
Abstract
Regulatory T (Treg) cells that express the transcription factor forkhead box protein P3 (FOXP3) play an essential role in enforcing immune tolerance to self tissues, regulating host-commensal flora interaction, and facilitating tissue repair. Their deficiency and/or dysfunction trigger unbridled autoimmunity and inflammation. A growing number of monogenic defects have been recognized that adversely impact Treg cell development, differentiation, and/or function, leading to heritable diseases of immune dysregulation and autoimmunity. In this article, we review recent insights into Treg cell biology and function, with particular attention to lessons learned from newly recognized clinical disorders of Treg cell deficiency.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
T lymphocytes have a fundamental role in fighting foreign pathogens by generating a diverse repertoire of antigen receptors through antigen receptor gene rearrangement [1]. Unfortunately, this diversity might lead to generation of T cell populations that recognize self-antigen and result in autoimmunity [2]. One way of protection from these self-reactive T cells is by the process of negative selection that prevents such harmful cells from maturation by successful inactivation or clonal deletion in thymic tissue. Several studies have shown high rates of autoimmunity in genetic mutations affecting thymic central tolerance [3, 4]. As some of these autoreactive T cells might escape medullary thymic epithelial tissue to the periphery or might be exclusively expressed in the peripheral organs, immune tolerance is necessary for the elimination of these self-reactive cells. For that reason, dominant tolerance ensured by regulatory T cells has been documented as an important strategy to maintain peripheral tolerance in human and mice [5, 6]. The importance of this population in mice have been delineated by neonatal thymectomy performed at day 3 of life that resulted in autoimmunity and production of autoantibodies, while transferring thymic or splenic T cells to these thymectomized mice from adult wild type prevented the development of immune-mediated inflammation and tissue damage [7, 8]. Compelling evidence has shown this thymic suppressive population expresses CD4 and IL-2 receptor α chain (CD25) and has been characterized as CD4+CD25+ regulatory T (Treg) cells [9]. Subsequently, forkhead box protein P3 (FOXP3) was identified as a transcription factor indispensable for Treg cell development and function [10–12]. Deleterious mutations in FOXP3 lead to immune dysregulation, polyendocrinopathy, and enteropathy X-linked (IPEX) syndrome in humans as well as fatal autoimmunity in scurfy mice [13, 14]. The aim of this review is to highlight current information on the defining features of T regulatory cells as well as their phenotypic and functional heterogeneity with particular emphasis on the consequences of this compartment deficiency and or dysfunction in the development of immune dysregulation and autoimmunity.
Treg Cell Subsets and Markers
Treg cells represent 5 to 10 % of the peripheral CD4+ T cell compartment in humans and in mice. The two key populations of Treg cells are those that develop in the thymus, referred to as natural or thymic Treg (nTreg or tTreg) cells and induced Treg that develop in the periphery from naïve conventional CD4+ T cells (iTreg or pTreg cells, respectively) [15].
In general, FOXP3+ Treg cells express high levels of interleukin-2 receptor α (CD25) and a low level of IL-7 receptor α (CD127) on the cell surface [16]. The majority of Treg cells constitutively express high levels of the inhibitory molecule cytotoxic T lymphocyte-associated antigen 4 (CTLA4) and the glucocorticoid-induced TNFR family related (GITR), as well as the regulatory cytokines IL-10 and transforming growth factor-beta (TGF-β) [17–20]. While FOXP3 staining is currently the best available marker for Treg cells, it may also be transiently induced at low levels in human (but not in mouse) T conventional (Tconv) cells upon their activation. Expression of other Treg cell markers such as CD25 and CTLA4, and down-regulation of CD127, may similarly be affected upon activation of Tconv cells. Accordingly, employment of combinatorial markers such as FOXP3highCD25highCD127low may better discriminate human Treg cells from otherwise activated Tconv cells. Human Treg cells can be further classified based on their activation profile using FOXP3 and CD45RA/RO. Resting Treg cells are CD45RA+FOXP3low, and activated Treg cells are CD45RA−FOXP3high while the CD45RA−FOXP3low population reflects effector cytokine-producing non-Treg cells [21].
Two markers have been used to discriminate nTreg from iTreg cells. Helios, a member of the Ikaros family of transcription factors, is highly enriched in nTreg as compared to iTreg cells and is commonly used as a marker of Treg cells of thymic origin [22]. Furthermore, neuropilin-1 is similarly enriched in nTreg versus iTreg cells. However, expression of both markers can be altered by T cell activation, and they should be judiciously used in discriminating those populations under conditions of inflammation or generalized T cell activation [23].
Finally, Treg cells that become unstable and lose their FOXP3 expression are referred to as ex-Treg cells [24]. They acquire effector functions and may contribute to pathology in inflammatory and autoimmune diseases [25•].
Treg Cell Development
nTreg cell development in the thymus proceeds through discrete steps including intermediate avidity interactions between self-reactive TCR on developing thymocytes and their cognate antigens presented in specialized thymic niches. These interactions, in the context of optimal input from co-stimulatory molecules and cytokines, enable the acquisition of CD25 expression, epigenetic modification of FOXP3, and other Treg cell-related genetic loci, leading to upregulation of FOXP3 and other Treg cell markers [26].
The interaction of the T cell receptor (TCR) with self-antigens in the thymus is pivotal for Treg cell differentiation. Typically, conventional thymocytes that receive high strength TCR signals undergo apoptosis while those that pass positive selection and receive low affinity signals will eventually develop into mature T cells. In contrast, the development of Treg cells in the thymus appears to require intermediate strength interactions between their TCRs and self-peptide/MHC ligands. These interactions, in the context of specialized niches in the thymic medulla, including medullary thymic epithelial cells (mTecs) and hematopoietic antigen presenting cells, lead to the upregulation of CD25 and also enabling subsequent developmental steps in thymic Treg cell development [27].
In addition to the TCR, co-stimulatory molecules, including CD28 and members of the tumor-necrosis factor receptor superfamily, including GITR, OX40, and TNFR2, all make important contributions to Treg cell differentiation [28•, 29]. These pathways converge on downstream signaling intermediates, most notably NF-κB, STAT5, mTOR, and others, to promote Treg cell development [30].
FOXP3 is upregulated at the terminal stage in thymic Treg cell differentiation under the action of IL-2 via the CD25 containing high-affinity IL-2R complex that provides the critical stimulus inducing its expression. FOXP3 itself is dispensable for thymic Treg cell development. However, it plays an indispensable role in enabling Treg cell function in the periphery. FOXP3 expression directly regulates a sizeable component of the Treg cell transcriptome, including further upregulation of CD25 and high expression of suppressor genes as well as repression of pro-inflammatory cytokines. It also shapes the Treg cell transcriptome by stabilizing the interaction of its different components, including those induced by TCR and cytokine signaling. On the other hand, a large fraction of the core Treg cell transcriptome is maintained in the absence of FOXP3, consistent with the FOXP3-independent development of Treg cells. Nevertheless, the mutant Treg cells acquire the phenotype and the relevant genetic circuitries of activated, unstable cytotoxic T cell-like cells that lack regulatory functions [31].
Besides FOXP3 expression, epigenetic (external DNA modifications that affect gene function without changing DNA sequencing) has an important impact on Treg cell development and function. These epigenetic marks show some heterogeneity between Treg and Tconv cells. Indeed, most nTreg cells have a completely demethylated CNS2 (conserved non-coding region 2) at the FOXP3 locus while the Tconv cells (CD4+CD25low) possess a partially methylated pattern even after transient FOXP3 upregulation. Treg-type DNA hypomethylation is exclusively imprinted in nTreg cells and found to be important in their suppressor function, lineage stability, and controlling Treg cell-specific genes expression [32–34, 35•].
Additionally, other transcription factors, including Helios and GATA3, play an important role in conferring suppressive functions on Treg cells, especially in the context of different inflammatory cues [36•, 37, 38]. Homeostatic proliferation of Treg cells in the periphery is dependent on cytokines, most notably IL-2, which act to maintain Treg cell fitness and maintain their stability and regulatory functions. In contrast, other signals including those delivered by the Notch family of receptors act to restrain Treg cell function in the periphery [39••].
In addition to nTreg cells, de novo generation of iTreg cells contributes to peripheral tolerance [40]. iTreg cells are particularly abundant at the mucosal interface, where they are induced in specialized niches by the action of tolerogenic antigen presenting cells (APCs) such CD103+ dendritic cells in the gut and CD11c+ macrophages in the lung. iTreg cell generation is dependent on TGF-β production by APCs and is potentiated by the production of retinoic acid, which acts to stabilize newly formed iTreg cells [41–43]. These cells possess a TCR repertoire distinct from that of nTreg cells, and the two populations synergize to maintain peripheral immunological tolerance [44]. iTreg cells are enriched at the environmental interfaces and are enriched in specificities directed at microbial antigens [45]. Microbial sensing by iTreg cells in the gut via the Toll-like receptor-associated adaptor protein MyD88 promotes their function and their differentiation into T follicular cells that regulate anti-commensal IgA production in Peyer’s patches [46•, 47•]. By regulating the anti-commensal IgA response, the Treg cells play an essential role in shaping a healthy commensal flora that contributes to peripheral tolerance.
Under conditions of sustained inflammation and/or lymphopenia, iTreg cells (and to a lesser extent nTreg cells), may acquire attributes of effector T cells and express cytokines and effector molecules that contribute to the inflammatory response such as food allergy [48•, 49•]. In extremis, this plasticity may lead to the loss of FOXP3 expression, leading as mentioned above to the generation of pathogenic ex-Treg cells that participate in disease pathology [25•]. Collectively, these findings indicate that Treg stability, function, and fate are influenced by multiple mechanisms that might be upstream of FOXP3.
Treg Cells: Regulatory Mechanisms and Functional Specialization
Regulatory T cells are central players in the area of the peripheral tolerance. By their immunosuppressive capability, they maintain immune homeostasis and prevent autoimmunity in diverse anatomical locations. The suppressive mechanisms of this population may proceed by contact-dependent suppressive mechanisms either through inhibitory receptors (e.g., CTLA-4, LAG3, Galectin-1) or by means of perforin and granzyme B-dependent, Treg cell-mediated cytotoxic target cell killing [50–53]. Treg cells may also mediate contact-independent suppression either by acting as IL-2 sink (through Treg cell-bound CD25) or by producing inhibitory cytokines (e.g., IL-10, TGF-β, IL-35) [54–56].
More than general suppressive activity, Treg cells might further differentiate in the periphery as a part of Treg plasticity to specialized fates that specifically control Th1, Th2, Th17, or T follicular helper (Tfh)-type immune responses by acquiring the transcriptional program of the specific effector cells they suppress, such as T-bet, IRF4, STAT3, or Bcl-6, respectively [57–60]. It is also important to note that the suppressive function of Treg cells might limit the beneficial effector responses against tumors and chronic infections [61].
Interestingly, Treg cells have an important role in tissue protection both directly by inducing tissue repair through amphiregulin production and indirectly by limiting tissue damage through the down-regulation of the inflammatory response [62••]. Beyond tissue protection, numerous reports have also suggested other Treg cell functions, including protection against allergic disorders, transplant rejection, and atherosclerosis as well as controlling metabolic disorders [63–65].
Monogenic Diseases Resulting in Treg Cell Deficiency/Dysfunction
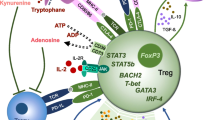
Treg cells play a key role in immune homeostasis by maintaining a balanced adaptive immune response. Human congenital defects that affect Treg cell number and/or function disrupt this balance and result in autoimmunity, lymphoproliferation, allergic dysregulation, and ongoing lymphocytic infiltration in different organs, which lead to disease progression and impact patient survival. The spectrum of manifestations due to Treg cell defect might range from mild allergy or autoimmunity to lethal immune dysregulation disorders. Interestingly, several human genetic disorders have been described recently and noted to have a tremendous impact on Treg cell development and functional activity. A loss of function mutation in FOXP3, the key transcriptional factor for Treg cell differentiation, leads to IPEX phenotype. Subsequently, a number of other gene defects have been described to cause IPEX-related phenotypes including loss of function mutations in CD25, STAT5b, LRBA, and CTLA4 (Fig. 1).

Defective Treg cell suppressive mechanisms in IPEX and IPEX-like disorders. Shown are key pathways for maintaining Treg cell homeostasis and function highlights human monogenic defects that lead to severe immune dysregulation due to altered Treg cell function. The engagement between IL-2 and IL-2R and the initiation of signal transduction through STAT5b phosphorylation are important for FOXP3 expression. While FOXP3 deficiency leads to IPEX, loss of function mutations in IL2Rα or STAT5b manifest with IPEX-like phenotype. LRBA-CTLA4 pathway is indispensible for Treg cell suppressive activity. LRBA controls CTLA4 expression, and the latter provides a negative feedback both directly by competing CD28 for binding to CD80/CD86 ligands and indirectly by down-regulating the co-stimulatory molecules on APCs. Both LRBA and CTLA4 deficiency cause IPEX-like disorder
IPEX
IPEX is a rare genetic disorder resulting from lack of functional Treg cells due to loss of function mutations in FOXP3. It exclusively affects males given its X-linked recessive pattern of inheritance and is often fatal within the first few years of life unless rescued with bone marrow transplantation [66]. Clinically, IPEX presents with a triad of autoimmune enteropathy, autoimmune endocrinopathy, and eczematous dermatitis. The most common manifestation is enteropathy followed by endocrinopathy, especially insulin-dependent type 1 diabetes mellitus. Additional described manifestations include immune-mediated cytopenias, which may present as neutropenia, anemia and/or thrombocytopenia, and autoimmune nephropathy, hepatitis, and lung disease. Food allergy with elevated serum IgE and peripheral eosinophilia are very common in this disorder, reflecting a breakdown in oral tolerance. Patients with IPEX usually have a wide range of autoantibodies due to adaptive immune dysregulation. As more than 60 FOXP3 mutations have been reported to date, it has been observed from the clinical phenotype reported for these mutations that there is genotype/phenotype relationships [67]. The only available curative treatment for this disease is allogeneic hematopoietic stem cell transplant with reduced-intensity chemotherapy. Before transplant, patients require nutritional support and immunosuppressive therapy, which may include glucocorticoids and/or steroid-sparing agents such as calcineurin inhibitors, the mechanistic target of rapamycin (mTOR) inhibitor, and others [68•].
IPEX-Like Disorders
IPEX-like disorders have been described in many patients, both males and females, who lack detectable mutations in FOXP3 [69]. Putative mutations in this syndrome may involve genes that adversely affect Treg cell differentiation and function and that present with an overlapping clinical picture with that of FOXP3 deficiency. To date, the most well-characterized IPEX-like disorders include mutations along the IL-2Rα/STAT5b and CTLA4/LRBA pathways, detailed below.
CD25 and STAT5b Deficiency
Fatal autoimmunity was initially described in mice lacking IL-2, IL-2Rα, IL-2Rβ, or STAT5 isoforms. Reconstitution of these mutant mice with Treg cells from wild-type mice rescues disease [70–73]. These observations confirmed the critical role for the IL-2R-STAT5 signaling pathway in Treg cell homeostasis and function. Interleukin-2 (IL-2) receptor is formed by three subunits namely α (CD25), β (CD122), and γ (CD132) subunit. Among those, CD25, the high-affinity IL-2 receptor, is a unique subunit that exclusively binds IL-2 and constitutively expressed at high levels by Treg cells. CD25 deficiency in human leads to both autoimmunity and immunodeficiency with recurrent infections. Features of CD25 deficiency that shared with IPEX include chronic eczema, enteropathy, lymphoproliferation, and autoimmunity disorders such as alopecia, diabetes mellitus, thyroiditis, and autoimmune hemolytic anemia [74–77]. CD25 deficiency is permissive to Treg cell differentiation, with normal count of FOXP3+ Treg cells found in circulation [78]. However, loss of CD25 expression impairs Treg cell suppressive function by several mechanisms. These include the defective production by Treg cells of the suppressive cytokine IL10, and their failure to provide an IL-2 “sink” that deprives Tconv cells of IL-2, leading to their apoptosis in a Bim-dependent manner [54, 75, 79]. Finally, the decreased sensitivity of CD25-deficient Treg cells to IL-2 impairs their metabolic fitness in the context of an immune response [80].
In contrast to FOXP3-deficient patients, CD25 deficiency is distinguished by chronic infections with members of the herpes family of viruses. The susceptibility to viral infections may reflect the importance of IL-2 signaling in generating effective cytotoxic CD8+ effector and memory T cell responses as well as NK cell activation [81–83]. Of note, CD25-deficient patients reported to date lack food allergies and significantly elevated IgE level, reflecting a distinct mechanisms for the control of oral allergic sensitization by Treg cells.
The transcriptional activating factor STAT5b, part of IL2/STAT5 axis, is required for signal transduction of gamma chain cytokines, growth hormone, erythropoietin, prolactin, and granulocyte colony-stimulating factor (G-CSF) [84]. STAT5b deficiency presents with growth failure, delayed puberty, prominent forehead, recurrent infections, chronic diarrhea, eczema, and lymphoid interstitial pneumonitis [85, 86]. Autoimmunity is a common manifestation in this monogenic defect due to abnormal Treg cell development and function, and most of the patients have hypergammaglobulinemia and increased percentages of memory T cells [87]. Finally, low IGF-1, low IGFBP-3, and high prolactin are usually present, reflecting defective growth hormone receptor signaling [88].
LRBA and CTLA4
Cytotoxic T lymphocyte antigen–4 (CTLA-4) is an inhibitory receptor expressed on both Treg and activated Tconv cells. CTLA4 expression on Treg cells is essential for their contact-dependent suppression. CTLA4 regulates the immune response by competing with CD28 for the ligands CD80/CD86 and also removing these ligands from antigen-presenting cells (APCs) via transendocytosis [89], which abrogates subsequent T effector cells activation. Recent report showed that CTLA4 trafficking and expression is regulated by LPS-responsive and beige-like anchor (LRBA) [90••]. Both CTLA4 haploinsufficiency and LRBA deficiency lead to severe immune dysregulation and fatal autoimmunity in human, and several patients who presented recently with IPEX-like phenotype were found to have mutations in LRBA [91••] or CTLA4 gene [92••, 93••]. Collectively, the clinical presentation of those two monogenic defects is similar, ranging from CVID phenotype to severe IPEX-like disorder. These features include recurrent infections, hypogammaglobulinemia, inflammatory bowel disease, lymphoproliferation with granulomatous lymphocytic infiltration (brain, lung, liver, kidney, bone marrow), solid tumors, intense autoantibody responses, and profound autoimmunity including autoimmune cytopenia, psoriasis, alopecia, arthritis, and autoimmune hepatitis [91••, 92••, 93••, 94–96, 97•]. There are many immunosuppressive medications that have been tried to suppress immune dysregulation and autoimmunity without good clinical improvement. Interestingly, it has been shown recently that medications targeting CTLA4 such as CTLA4 fusion proteins and hydroxychloroquine (lysosomal degradation inhibitor) are highly effective in controlling the profound autoimmunity in these disorders [90••, 98••]. Finally, the curative treatment option is still hematopoietic stem cell transplantation despite the high risk of mortality and possible recurrence of autoimmunity post transplant [99].
Conclusion
It is clear that Treg cells play a critical role not only in maintaining peripheral tolerance and preventing autoimmunity (negative regulation) but also in promoting tissue repair, intestinal IgA response, and healthy commensalism (positive regulation). Exploring the molecular mechanisms involved in Treg cell dysfunction in IPEX and IPEX-like disorders provides insight into the biology of Treg cells and their role in common autoimmune and immune dysregulatory diseases. This understanding enables the development of successful therapeutic interventions in these disorders.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Sleckman BP, Gorman JR, Alt FW. Accessibility control of antigen-receptor variable-region gene assembly: role of cis-acting elements. Annu Rev Immunol. 1996;14:459–81.
Laufer TM, Fan L, Glimcher LH. Self-reactive T cells selected on thymic cortical epithelium are polyclonal and are pathogenic in vivo. J Immunol. 1999;162(9):5078–84.
Peterson P et al. APECED: a monogenic autoimmune disease providing new clues to self-tolerance. Immunol Today. 1998;19(9):384–6.
Ohashi PS. Negative selection and autoimmunity. Curr Opin Immunol. 2003;15(6):668–76.
Lio CW, Hsieh CS. Becoming self-aware: the thymic education of regulatory T cells. Curr Opin Immunol. 2011;23(2):213–9.
Klein L, Jovanovic K. Regulatory T cell lineage commitment in the thymus. Semin Immunol. 2011;23(6):401–9.
Bonomo A et al. Pathogenesis of post-thymectomy autoimmunity. Role of syngeneic MLR-reactive T cells. J Immunol. 1995;154(12):6602–11.
Asano M et al. Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J Exp Med. 1996;184(2):387–96.
Sakaguchi S et al. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155(3):1151–64.
Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4 + CD25+ regulatory T cells. Nat Immunol. 2003;4(4):330–6.
Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299(5609):1057–61.
Zheng Y, Rudensky AY. Foxp3 in control of the regulatory T cell lineage. Nat Immunol. 2007;8(5):457–62.
Bennett CL et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27(1):20–1.
Clark LB et al. Cellular and molecular characterization of the scurfy mouse mutant. J Immunol. 1999;162(5):2546–54.
Komatsu N et al. Heterogeneity of natural Foxp3+ T cells: a committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc Natl Acad Sci U S A. 2009;106(6):1903–8.
Abbas AK et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol. 2013;14(4):307–8.
Maynard CL et al. Regulatory T cells expressing interleukin 10 develop from Foxp3+ and Foxp3− precursor cells in the absence of interleukin 10. Nat Immunol. 2007;8(9):931–41.
Gutcher I et al. Autocrine transforming growth factor-beta1 promotes in vivo Th17 cell differentiation. Immunity. 2011;34(3):396–408.
Takahashi T et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192(2):303–10.
McHugh RS et al. CD4(+)CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16(2):311–23.
Miyara M et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. 2009;30(6):899–911.
Himmel ME et al. Helios+ and Helios− cells coexist within the natural FOXP3+ T regulatory cell subset in humans. J Immunol. 2013;190(5):2001–8.
Yadav M et al. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J Exp Med. 2012;209(10):1713–22. S1-19.
Zhou X et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. 2009;10(9):1000–7.
Komatsu N et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med. 2014;20(1):62–8. This article shows production of pathogenic TH17 cells due to Foxp3 instability and their contribution to the pathogenesis of autoimmunity.
Sawant DV, Vignali DA. Once a Treg, always a Treg? Immunol Rev. 2014;259(1):173–91.
Jordan MS et al. Thymic selection of CD4 + CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol. 2001;2(4):301–6.
Mahmud SA et al. Costimulation via the tumor-necrosis factor receptor superfamily couples TCR signal strength to the thymic differentiation of regulatory T cells. Nat Immunol. 2014;15(5):473–81. This study delineates the importance of the high expression of GITR, OX40, and TNFR2 on Treg cell progenitors to undergo successful maturation.
Tai X et al. CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat Immunol. 2005;6(2):152–62.
Hsieh CS, Lee HM, Lio CW. Selection of regulatory T cells in the thymus. Nat Rev Immunol. 2012;12(3):157–67.
Gavin MA et al. Single-cell analysis of normal and FOXP3-mutant human T cells: FOXP3 expression without regulatory T cell development. Proc Natl Acad Sci U S A. 2006;103(17):6659–64.
Floess S et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007;5(2), e38.
Lal G et al. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J Immunol. 2009;182(1):259–73.
Ohkura N et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity. 2012;37(5):785–99.
Kitagawa Y, Wing JB, Sakaguchi S. Transcriptional and epigenetic control of regulatory T cell development. Prog Mol Biol Transl Sci. 2015;136:1–33. This publication discusses key transcriptional and epigenetic factors that are important for Treg cell genetic profile.
Kim HJ et al. Stable inhibitory activity of regulatory T cells requires the transcription factor Helios. Science. 2015;350(6258):334–9. This experimental study provides evidence that Helios is important factor in Treg cell suppressive activity.
Wohlfert EA et al. GATA3 controls Foxp3(+) regulatory T cell fate during inflammation in mice. J Clin Invest. 2011;121(11):4503–15.
Wang Y, Su MA, Wan YY. An essential role of the transcription factor GATA-3 for the function of regulatory T cells. Immunity. 2011;35(3):337–48.
Charbonnier LM et al. Control of peripheral tolerance by regulatory T cell-intrinsic Notch signaling. Nat Immunol. 2015;16(11):1162–73. This publication shows the critical role for Notch signaling in controlling peripheral Treg cell function.
Bilate AM, Lafaille JJ. Induced CD4 + Foxp3+ regulatory T cells in immune tolerance. Annu Rev Immunol. 2012;30:733–58.
Chen W et al. Conversion of peripheral CD4+ CD25− naive T cells to CD4+ CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198(12):1875–86.
Coombes JL et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204(8):1757–64.
Sun CM et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. 2007;204(8):1775–85.
Haribhai D et al. A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity. 2011;35(1):109–22.
Lathrop SK et al. Peripheral education of the immune system by colonic commensal microbiota. Nature. 2011;478(7368):250–4.
Wang S et al. MyD88 adaptor-dependent microbial sensing by regulatory T cells promotes mucosal tolerance and enforces commensalism. Immunity. 2015;43(2):289–303. This study demonstrates the important role for MyD88-dependent microbial sensing by Treg cells in promoting immunological tolerance by anti-microbial IgA responses.
Kawamoto S et al. Foxp3(+) T cells regulate immunoglobulin a selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity. 2014;41(1):152–65. This article reviews the contribution of Foxp3 + T cells in diversification of gut microbiota.
Pandiyan P, Zhu J. Origin and functions of pro-inflammatory cytokine producing Foxp3+ regulatory T cells. Cytokine. 2015;76(1):13–24. This study reviews the mechanisms of induction of effector cytokines in Foxp3 + Treg cells.
Noval Rivas M et al. Regulatory T cell reprogramming toward a Th2-cell-like lineage impairs oral tolerance and promotes food allergy. Immunity. 2015;42(3):512–23. Study shows reprogramming of Treg cells into Th2-like cells under the action of IL-4R signaling. Interruption of this process might provide candidate therapeutic strategies in food allergy.
Wing K et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322(5899):271–5.
Huang CT et al. Role of LAG-3 in regulatory T cells. Immunity. 2004;21(4):503–13.
Garin MI et al. Galectin-1: a key effector of regulation mediated by CD4 + CD25+ T cells. Blood. 2007;109(5):2058–65.
Grossman WJ et al. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity. 2004;21(4):589–601.
Pandiyan P et al. CD4 + CD25 + Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol. 2007;8(12):1353–62.
Collison LW et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450(7169):566–9.
Li MO et al. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146.
Koch MA et al. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. 2009;10(6):595–602.
Zheng Y et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature. 2009;458(7236):351–6.
Chaudhry A et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science. 2009;326(5955):986–91.
Chung Y et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med. 2011;17(8):983–8.
Rouse BT, Sarangi PP, Suvas S. Regulatory T cells in virus infections. Immunol Rev. 2006;212:272–86.
Arpaia N et al. A distinct function of regulatory T cells in tissue protection. Cell. 2015;162(5):1078–89. This providing a new role for Treg cells in tissue protection.
Fyhrquist N et al. Foxp3+ cells control Th2 responses in a murine model of atopic dermatitis. J Investig Dermatol. 2012;132(6):1672–80.
Vudattu NK, Herold KC. Delayed anti-CD3 therapy in a mouse heart transplant model induced tolerance and long-term survival of allograft: achieving tolerance. Immunotherapy. 2013;5(11):1173–6.
Ait-Oufella H et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med. 2006;12(2):178–80.
Torgerson TR, Ochs HD. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked: forkhead box protein 3 mutations and lack of regulatory T cells. J Allergy Clin Immunol. 2007;120(4):744–50. quiz 751–2.
Gambineri E et al. Clinical and molecular profile of a new series of patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome: inconsistent correlation between forkhead box protein 3 expression and disease severity. J Allergy Clin Immunol. 2008;122(6):1105–12. e1.
Kucuk ZY, et al. A challenging undertaking: Stem cell transplantation for immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. J Allergy Clin Immunol. 2015. doi:10.1016/j.jaci.2015.09.030. This publication highlights HSCT long-term outcomes in patients with IPEX syndrome.
Verbsky JW, Chatila TA. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) and IPEX-related disorders: an evolving web of heritable autoimmune diseases. Curr Opin Pediatr. 2013;25(6):708–14.
Sadlack B et al. Generalized autoimmune disease in interleukin-2-deficient mice is triggered by an uncontrolled activation and proliferation of CD4+ T cells. Eur J Immunol. 1995;25(11):3053–9.
Sadlack B et al. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell. 1993;75(2):253–61.
Suzuki H et al. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor beta. Science. 1995;268(5216):1472–6.
Snow JW et al. Loss of tolerance and autoimmunity affecting multiple organs in STAT5A/5B-deficient mice. J Immunol. 2003;171(10):5042–50.
Sharfe N et al. Human immune disorder arising from mutation of the alpha chain of the interleukin-2 receptor. Proc Natl Acad Sci U S A. 1997;94(7):3168–71.
Caudy AA et al. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 lymphocytes. J Allergy Clin Immunol. 2007;119(2):482–7.
Goudy K et al. Human IL2RA null mutation mediates immunodeficiency with lymphoproliferation and autoimmunity. Clin Immunol. 2013;146(3):248–61.
Bezrodnik L et al. Follicular bronchiolitis as phenotype associated with CD25 deficiency. Clin Exp Immunol. 2014;175(2):227–34.
Fontenot JD et al. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6(11):1142–51.
Barron L et al. Cutting edge: mechanisms of IL-2-dependent maintenance of functional regulatory T cells. J Immunol. 2010;185(11):6426–30.
Maloy KJ, Powrie F. Fueling regulation: IL-2 keeps CD4+ Treg cells fit. Nat Immunol. 2005;6(11):1071–2.
Williams MA, Tyznik AJ, Bevan MJ. Interleukin-2 signals during priming are required for secondary expansion of CD8+ memory T cells. Nature. 2006;441(7095):890–3.
Pipkin ME et al. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity. 2010;32(1):79–90.
Felices M et al. Functional NK cell repertoires are maintained through IL-2R alpha and Fas ligand. J Immunol. 2014;192(8):3889–97.
Cui Y et al. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol Cell Biol. 2004;24(18):8037–47.
Bernasconi A et al. Characterization of immunodeficiency in a patient with growth hormone insensitivity secondary to a novel STAT5b gene mutation. Pediatrics. 2006;118(5):e1584–92.
Nadeau K, Hwa V, Rosenfeld RG. STAT5b deficiency: an unsuspected cause of growth failure, immunodeficiency, and severe pulmonary disease. J Pediatr. 2011;158(5):701–8.
Bezrodnik L et al. Long-term follow-up of STAT5B deficiency in three argentinian patients: clinical and immunological features. J Clin Immunol. 2015;35(3):264–72.
Kofoed EM et al. Growth hormone insensitivity associated with a STAT5b mutation. N Engl J Med. 2003;349(12):1139–47.
Qureshi OS et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332(6029):600–3.
Lo B et al. AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science. 2015;349(6246):436–40. It is providing a mechanistic view of LRBA in controlling CTLA4 expression and highlighting response to Abetacept in LRBA deficient patients.
Charbonnier LM et al. Regulatory T-cell deficiency and immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like disorder caused by loss-of-function mutations in LRBA. J Allergy Clin Immunol. 2015;135(1):217–27. Study shows increased TFH and decreased TFR cell in LRBA-deficient patients and their implication in the development of autoantibodies.
Kuehn HS et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science. 2014;345(6204):1623–7. This article demonstrates that CTLA4 haploinsufficiency in human might present with IPEX like phenotype.
Schubert D et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med. 2014;20(12):1410–6. This study reported a spectrum of genetic alterations leading to defective CTLA-4 function.
Lopez-Herrera G et al. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet. 2012;90(6):986–1001.
Alangari A et al. LPS-responsive beige-like anchor (LRBA) gene mutation in a family with inflammatory bowel disease and combined immunodeficiency. J Allergy Clin Immunol. 2012;130(2):481–8. e2.
Serwas NK et al. Atypical manifestation of LRBA deficiency with predominant IBD-like phenotype. Inflamm Bowel Dis. 2015;21(1):40–7.
Revel-Vilk S et al. Autoimmune lymphoproliferative syndrome-like disease in patients with LRBA mutation. Clin Immunol. 2015;159(1):84–92. This study emphasizes that LRBA deficiency should be considered in patients presenting with ALPS like phenotype.
Lee S, et al. Abatacept alleviates severe autoimmune symptoms in a patient carrying a de novo variant in CTLA-4. J Allergy Clin Immunol. 2015. This article shows the positive effect of abatacept in CTLA4 haploinsuuficiency.
Seidel MG et al. Long-term remission after allogeneic hematopoietic stem cell transplantation in LPS-responsive beige-like anchor (LRBA) deficiency. J Allergy Clin Immunol. 2015;135(5):1384–90 e1-8.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Talal Chatila and Fayhan Alroqi declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by the authors.
Additional information
This article is part of the Topical Collection on Immune Deficiency and Dysregulation
Rights and permissions
About this article

Cite this article
Alroqi, F.J., Chatila, T.A. T Regulatory Cell Biology in Health and Disease. Curr Allergy Asthma Rep 16, 27 (2016). https://doi.org/10.1007/s11882-016-0606-9
Published:
DOI: https://doi.org/10.1007/s11882-016-0606-9