-specific symptoms when presented to a physician. Such lack of specificity and insidious onset make this entity even harder to diagnose. As a result, there is often a delay greater than 5 years from the time a patient first presents with symptoms to the time a correct diagnosis is confirmed [50].
There are several methods defining diagnostic workup specific to this disorder. Measuring AAT levels evaluates the serum concentration of AAT and allows for a rough classification into the normal predicted range, lower than normal, and lower than the “protective threshold.” There are multiple measurement techniques, with nephelometry representing the standard of care today. However, ATT level alone can be insufficient to identify individuals at risk for emphysema or liver disease as levels can change in the setting of inflammation, pregnancy, and during oral contraceptive use [51]. Furthermore, the normal range of AAT level by genotype is wide. Thus, a level within the normal range does not necessarily mean a normal genotype, which highlights the need to perform more than just AAT level testing when trying to establish a diagnosis of AATD.
Alpha-1 antitrypsin phenotyping is based on the measurement of AAT protein from the blood through isoelectric focusing electrophoresis. This form of testing can help identify most of the mutations associated with AATD based on the pattern of migration of proteins down an electrophoretic field. Isoelectric focusing, however, requires technical skills to interpret the results as AAT phenotype is determined by visual inspection and comparison to known patterns. AAT genotyping is able to distinguish between specific alleles at the DNA level. This technique is often utilized and is based on using PCR primers to identify common deficiency variants such as the Z and S alleles. It is simple and widely applicable; nevertheless, it can miss rare alleles as it is limited to diagnose only the alleles for which specific PCR primers are included in the test. AAT gene sequencing allows for precise identification of AAT mutations, especially rare variants, by looking at the specific sequence of DNA nucleotides. It is the most accurate way to determine a specific AAT variant [52, 53]. With advancing accessibility to commercial gene testing, the number of individuals recognized as AAT deficient is increasing and additional testing methods, which bypass health care systems, are anticipated to play a more significant role in the diagnosis of AATD.
According to the American Thoracic Society (ATS) guidelines published in 2003, diagnostic testing for AATD is highly recommended in individuals with emphysema, COPD, asthma, and airflow obstruction that is not completely reversible after administration of inhaled bronchodilators; individuals with unexplained liver disease; asymptomatic individuals with persistent obstruction on spirometry with identifiable risk factors (cigarette smoking, occupational exposures); and individuals with necrotizing panniculitis. A lower level of evidence was assigned to other indications for testing [11]. As such, diagnostic testing is often pursued only in individuals who are either symptomatic or have airflow obstruction on spirometry. Unfortunately, testing is performed at a significantly lower rate than recommended. Recent analysis of the knowledge and applicability of ATS/ERS recommendations on Alpha-1 testing revealed that, even in countries offering augmentation therapy to AAT patients (which most countries still do not), only 18–25% of physicians tested COPD patients. Furthermore, familiarity with these guidelines is low, ranging from poor among pulmonologists to worse among internal medicine physicians [54].
Currently, within the USA, there are no targeted detection or screening programs to aid in the identification of individuals with AATD. Between 1972 and 1974, Sweden conducted a national neonatal screening program which identified 127 Pi*ZZ, 2 Pi*Z-null, 54 Pi*SZ, and 1 Pi*S-null infants out of 200,000 total screened [55]. Analysis 38 years later revealed that only 4.3% of Pi*ZZ individuals were current smokers but had significantly worse FEV1, quality of life, as measured by Saint-George’s Respiratory Questionnaire, and were more symptomatic compared to never-smokers. Not only do these findings highlight the impact life-style factors can have on disease development, but also suggest the impact that early screening and timely diagnosis can have on life-style modifications since the incidence of current smoking in this early diagnosed population was lower than AAT non-deficient Pi*MM controls [55].
Treatment of Alpha-1 Antitrypsin Deficiency
Management of AAT deficiency is complex, but it is roughly directed toward (1) prevention of disease development, (2) treatment of the condition which is a result of this deficiency, and (3) management of AAT deficiency/dysfunction. Since lung disease and, to a lesser degree, liver disease represent the most important pathological features of AATD, we will discuss the currently recommended and clinically investigated approaches primarily targeting the management of these organ systems.
As we increase our ability to diagnose AAT deficiency, early implementation of preventive measures will grow in importance (Table 1). Appropriate education, recognition of other family members who may be at risk, modification of environmental and habitual risks, and application of good preventive medicine is key to avoid the development of clinically significant disease in AAT deficiency.
The most frequent presentation of AATD is COPD/emphysema. Therefore, treating AATD is, before all, treating COPD. Its management encompasses a set of recommended pharmacological and non-pharmacological interventions [56••]. Applying these measures should be the primary approach to newly diagnosed patients with AATD-related lung disease (Table 2). Bronchodilator therapy represents the cornerstone of treatment [57, 58], with inhaled corticosteroids (ICS), anti-inflammatory medications such as roflumilast and azithromycin, and mucolytics encompassing recommended pharmacologic approaches. Non-pharmacologic approaches include physical activity, pulmonary rehabilitation, nutrition, and survival-improving measures such as supplemental oxygen, as well as smoking cessation. Non-pharmacologic interventional approaches include surgical methods that range from minor palliative procedures to lung transplantation, the latter accounting for 3.2% of all adult lung transplants and 10% of all transplants for emphysema [59••, 60]. In addition, recent studies offer promising data on endobronchial therapeutic approaches which include bronchoscopic lung volume reduction (BLVR) [61, 62]. This approach, while novel, carries a lot of promise in select patients with advanced disease and offers the possibility of improvement in quality of life [63].
In terms of liver disease management, early recognition and education along with preventive measures such as hepatitis A and B vaccinations, diet, and lifestyle modifications represent major interventions. No specific therapies have been approved for the prevention of cirrhosis development. In the setting of advanced liver disease and cirrhosis, liver transplant remains a viable and quite readily available option in the USA. In contrast to lung transplantation, which does not resolve the problem of low circulating AAT levels, liver transplantation offers complete normalization of AAT levels in the blood and lung microenvironment, thus reducing the chance of further accelerated lung function decline.
The only currently available approach to the specific management of AATD is augmentation therapy. Since the 1980s, intravenous infusion of purified, pooled human AAT is the only treatment available that addresses the underlying cause of lung disease in AATD [64]. Food and Drug Administration (FDA) approval of augmentation therapy came after Wewers et al. demonstrated its safety, tolerability, and efficacy (improvement in AAT levels in serum and bronchoalveolar lavage fluid) at a dose of 60 mg/kg/week [65]. Intravenous AAT augmentation products available in the USA and Europe are presented in Table 3. Whole acquisition cost (manufacturer’s list price) for augmentation therapies available within the USA are as follows: Prolastin-C $0.51/mg, Zemaira $0.52/mg, Aralast NP $0.55/mg, and Glassia $0.56/mg [66]. The typically utilized average monthly dose for an adult patient is around 20,000 mg.
The biochemical efficacy of 60 mg/kg AAT augmentation therapy has been well established and proven to raise serum levels consistently above the theoretical protective threshold of 11 μM with a half-life of 212.7 h [65, 67]. From the clinical perspective, AAT therapy can slow down FEV1 decline. Nevertheless, this effect has been consistently proven only within those individuals with moderate and severe obstruction [68]. Pooling the data from five relevant studies which included over 1500 subjects with AATD and exploring the effect of augmentation therapy on FEV1 decline, the largest effect has been seen in the population of individuals with FEV1 ranging between 30 and 65% predicted. Augmentation therapy was shown to slow the rate of decline of FEV1 by 23% in comparison with individuals not on augmentation therapy [69]. CT lung densitometry, which has been shown to correlate with both pathological changes in the lungs and mortality, was also explored as a clinical end-point [64]. In a randomized controlled trial, 180 AAT-deficient individuals were randomized to receive replacement therapy or placebo [70]. While the primary composite-outcome measure (total lung capacity + functional residual capacity) did not reach statistical significance, significant reduction in lung density decline rate was observed at total lung capacity in those receiving augmentation therapy. This supports the beneficial effect of augmentation therapy and supports the use of quantitative imaging in airway disease-focused studies [64]. The effect of augmentation therapy on acute exacerbations has not been confirmed, although several smaller studies reported less severe acute exacerbations [71, 72] and improvement in the qualitative severity of exacerbations following AAT augmentation [73]. Mortality benefit of AAT therapy has not been confirmed. Nevertheless, in a select population of patients with FEV1 < 50% predicted, mortality rate was statistically lower in patients receiving AAT therapy compared to individuals not on therapy [68].
In 2018, it was estimated that there were over 8000 patients within the USA on AAT augmentation therapy [74]. Despite the multiple benefits of augmentation therapy, there are some downsides associated with it. Augmentation therapy is contraindicated in IgA-deficient patients due to the potential of anaphylaxis, and its administration carries a (very low) risk of transmission of bloodborne infections since it represents a human serum-derived product. Side effects are minor but are observed at a rate of > 5% of upper respiratory tract infections. Therapy is also expensive; the annual cost of therapy in the USA can total a little over $127,000, with the majority of the cost to insurers driven by physician visits (12.3% of the cost) and augmentation therapy (66.7% of cost) [75]. Augmentation therapy can be time-consuming, significantly affecting lifestyle due to the need for weekly infusions. It is subject to possible supply limitations and shortages given its dependence on blood donors. Most importantly, it is approved for only significantly diseased (moderate or severe COPD) individuals, thus missing thousands of people in the USA with various degrees of AAT deficiency or lung disease.
Given the expense and time-consuming nature of intravenous augmentation therapy, there has been an increased interest in alternative options for the management of AAT deficiency. While this review does not intend to discuss ongoing clinical trial efforts, a brief discussion of tested concepts may help understand a wide variety of promising approaches (Table 4).
Higher doses of intravenous AAT (120 mg/kg) have been investigated [76] with hope that higher doses may allow for normalization of AAT levels to be carried over a longer period of time that could translate into clinical benefit. Current augmentation regimens are intended to maintain levels > 11 μM over more than 7 days. However, AAT levels decrease below the lower limit of normal by day 3 [77]. Attempts to create recombinant AAT have been ongoing for years, and recent studies [78] demonstrate progress with a hope to prove increased stability and optimal pharmacokinetics which could improve cost, dosing regimens, and safety. AAT replacement therapy delivery via nebulizer has also been investigated [79]. Advantages of this approach include the need for less product (roughly a quarter of the intravenous dose) and lack of need for IV access and infusion setup. Direct intrapleural delivery of both purified human and recombinant products has also been tested [80].
Other approaches, such as neutrophilic elastase blockade which aims to establish more optimal protease-antiprotease balance within the respiratory system [81], gene therapies [82, 83], or approaches targeting stabilization and clearance of Z protein [84], have been actively tested. Other studies have focused more on liver disease in AATD such as RNA silencing targeting Z-gene expression suppression in order to stabilize liver function decline [85, 86] and help clearance of Z granules from the liver [87].
Open Questions
Despite significant progress over more than 50 years since AATD was first described, it is important to acknowledge that our current understanding of the roles of AAT beyond its antagonistic activity toward ELANE remains modest. The ability to predict disease development in AAT-deficient individuals is limited, and clear understanding of the clinical relevance of moderate AAT deficiency due to Pi heterozygosity is less than optimal. Finally, our only approved therapeutic approach is limited by its availability, expense, risks, and burden on patient’s lifestyle. The full potential of augmentation is not yet completely understood. In addition, current management approaches do not target complete normalization of AAT levels nor a full correction of protease-antiprotease imbalance in healthy or diseased states, but rather target increasing levels above a protective threshold only. The research focused on finding solutions to these limitations will allow for the scientific progress in the management of this disorder (Fig. 1), and even allow us to recognize the possible therapeutic potential of AAT supplementation in acute inflammatory disorders.

Therapeutic approaches in AAT deficiency may evolve along with the development of deeper understanding of the function and roles of AAT and development of new pharmacotherapies. Long-term goal may target changing the paradigm of treating only chronically ill individuals with severe deficiency as currently recommended, to actually correcting AAT levels both at baseline and in the acute phase of inflammation, in all AAT-deficient individuals, and exploring the therapeutic benefits of AAT in acute inflammatory disorders where relative AAT levels may be suboptimal
Conclusion
Alpha-1 antitrypsin deficiency is the only relatively well-described hereditary cause of obstructive lung disease, and one of the most frequent inheritable etiologies of liver failure. While often considered a rare genetic disease, we argue here that it is neither rare nor a disease per se, but rather a potential for a variety of different clinical diseases. Early diagnosis, adequate preventive measures, and contemporary management of the presenting clinical phenotype can significantly improve clinical outcomes. AAT augmentation therapy allows for better disease control in select patients, while ongoing research efforts are focused on developing new strategies which aim to allow for AATD to be perceived as a correctable inherited disorder rather than a fatal disease.
Change history
12 September 2020
The original version of this article contained errors in Table 3 on page 6.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Laurell CB, Eriksson S. The electrophoretic alpha1-globulin pattern of serum in alpha1-antitrypsin deficiency. 1963. COPD. 2013;10(Suppl 1):3–8.
Greene CM, Marciniak SJ, Teckman J, Ferrarotti I, Brantly ML, Lomas DA, et al. alpha1-Antitrypsin deficiency. Nat Rev Dis Primers. 2016;2:16051.
Stoller JK, Aboussouan LS. A review of alpha1-antitrypsin deficiency. Am J Respir Crit Care Med. 2012;185(3):246–59.
Mahadeva R, Chang WSW, Dafforn TR, Oakley DJ, Foreman RC, Calvin J, et al. Heteropolymerization of S, I, and Z alpha1-antitrypsin and liver cirrhosis. J Clin Invest. 1999;103(7):999–1006.
Ferrarotti I, Thun GA, Zorzetto M, Ottaviani S, Imboden M, Schindler C, et al. Serum levels and genotype distribution of alpha1-antitrypsin in the general population. Thorax. 2012;67(8):669–74.
•• Al Ashry HS, Strange C. COPD in individuals with the PiMZ alpha-1 antitrypsin genotype. Eur Respir Rev. 2017;26(146) COMMENT: This review highlights the risk of emphysema in MZ individuals especially in the setting of smoking.
• Ortega VE, et al. The effects of rare SERPINA1 variants on lung function and emphysema in SPIROMICS. Am J Respir Crit Care Med. 2020;201(5):540–54 COMMENT: This study demonstrates the effects of Pi Z heterozygote and compound heterozygote genotypes have on lung function within a cohort of smokers.
Crystal RG. Alpha 1-antitrypsin deficiency, emphysema, and liver disease. Genetic basis and strategies for therapy. J Clin Invest. 1990;85(5):1343–52.
Janciauskiene S. Conformational properties of serine proteinase inhibitors (serpins) confer multiple pathophysiological roles. Biochim Biophys Acta. 2001;1535(3):221–35.
Stoller JK, et al. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Pneumologie. 2005;59(1):36–68.
American Thoracic, S. and S. European Respiratory, American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med, 2003. 168(7): p. 818–900.
Blanco I, Bueno P, Diego I, Pérez-Holanda S, Lara B, Casas-Maldonado F, et al. Alpha-1 antitrypsin Pi*SZ genotype: estimated prevalence and number of SZ subjects worldwide. Int J Chron Obstruct Pulmon Dis. 2017;12:1683–94.
Strange C. Anti-proteases and alpha-1 antitrypsin augmentation therapy. Respir Care. 2018;63(6):690–8.
•• Janciauskiene S, Welte T. Well-known and less well-known functions of alpha-1 antitrypsin. Its role in chronic obstructive pulmonary disease and other disease developments. Ann Am Thorac Soc. 2016;13(Suppl 4):S280–8 COMMENT: This article provides a comprehensive review of the lesser known functions of AAT and its role in disease management of AATD as well as other diseases not related to AATD.
Janciauskiene SM, Bals R, Koczulla R, Vogelmeier C, Köhnlein T, Welte T. The discovery of alpha1-antitrypsin and its role in health and disease. Respir Med. 2011;105(8):1129–39.
Tonelli AR, Brantly ML. Augmentation therapy in alpha-1 antitrypsin deficiency: advances and controversies. Ther Adv Respir Dis. 2010;4(5):289–312.
Mahadeva R, Lomas DA. Genetics and respiratory disease. 2. Alpha 1-antitrypsin deficiency, cirrhosis and emphysema. Thorax. 1998;53(6):501–5.
Sanders CL, Ponte A, Kueppers F. The effects of inflammation on alpha 1 antitrypsin levels in a national screening cohort. COPD. 2018;15(1):10–6.
Law RH, Zhang Q, McGowan S, Buckle AM, Silverman GA, Wong W, et al. An overview of the serpin superfamily. Genome Biol. 2006;7(5):216.
Duvoix A, Roussel BD, Lomas DA. Molecular pathogenesis of alpha-1-antitrypsin deficiency. Rev Mal Respir. 2014;31(10):992–1002.
Hunt JM, Tuder R. Alpha 1 anti-trypsin: one protein, many functions. Curr Mol Med. 2012;12(7):827–35.
Janciauskiene S, Larsson S, Larsson P, Virtala R, Jansson L, Stevens T. Inhibition of lipopolysaccharide-mediated human monocyte activation, in vitro, by alpha1-antitrypsin. Biochem Biophys Res Commun. 2004;321(3):592–600.
Song S. Alpha-1 antitrypsin therapy for autoimmune disorders. Chronic Obstr Pulm Dis. 2018;5(4):289–301.
Cosio MG, Bazzan E, Rigobello C, Tinè M, Turato G, Baraldo S, et al. Alpha-1 antitrypsin deficiency: beyond the protease/antiprotease paradigm. Ann Am Thorac Soc. 2016;13(Suppl 4):S305–10.
Lewis EC. Expanding the clinical indications for alpha(1)-antitrypsin therapy. Mol Med. 2012;18:957–70.
Marcondes AM, Li X, Tabellini L, Bartenstein M, Kabacka J, Sale GE, et al. Inhibition of IL-32 activation by alpha-1 antitrypsin suppresses alloreactivity and increases survival in an allogeneic murine marrow transplantation model. Blood. 2011;118(18):5031–9.
Toldo S, Seropian IM, Mezzaroma E, van Tassell BW, Salloum FN, Lewis EC, et al. Alpha-1 antitrypsin inhibits caspase-1 and protects from acute myocardial ischemia-reperfusion injury. J Mol Cell Cardiol. 2011;51(2):244–51.
Guttman O, et al. alpha1-Antitrypsin modifies general NK cell interactions with dendritic cells and specific interactions with islet beta-cells in favor of protection from autoimmune diabetes. Immunology. 2014.
Rivera-Nieves J, Bamias G, Vidrich A, Marini M, Pizarro TT, McDuffie MJ, et al. Emergence of perianal fistulizing disease in the SAMP1/YitFc mouse, a spontaneous model of chronic ileitis. Gastroenterology. 2003;124(4):972–82.
Grimstein C, Choi YK, Wasserfall CH, Satoh M, Atkinson MA, Brantly ML, et al. Alpha-1 antitrypsin protein and gene therapies decrease autoimmunity and delay arthritis development in mouse model. J Transl Med. 2011;9:21.
Daemen MA, et al. Functional protection by acute phase proteins alpha(1)-acid glycoprotein and alpha(1)-antitrypsin against ischemia/reperfusion injury by preventing apoptosis and inflammation. Circulation. 2000;102(12):1420–6.
Hersh CP. Diagnosing alpha-1 antitrypsin deficiency: the first step in precision medicine. F1000Res. 2017, 2049;6.
de Serres F, Blanco I. Role of alpha-1 antitrypsin in human health and disease. J Intern Med. 2014;276(4):311–35.
Campos MA, Wanner A, Zhang G, Sandhaus RA. Trends in the diagnosis of symptomatic patients with alpha1-antitrypsin deficiency between 1968 and 2003. Chest. 2005;128(3):1179–86.
de Serres FJ. Worldwide racial and ethnic distribution of alpha1-antitrypsin deficiency: summary of an analysis of published genetic epidemiologic surveys. Chest. 2002;122(5):1818–29.
Stockley RA, et al. Individualized lung function trends in alpha-1-antitrypsin deficiency: a need for patience in order to provide patient centered management? Int J Chron Obstruct Pulmon Dis. 2016;11:1745–56.
Tanash HA, et al. Survival in individuals with severe alpha 1-antitrypsin deficiency (PiZZ) in comparison to a general population with known smoking habits. Eur Respir J. 2017:50(3).
Hersh CP, Campbell EJ, Scott LR, Raby BA. Alpha-1 antitrypsin deficiency as an incidental finding in clinical genetic testing. Am J Respir Crit Care Med. 2019;199(2):246–8.
Tanash HA, Nilsson PM, Nilsson JÅ, Piitulainen E. Survival in severe alpha-1-antitrypsin deficiency (PiZZ). Respir Res. 2010;11:44.
Larsson C. Natural history and life expectancy in severe alpha1-antitrypsin deficiency, Pi Z. Acta Med Scand. 1978;204(5):345–51.
Stoller JK, Tomashefski J Jr, Crystal RG, Arroliga A, Strange C, Killian DN, et al. Mortality in individuals with severe deficiency of alpha1-antitrypsin: findings from the National Heart, Lung, and Blood Institute Registry. Chest. 2005;127(4):1196–204.
Mahr AD, Edberg JC, Stone JH, Hoffman GS, St.Clair EW, Specks U, et al. Alpha(1)-antitrypsin deficiency-related alleles Z and S and the risk of Wegener’s granulomatosis. Arthritis Rheum. 2010;62(12):3760–7.
Davis ID, Burke B, Freese D, Sharp HL, Kim Y. The pathologic spectrum of the nephropathy associated with alpha 1-antitrypsin deficiency. Hum Pathol. 1992;23(1):57–62.
Yang P, Tremaine WJ, Meyer RL, Prakash UBS. Alpha1-antitrypsin deficiency and inflammatory bowel diseases. Mayo Clin Proc. 2000;75(5):450–5.
Lomas DA. The selective advantage of alpha1-antitrypsin deficiency. Am J Respir Crit Care Med. 2006;173(10):1072–7.
Townsend SA, Edgar RG, Ellis PR, Kantas D, Newsome PN, Turner AM. Systematic review: the natural history of alpha-1 antitrypsin deficiency, and associated liver disease. Aliment Pharmacol Ther. 2018;47(7):877–85.
Mitchell EL, Khan Z. Liver disease in alpha-1 antitrypsin deficiency: current approaches and future directions. Curr Pathobiol Rep. 2017;5(3):243–52.
Beiko T, Strange C. Anxiety and depression in patients with alpha-1 antitrypsin deficiency: current insights and impact on quality of life. Ther Clin Risk Manag. 2019;15:959–64.
Zacherle E, N.J, Runken MC, Blanchette CM. Health care cost and utilization associated with alpha-1 antitrypsin deficiency among a cohort of medicare beneficiaries with COPD. Value in Health. 2015.
Chorostowska-Wynimko J, Barrecheguren M, Ferrarotti I, Greulich T, Sandhaus RA, Campos M. New patient-centric approaches to the management of alpha-1 antitrypsin deficiency. Int J Chron Obstruct Pulmon Dis. 2020;15:345–55.
Sandhaus RA, Turino G, Brantly ML, Campos M, Cross CE, Goodman K, et al. The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chronic Obstr Pulm Dis. 2016;3(3):668–82.
Craig TJ. Suspecting and testing for alpha-1 antitrypsin deficiency-an allergist’s and/or immunologist’s perspective. J Allergy Clin Immunol Pract. 2015;3(4):506–11.
McElvaney NG. Diagnosing alpha1-antitrypsin deficiency: how to improve the current algorithm. Eur Respir Rev. 2015;24(135):52–7.
Greulich T, Ottaviani S, Bals R, Lepper PM, Vogelmeier C, Luisetti M, et al. Alpha1-antitrypsin deficiency - diagnostic testing and disease awareness in Germany and Italy. Respir Med. 2013;107(9):1400–8.
Piitulainen E, Mostafavi B, Tanash HA. Health status and lung function in the Swedish alpha 1-antitrypsin deficient cohort, identified by neonatal screening, at the age of 37-40 years. Int J Chron Obstruct Pulmon Dis. 2017;12:495–500.
•• Singh D, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease: the GOLD science committee report 2019. Eur Respir J. 2019;53(5) COMMENT: This article focuses on new research within the field of COPD and offers updated recommendations regarding its management.
Cooper CB, Barjaktarevic I. A new algorithm for the management of COPD. Lancet Respir Med. 2015;3(4):266–8.
Barjaktarevic IZ, Arredondo AF, Cooper CB. Positioning new pharmacotherapies for COPD. Int J Chron Obstruct Pulmon Dis. 2015;10:1427–42.
•• Lomas DA. New therapeutic targets for alpha-1 antitrypsin deficiency. Chronic Obstr Pulm Dis. 2018;5(4):233–43 COMMENT: This article provides a comprehensive review of the new approaches to treatment of liver disease in AATD.
Zamora M. Surgery for patients with alpha 1 antitrypsin deficiency: a review. Am J Surg. 2019;218(3):639–47.
Endoscopic lung volume reduction in patients with advanced emphysema due to alpha1 antitrypsin deficiency. https://clinicaltrials.gov/ct2/show/NCT01357460?type=Intr&cond=Alpha+1-Antitrypsin+Deficiency&draw=2&rank=15.
Lung volume reduction coils for emphysema in alpha-1 antitrypsin deficiency (LuReCAA). https://clinicaltrials.gov/ct2/show/NCT02273349?type=Intr&cond=Alpha+1-Antitrypsin+Deficiency&draw=2&rank=49.
Perotin JM, Leroy S, Marquette CH, Mal H, Dutau H, Bourdin A, et al. Endobronchial coil treatment in severe emphysema patients with alpha-1 antitrypsin deficiency. Int J Chron Obstruct Pulmon Dis. 2018;13:3645–9.
Brantly ML, Lascano JE, Shahmohammadi A. Intravenous alpha-1 antitrypsin therapy for alpha-1 antitrypsin deficiency: the current state of the evidence. Chronic Obstr Pulm Dis. 2018;6(1):100–14.
Wewers MD, Casolaro MA, Sellers SE, Swayze SC, McPhaul KM, Wittes JT, et al. Replacement therapy for alpha 1-antitrypsin deficiency associated with emphysema. N Engl J Med. 1987;316(17):1055–62.
RED BOOK® database. IBM Micromedex®, 2020.
Campos MA, Kueppers F, Stocks JM, Strange C, Chen J, Griffin R, et al. Safety and pharmacokinetics of 120 mg/kg versus 60 mg/kg weekly intravenous infusions of alpha-1 proteinase inhibitor in alpha-1 antitrypsin deficiency: a multicenter, randomized, double-blind, crossover study (SPARK). COPD. 2013;10(6):687–95.
Survival and FEV1 decline in individuals with severe deficiency of alpha1-antitrypsin. The Alpha-1-Antitrypsin Deficiency Registry Study Group. Am J Respir Crit Care Med, 1998. 158(1): p. 49–59.
Chapman KR, Stockley RA, Dawkins C, Wilkes MM, Navickis RJ. Augmentation therapy for alpha1 antitrypsin deficiency: a meta-analysis. COPD. 2009;6(3):177–84.
Chapman KR, Burdon JGW, Piitulainen E, Sandhaus RA, Seersholm N, Stocks JM, et al. Intravenous augmentation treatment and lung density in severe alpha1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;386(9991):360–8.
Barros-Tizon JC, et al. Reduction of severe exacerbations and hospitalization-derived costs in alpha-1-antitrypsin-deficient patients treated with alpha-1-antitrypsin augmentation therapy. Ther Adv Respir Dis. 2012;6(2):67–78.
Lieberman J. Augmentation therapy reduces frequency of lung infections in antitrypsin deficiency: a new hypothesis with supporting data. Chest. 2000;118(5):1480–5.
Dirksen A, Piitulainen E, Parr DG, Deng C, Wencker M, Shaker SB, et al. Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha1-antitrypsin deficiency. Eur Respir J. 2009;33(6):1345–53.
US Plasma Proteins Market Report The Marketing Research Bureau (MRB) Inc. , Aug 2019.
Sieluk J, Levy J, Sandhaus RA, Silverman H, Holm KE, Mullins CD. Costs of medical care among augmentation therapy users and non-users with alpha-1 antitrypsin deficiency in the United States. Chronic Obstr Pulm Dis. 2018;6(1):6–16.
Campos MA, Geraghty P, Holt G, Mendes E, Newby PR, Ma S, et al. The biological effects of double-dose alpha-1 antitrypsin augmentation therapy. A pilot clinical trial. Am J Respir Crit Care Med. 2019;200(3):318–26.
Stocks JM, Brantly ML, Wang-Smith L, Campos MA, Chapman KR, Kueppers F, et al. Pharmacokinetic comparability of Prolastin(R)-C to Prolastin(R) in alpha(1)-antitrypsin deficiency: a randomized study. BMC Clin Pharmacol. 2010;10:13.
Phase 1 study to assess the safety, PK and PD of INBRX-101 in adults with alpha-1 antitrypsin deficiency (rhAAT-Fc). https://clinicaltrials.gov/ct2/show/NCT03815396?type=Intr&cond=Alpha+1-Antitrypsin+Deficiency&draw=2&rank=2.
Evaluate efficacy and safety of "Kamada-AAT for Inhalation" in patients with AATD (InnovAATe). https://clinicaltrials.gov/ct2/show/NCT04204252?type=Intr&cond=Alpha+1-Antitrypsin+Deficiency&draw=2&rank=8.
Safety study of an aerosolized, recombinant alpha 1-antitrypsin in subjects with alpha 1-antitrypsin deficiency. https://clinicaltrials.gov/ct2/show/NCT00161707?type=Intr&cond=Alpha+1-Antitrypsin+Deficiency&draw=2&rank=11.
A 12-week study treating participants who have alpha1-antitrypsin-related COPD with alvelestat (MPH966) or placebo. (ASTRAEUS). https://clinicaltrials.gov/ct2/show/NCT03636347?type=Intr&cond=Alpha+1-Antitrypsin+Deficiency&draw=2&rank=50.
Safety dose finding study of ADVM-043 gene therapy to treat alpha-1 antitrypsin (A1AT) deficiency (ADVANCE). https://clinicaltrials.gov/ct2/show/NCT02168686?type=Intr&cond=Alpha+1-Antitrypsin+Deficiency&draw=2&rank=17.
Experimental gene transfer procedure to treat alpha 1-antitrypsin (AAT) deficiency (AAT). https://clinicaltrials.gov/ct2/show/NCT00430768?type=Intr&cond=Alpha+1-Antitrypsin+Deficiency&draw=2&rank=40.
Evaluation of the efficacy and safety of VX-814 in subjects with the PiZZ genotype. https://clinicaltrials.gov/ct2/show/NCT04167345?type=Intr&cond=Alpha+1-Antitrypsin+Deficiency&draw=2&rank=39.
Study of ARO-AAT in normal adult volunteers. https://clinicaltrials.gov/ct2/show/NCT03362242?type=Intr&cond=Alpha+1-Antitrypsin+Deficiency&draw=2&rank=38.
A study of an investigational drug, ALN-AAT, in healthy adult subjects and patients with ZZ type alpha-1 antitrypsin deficiency liver disease. https://clinicaltrials.gov/ct2/show/NCT02503683?type=Intr&cond=Alpha+1-Antitrypsin+Deficiency&draw=2&rank=41.
Carbamazepine in severe liver disease due to alpha-1 antitrypsin deficiency (CBZ). https://clinicaltrials.gov/ct2/show/NCT01379469?cond=Alpha+1-Antitrypsin+Deficiency&draw=2&rank=30.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Dr. Barjaktarevic reports grants and personal fees from GE Healthcare, Asta Zeneca, personal fees from GSK, grants and personal fees from Theravance, grants and personal fees from Mylan, grants from Amgen, personal fees from Boehringer Ingelheim, personal fees from Verona Pharma, personal fees from Grifols, and personal fees from CSL Behring, outside the submitted work. Dr. Cortes Lopez reports travel reimbursement from Vertex Pharmaceuticals.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Immune Deficiency and Dysregulation
Rights and permissions
About this article

Cite this article
Cortes-Lopez, R., Barjaktarevic, I. Alpha-1 Antitrypsin Deficiency: a Rare Disease?. Curr Allergy Asthma Rep 20, 51 (2020). https://doi.org/10.1007/s11882-020-00942-4
Published:
DOI: https://doi.org/10.1007/s11882-020-00942-4