
Abstract
Since the first discovery of coronin in the amoeba Dictyostelium discoideum, remarkable insights have been gained regarding the structure and function of coronins, highly conserved from yeast to humans. It has been speculated that coronins have evolved from actin-binding molecules in lower eukaryotes to regulators of various cellular processes in mammals. Indeed, coronins are not only involved in cytokinesis, cell motility, and other actin-related processes but they are also implicated in immune homeostasis and calcium–calcineurin signaling. Most strikingly, coronin 1 deficiencies give rise to immune deficiencies in mice and humans that are characterized by severe T lymphocytopenia. Whereas complete absence of coronin 1A is associated with severe combined immunodeficiency in humans, hypomorphic mutations lead to a profound defect in naïve T cells, expansion of oligoclonal memory T cells, and exquisite susceptibility to EBV-associated B cell lymphoproliferation. Recent publications show that coronin 1A also plays a role in natural killer cell cytotoxic function and in neurobehavioral processes. It can be expected that future identification of coronin 1A-deficient patients will further extend the phenotypic spectrum thereby increasing our knowledge of this fascinating molecule.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Coronin was initially isolated in the amoeba Dictyostelium discoideum as a soluble component that binds to actin–myosin complexes [1]. Antibodies against coronin demonstrated a crown-like localization of the coronin protein on the dorsal structure of the cells. This gave rise to the name coronin from the Latin “corona” meaning “crown” [2]. Dictyostelium mutants lacking coronin have been shown to be defective in cytokinesis, cell motility, and actin-related processes [3]. Therefore, coronin was designated as an actin-binding protein. It belongs to a large family of highly conserved actin regulatory proteins. Through a recent bioinformatical query 723 coronin molecules have been found in 358 species [4]. Coronins are truly ancient proteins that are conserved from yeast to humans [5], whereas they are not found in bacteria.
Mammalian coronin 1 is the best-known member of the highly conserved coronin family [6]. Most coronins have a similar structure. The overall structure of type I/type II coronin protein is represented schematically in Fig. 1 through the example of human coronin 1A. It comprises an amino-terminal tryptophan–aspartate (WD) repeat-containing region, a unique region (U) of variable length, and a carboxy-terminal coiled coil domain (CC) that is required for oligomerization [7] [8]. WD repeats are minimally conserved regions of around 40 amino acids. They are thought to facilitate the formation of heterotrimeric or multiprotein complexes. The WD repeat of coronin 1 forms a beta propeller [9], a structural motif found in many signaling and adaptor proteins that might function as a scaffold protein [2]. The so-called type III coronins lack the coiled coil domains. As they contain two core WD repeat-containing regions fused to one another, they are also named “tandem” coronins [2, 6]. Seven mammalian coronin genes have been identified so far. Coronin 1 to 6 present the classical coronin structure, whereas coronin 7 differs significantly from other mammalian coronins in its domain architecture. It is a tandem coronin molecule without coiled coil domain (Table 1) [2]. Coronin 7 is the first coronin protein proven to localize to the Golgi membrane. It is thought to play a role in the organization of intracellular membrane compartments and vesicular trafficking rather than in remodeling the cytoskeleton [10].

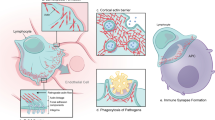
a Schematic representation of CORO1A. Mutations identified in mice and humans are indicated above and below the diagram of the protein, respectively. The major structural protein domains are depicted: CC coiled coil domain, CE C-terminal extension, NE N-terminal extension, U unique region. References for depicted mutations: 29,39 32. b Top and side view (left and right, respectively) of the beta propeller of murine coronin 1. This ribbon representation of the three-dimensional structure was generated through the Protein Data bank (pdb-ID 2B4E) 9
Structure and Function of Coronin 1A
Human coronin 1A (CORO1A) is also known as TACO (for tryptophan–aspartate-containing coat protein), Clipin A, or p57 [11]. The human CORO1A gene maps to chr. 16p11.2 and consists of 11 exons. It codes for coronin 1A, a highly conserved 57-kDa actin-binding protein of 461aa [11] that is expressed in a variety of hematopoietic and immune cells [12]. There is a 95 % sequence homology on the protein level between human and mouse (Table 1) [13]. The murine coronin 1 molecules are coiled coil-mediated homotrimeric complexes and associate with the plasma membrane and with the cytoskeleton via two distinct domains [7]. Their WD repeat domain forms a seven-bladed beta propeller (Fig. 1) [9].
Coronin 1 has been shown to associate with filamentous (F)-actin and the actin-related protein 2/3 complex (ARP2/3 complex) [14, 15]. It has been suggested that the N-terminal WD repeat-containing domain mediates the membrane interaction [7]. With regard to other mammalian coronins, coronin 4 also associates with F-actin [16]. There are some reports indicating that coronin 2 might bind to F-actin [17], but other studies demonstrate that it fails to interact with F-actin in cells [18]. This also holds true for coronin 7, the first coronin protein that has been shown to localize to the Golgi membrane [10]. It has therefore been speculated that coronin 7 might take part in the organization of intracellular membrane compartments and vesicular trafficking rather than in remodeling the cytoskeleton [10]. Coronins are thought to have evolved from actin-binding molecules in lower eukaryotes to become regulators of diverse cellular processes in mammals [19••].
Role of Coronin 1 in Innate Immune Cells
The first indications to the function of coronin 1 in mammals were revealed through studies of the immune evasion mechanisms used by Mycobacterium tuberculosis. In macrophages, a WD repeat protein was recruited and actively retained at the cytoplasmic side of phagosomes that contained living but not dead mycobacteria [20]. This protein was termed TACO for “tryptophan–aspartate-containing coat protein” and had been shown to be a component of the phagosome coat that is released prior to phagosome fusion with or maturation into lysosomes. By peptide sequence analysis and cDNA cloning, TACO was shown to be homologous to p57, a previously identified protein of unknown function that had been co-purified with phospholipase C from human leukocytes [11]. TACO is now usually referred to as coronin 1A. In mice, coronin 1 promotes calcium–calcineurin signaling upon infection of macrophages by pathogenic mycobacteria [21]. It has also been shown to localize to phagosomes that contain Mycobacterium leprae [22] and is implicated in immune evasion of virulent strains of Helicobacter pylori [23].
In contrast to the well-documented role of coronin 1 in macrophages, there seems to be no discernable phenotype in other innate immune cells in coronin 1-deficient mice. It has been shown that neutrophil populations and dendritic cells develop normally in mice lacking coronin 1 [24, 25•]. Coronin 1-deficient neutrophils have a normal function regarding adherence, membrane dynamics, migration, phagocytosis, and oxidative burst [24]. Likewise, the function of dendritic cells was not impaired in coronin 1-deficient mice, in which antigen processing and presentation by dendritic cells was comparable to the one found in wild-type mice [25•].
Role of Coronin 1 in Lymphocytes
Coronin 1-deficient mice show important T lymphocytopenia, while B and natural killer (NK) cells are present in normal numbers [15, 26–29]. Coronin 1 has been shown important for T cell homeostasis [15, 27] and T cell receptor signaling [19••, 28, 30].
Human Coronin 1A Deficiency
Human coronin 1A deficiency (CORO1A, OMIM 605000) has been first identified in a girl who presented with a phenotype of severe combined immunodeficiency (SCID) (Table 2) [29]. Since the first year of life, the patient experienced recurrent respiratory infections and oral thrush. At 13 months of age, she developed severe mucocutaneous chickenpox after live attenuated varicella vaccine [29, 31]. The immunophenotype evidenced reduced T cell and very low naïve T cell counts, whereas B and NK cells were present in normal numbers. Further immunological work-up revealed low proliferative responses to mitogens and absent proliferation upon stimulation with antigens. Total levels of serum immunoglobulins were normal for age, but specific antibodies were poor despite regular vaccination. The immunophenotype resembled T-negative SCID; therefore, the indication for hematopoietic stem cell transplantation (HSCT) was made [31].
Recently, the phenotypic spectrum of coronin 1A deficiency has been extended through the description of hypomorphic CORO1A mutations in three siblings from a consanguineous family (Table 2) [32•]. All three patients presented severe CD4+ T cell lymphopenia with massively reduced numbers of CD4+ and CD8+ naïve T cells and aggressive EBV-associated B cell lymphoproliferation at an early age. Normal T cell proliferation was observed upon stimulation with mitogens in sibling 1, slightly reduced proliferation in sibling 3. Due to an absent material, sibling 2 could not be explored. Proliferation to OKT3 and antigens was normal in sibling 1, absent despite normal vaccination in sibling 3. Additional immunological features were the impaired development of a diverse T cell repertoire, near to absent invariant natural killer T (iNKT) cells and severely diminished mucosal-associated invariant T (MAIT) cells. All three patients presented with recurrent ear, nose, and throat, as well as upper respiratory infections, and sibling 1 developed bronchiectasis. Two of them had uneventful chickenpox at 4 years and 4 months, respectively, and sibling 1 presented with leishmaniasis at 7 years of age. Intriguingly, all three of them developed aggressive EBV-associated B cell lymphoproliferation at an early age (12, 7.5, and 14 months, respectively) [32•]. The oldest sibling is still alive and in remission from his aggressive B cell lymphoproliferation. His two younger sisters died; one in the course of chemotherapy for disseminated B cell lymphoproliferation, the other due to overwhelming graft-versus-host disease after phenoidentical parental HSCT.
Molecular Findings and Clinical Implications
The first reported patient [29, 31] presented a heterozygous 2-bp deletion in exon 3 of the CORO1A gene (c.248_249delCT) leading to a frameshift and premature truncation (p.83RfsX10) inherited from the unaffected father. On the other allele, there was a heterozygous 600-kb de novo deletion of chromosome 16p11.2 encompassing 24 genes, including CORO1A. This patient presents thus a complete absence of functional coronin 1A.
Genome-wide homozygosity mapping and whole-exome sequencing studies allowed the identification of a homozygous V134M mutation in CORO1A in three siblings of a consanguineous family [32•]. A residual expression of the mutated protein has been shown suggesting a hypomorphic mutation [32•].
There seems to be a genotype–phenotype correlation in the patients reported so far. The CORO1A-deficient patient reported by Shiow et al. had a significantly more severe clinical phenotype than the hypomorphic patients. This patient presented failure to thrive and developed a severe varicella infection upon attenuated live vaccination. She had also repeated infections despite prophylaxis with trimethoprim, sulfamethoxazole, and intravenous immunoglobulin. HSCT was performed at the age of 4 years [29, 31]. EBV-induced B cell lymphoproliferation was not reported, but this patient may not have encountered EBV prior to HSCT.
In contrast to this patient with complete absence of coronin 1A, the three siblings with the homozygous V134M mutation had a significantly diminished but still detectable protein expression as evidenced in FACS and Western blot analysis [32•]. Thus, residual protein function may account for the milder immunological phenotype when compared to the first patient [29, 31]. The expected identification of other mutations in the CORO1A gene in the future may considerably extend the clinical phenotype and thereby contribute to further elucidate the genotype–phenotype correlation. Recently, Mace et al. reported a new patient who was compound heterozygous for two mutations in the CORO1A gene: the P83RfsX10 mutation that has already been described by Shiow et al. [29] and a new Q360EfsX44 mutation.
Interestingly, and in sharp contrast to patients with severe combined immunodeficiencies due to other genetic conditions who have an aplastic thymus, the thymus size was found normal in all reported CORO1A-deficient patients despite the extremely low naïve T cell counts [29, 31, 32•].
It was therefore initially speculated that CORO1A-immunodeficiency might result from a thymus egress defect [29, 33]. This hypothesis was based on the former observation of a thymic egress anomaly found in a mouse strain with profound peripheral T cell deficiency (Ptcd) [34]. The Ptcd locus was later reported to harbor a mutation (E26K) in the Coro1a gene [29]. However, the E26K mutation is a gain of function mutation. Therefore, it is rather unlikely that the defective thymic egress observed in the Ptcd mice is the cause for the T cell immunodeficiency in patients, as these patients carry loss of function mutations of CORO1A. Instead, a survival deficiency mechanism could explain the T cell immunodeficiency. If this hypothesis is correct, coronin 1A deficiency may belong, together with Dedicator of cytokinesis 8 protein (DOCK8) and mammalian sterile 20-like kinase 1 (MST1)/serine–threonine protein kinase 4 (STK4) deficiencies [35–38] to a group of T cell immunodeficiencies that are characterized by the loss of naïve T cells due to survival defects. It remains to be elucidated if there exists a functional link between the three molecules CORO1A, DOCK8, and MST1/STK4.
Immunological Phenotype of the Homozygous V134M CORO1A Deficiency
The three siblings with the homozygous V134M CORO1A mutation show a very particular T cell immunodeficiency. They have a profound defect in naïve T cells and an expansion of oligoclonal memory T cells associated with a particular susceptibility to EBV infection. It has been shown previously that null and hypomorphic mutation of coronin 1 in mice are associated with defects in T cell survival and migration [15, 29]. The preserved thymus size despite the virtual absence of circulating naïve T cells in the patients may suggest that coronin 1A deficiency is predominantly a T cell immunodeficiency caused by impaired survival of mature T cells hereby impacting on lymphocyte homeostasis, repertoire selection, and lineage commitment. The fact that the patients show an abnormal T cell antigen receptor (TCR) repertoire is supporting this hypothesis [32•].
There have been different suggestions in order to explain the altered T cell survival in coronin 1A deficiency. It had been speculated that it might be due to defective TCR-mediated activation notably defective calcium signaling [30]. Otherwise, it has been proposed that coronin 1A inhibits branched F-actin formation through binding of ARP2/3 [29] and contributes thereby to TCR-induced immunological synapse formation and signaling [28]. Thus, conversely, accumulation of F-actin may eventually lead to cell death in mature T cells in coronin 1A deficiency [15], although accumulation of F-actin alone has been shown not sufficient for inducing apoptosis [26].
Recently, the role of coronin 1A in natural killer (NK) cells has been elucidated through the careful study of NK cells derived from a CORO1A-deficient patient and in NK cell lines [39•]. Notably, it has been shown that coronin 1A is required for NK cell cytotoxicity. In fact, coronin 1A mediates the disassembly of F-actin at the immunological synapse and thereby facilitates the effective lytic granule secretion in NK cells.
Associated Clinical Features
Intriguingly, the only surviving sibling carrying the homozygous V134M mutation [32•] [40••] and the compound heterozygous patient described by Shiow et al. [31] present both a delay in language acquisition as well as behavioral and cognitive impairment. Interestingly, 16p11.2 microdeletions and duplications have been previously described to be associated with a high frequency of cognitive, developmental, and speech delay as well as behavior abnormalities including attention deficit hyperactivity disorder (ADHD) and autism spectrum disorders [41–44].
Further research is needed to study the possible relation of these cognitive problems to CORO1A deficiencies in humans. However, recent studies in coronin 1-deficient mice have shown that coronin 1 has an essential role for the modulation of the cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA) signaling [40••]. The absence of coronin 1 in mice results in severe functional defects at excitatory synapses and considerable neurobehavioral disabilities. Treatment with a membrane-permeable analog of cAMP was able to restore the synaptic plasticity and the behavioral defects in mice lacking coronin 1 [40••]. Furthermore, a very recent study showed that coronin is implicated in regulation of actin organization and cell morphology during postembryonic neuroblast migration and neuritogenesis in Caenorhabditis elegans [45••].
Treatment and Long-Term Survival
Hematopoietic stem cell transplantation may be a curative treatment option to be considered in patients with severe T cell immunodeficiency. So far, HSCT was performed successfully only in the first patient who received a matched unrelated cord blood hematopoietic cell transplant following cytoreductive conditioning with fludarabine, melphalan, and monoclonal anti-hCD52 (alemtuzumab) at 4 years of age [29, 31]. This patient reached full myeloid and lymphoid donor chimerism and complete immune reconstitution [46]. Out of the three affected siblings carrying the homozygous V134M mutation, only the oldest sibling survived. He had not been considered for HSCT. He is currently 15 years old, in complete remission from his B cell lymphoproliferation, and does not present any major infectious complications. His respiratory function stabilized under prophylactic treatment with trimethoprim/sulfamethoxazole, azithromycin, and intravenous immunoglobulins. His younger sisters both died, one due to GVHD after a HSCT from her phenoidentical mother, the other due to complications of chemotherapy administered for the disseminated EBV-associated B cell lymphoproliferation [32•].
Conclusion
The identification of more CORO1A-deficient patients will undoubtedly broaden the phenotypic spectrum of clinical manifestations and help the clinicians to establish precise treatment recommendations. Whereas complete absence of CORO1A seems to be associated with a T− SCID requiring HSCT, the indication for HSCT in hypomorphic patients depends clearly on the precise clinical and immunological phenotype. Prophylaxis with trimethoprim, sulfamethoxazole, and intravenous immunoglobulin are required. EBV (primo) infection needs to be monitored carefully as CORO1A-deficient patients are at major risk to develop EBV-related B cell lymphoproliferation/lymphoma requiring prompt immunotherapy and chemotherapy, whenever necessary.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
de Hostos EL, Bradtke B, Lottspeich F, Guggenheim R, Gerisch G. Coronin, an actin binding protein of Dictyostelium discoideum localized to cell surface projections, has sequence similarities to G protein beta subunits. EMBO J. 1991;10(13):4097–104.
Chan KT, Creed SJ, Bear JE. Unraveling the enigma: progress towards understanding the coronin family of actin regulators. Trends Cell Biol. 2011;21(8):481–8.
de Hostos EL, Rehfuess C, Bradtke B, et al. Dictyostelium mutants lacking the cytoskeletal protein coronin are defective in cytokinesis and cell motility. J Cell Biol. 1993;120(1):163–73.
Eckert C, Hammesfahr B, Kollmar M. A holistic phylogeny of the coronin gene family reveals an ancient origin of the tandem-coronin, defines a new subfamily, and predicts protein function. BMC Evol Biol. 2011;11:268.
Xavier CP, Eichinger L, Fernandez MP, Morgan RO, Clemen CS. Evolutionary and functional diversity of coronin proteins. Sub-cellular Biochem. 2008;48:98–109.
de Hostos EL. The coronin family of actin-associated proteins. Trends Cell Biol. 1999;9(9):345–50.
Gatfield J, Albrecht I, Zanolari B, Steinmetz MO, Pieters J. Association of the leukocyte plasma membrane with the actin cytoskeleton through coiled coil-mediated trimeric coronin 1 molecules. Mol Biol Cell. 2005;16(6):2786–98.
Kammerer RA, Kostrewa D, Progias P, et al. A conserved trimerization motif controls the topology of short coiled coils. Proc Natl Acad Sci U S A. 2005;102(39):13891–6.
Appleton BA, Wu P, Wiesmann C. The crystal structure of murine coronin-1: a regulator of actin cytoskeletal dynamics in lymphocytes. Structure. 2006;14(1):87–96.
Rybakin V, Stumpf M, Schulze A, Majoul IV, Noegel AA, Hasse A. Coronin 7, the mammalian POD-1 homologue, localizes to the Golgi apparatus. FEBS Lett. 2004;573(1–3):161–7.
Suzuki K, Nishihata J, Arai Y, et al. Molecular cloning of a novel actin-binding protein, p57, with a WD repeat and a leucine zipper motif. FEBS Lett. 1995;364(3):283–8.
Oku T, Itoh S, Okano M, et al. Two regions responsible for the actin binding of p57, a mammalian coronin family actin-binding protein. Biol Pharm Bull. 2003;26(4):409–16.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10.
Rybakin V, Clemen CS. Coronin proteins as multifunctional regulators of the cytoskeleton and membrane trafficking. BioEssays News Rev Mol Cell Dev Biol. 2005;27(6):625–32.
Foger N, Rangell L, Danilenko DM, Chan AC. Requirement for coronin 1 in T lymphocyte trafficking and cellular homeostasis. Science. 2006;313(5788):839–42.
Huang W, Ghisletti S, Saijo K, et al. Coronin 2A mediates actin-dependent de-repression of inflammatory response genes. Nature. 2011;470(7334):414–8.
Cai L, Makhov AM, Bear JE. F-actin binding is essential for coronin 1B function in vivo. J Cell Sci. 2007;120(Pt 10):1779–90.
Cai L, Marshall TW, Uetrecht AC, Schafer DA, Bear JE. Coronin 1B coordinates Arp2/3 complex and cofilin activities at the leading edge. Cell. 2007;128(5):915–29.
Pieters J, Muller P, Jayachandran R. On guard: coronin proteins in innate and adaptive immunity. Nat Rev Immunol. 2014;13(7):510–8. A very recent and comprehensive review on the coronin proteins and the many facettes of their functions.
Ferrari G, Langen H, Naito M, Pieters J. A coat protein on phagosomes involved in the intracellular survival of mycobacteria. Cell. 1999;97(4):435–47.
Jayachandran R, Sundaramurthy V, Combaluzier B, et al. Survival of mycobacteria in macrophages is mediated by coronin 1-dependent activation of calcineurin. Cell. 2007;130(1):37–50.
Suzuki K, Takeshita F, Nakata N, Ishii N, Makino M. Localization of CORO1A in the macrophages containing Mycobacterium leprae. Acta Histochem Cytochemica. 2006;39(4):107–12.
Zheng PY, Jones NL. Helicobacter pylori strains expressing the vacuolating cytotoxin interrupt phagosome maturation in macrophages by recruiting and retaining TACO (coronin 1) protein. Cell Microbiol. 2003;5(1):25–40.
Combaluzier B, Pieters J. Chemotaxis and phagocytosis in neutrophils is independent of coronin 1. J Immunol. 2009;182(5):2745–52.
Westritschnig K, BoseDasgupta S, Tchang V, Siegmund K, Pieters J. Antigen processing and presentation by dendritic cells is independent of coronin 1. Mol Immunol. 2013;53(4):379–86. First report on the role of coronin 1 in dendritic cells.
Haraldsson MK, Louis-Dit-Sully CA, Lawson BR, et al. The lupus-related Lmb3 locus contains a disease-suppressing Coronin-1A gene mutation. Immunity. 2008;28(1):40–51.
Mueller P, Massner J, Jayachandran R, et al. Regulation of T cell survival through coronin-1-mediated generation of inositol-1,4,5-trisphosphate and calcium mobilization after T cell receptor triggering. Nat Immunol. 2008;9(4):424–31.
Mugnier B, Nal B, Verthuy C, et al. Coronin-1A links cytoskeleton dynamics to TCR alpha beta-induced cell signaling. PLoS One. 2008;3(10):e3467.
Shiow LR, Roadcap DW, Paris K, et al. The actin regulator coronin 1A is mutant in a thymic egress-deficient mouse strain and in a patient with severe combined immunodeficiency. Nat Immunol. 2008;9(11):1307–15.
Mueller P, Liu X, Pieters J. Migration and homeostasis of naive T cells depends on coronin 1-mediated prosurvival signals and not on coronin 1-dependent filamentous actin modulation. J Immunol. 2011;186(7):4039–50.
Shiow LR, Paris K, Akana MC, Cyster JG, Sorensen RU, Puck JM. Severe combined immunodeficiency (SCID) and attention deficit hyperactivity disorder (ADHD) associated with a Coronin-1A mutation and a chromosome 16p11.2 deletion. Clin Immunol. 2009;131(1):24–30.
Moshous D, Martin E, Carpentier W, et al. Whole-exome sequencing identifies Coronin-1A deficiency in 3 siblings with immunodeficiency and EBV-associated B-cell lymphoproliferation. J Allergy Clin Immunol. 2013;131(6):1594–603. In this study, the second kindred with CORO1A deficiency is described. The patients, three siblings, presented a hypomorphic mutation in Coro1a leading to a combined immunodeficiency and particular predisposition to develop EBV-associated B-cell lymphoproliferation at an very early age.
Hogquist KA. Immunodeficiency: when T cells are stuck at home. Nat Immunol. 2008;9(11):1207–8.
Yagi H, Matsumoto M, Nakamura M, et al. Defect of thymocyte emigration in a T cell deficiency strain (CTS) of the mouse. J Immunol. 1996;157(8):3412–9.
Engelhardt KR, McGhee S, Winkler S, et al. Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. J Allergy Clin Immunol. 2009;124(6):1289–302 e4.
Nehme NT, Pachlopnik Schmid J, Debeurme F, et al. MST1 mutations in autosomal recessive primary immunodeficiency characterized by defective naive T-cell survival. Blood. 2012;119(15):3458–68.
Abdollahpour H, Appaswamy G, Kotlarz D, et al. The phenotype of human STK4 deficiency. Blood. 2012;119(15):3450–7.
Mou F, Praskova M, Xia F, et al. The Mst1 and Mst2 kinases control activation of rho family GTPases and thymic egress of mature thymocytes. J Exp Med. 2012;209(4):741–59.
Mace EM, Orange JS. Lytic immune synapse function requires filamentous actin deconstruction by Coronin 1A. Proc Natl Acad Sci U S A. 2014;111(18):6708–13. This is the first report on the role of Coronin 1A for NK cell function.
Jayachandran R, Liu X, Bosedasgupta S, et al. Coronin 1 regulates cognition and behavior through modulation of cAMP/protein kinase A signaling. PLoS Biol. 2014;12(3):e1001820. Important work showing the critical role for coronin 1 in neurobehavior in mice and humans through regulating the transmission of signals within cells. It is shown that coronin 1 modulates the cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA) signaling.
Weiss LA, Shen Y, Korn JM, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358(7):667–75.
Rosenfeld JA, Coppinger J, Bejjani BA, Girirajan S, Eichler EE. Speech delays and behavioral problems are the predominant features in individuals with developmental delays and 16p11.2 microdeletions and microduplications. J Neurodev Disord. 2010;2:26–38.
Kumar RA, KaraMohamed S, Sudi J, et al. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet. 2008;17(4):628–38.
Shinawi M, Liu P, Kang SH, et al. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J Med Genet. 2010;47(5):332–41.
Shen Z, Zhang X, Chai Y, et al. Conditional knockouts generated by engineered crispr-cas9 endonuclease reveal the roles of coronin in C. elegans neural development. Dev Cell. 2014. doi:10.1016/j.devcel.2014.07.017. A very recent study showing very elegantly through a conditional knockout strategy that Coronin is implicated in regulation of actin organization and cell morphology during postembryonic neuroblast migration and neuritogenesis in C. elegans.
Shiow LR, Paris K, Puck JM. Severe combined immunodeficiency due to absent coronin-1a. Primary immunodeficiency diseases a molecular and genetic approach 2014; 3rd ed. In: Ochs HD, Smith E, and Puck, JM, editors. 295.
Okumura M, Kung C, Wong S, Rodgers M, Thomas ML. Definition of family of coronin-related proteins conserved between humans and mice: close genetic linkage between coronin-2 and CD45-associated protein. DNA Cell Biol. 1998;17(9):779–87.
Acknowledgments
This work was supported by institutional grants from INSERM, Ligue Nationale contre le Cancer (Equipe Labellisée La Ligue), INCa, Institut Imagine, and the European Research Council (PIDIMMUN grant no. 249816).
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Despina Moshous and Jean-Pierre de Villartay report no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Immune Deficiency and Dysregulation
Rights and permissions
About this article

Cite this article
Moshous, D., de Villartay, JP. The Expanding Spectrum of Human coronin 1A deficiency. Curr Allergy Asthma Rep 14, 481 (2014). https://doi.org/10.1007/s11882-014-0481-1
Published:
DOI: https://doi.org/10.1007/s11882-014-0481-1
Keywords
- Coronin 1A
- Coronin
- Actin-binding molecules
- Actin-related protein 2/3 (ARP2/3) complex
- F-actin
- Immune homeostasis
- Calcium–calcineurin signaling
- Primary immunodeficiency
- T cell immunodeficiency
- Severe combined immune deficiency
- EBV-associated B cell lymphoproliferation
- Thymus
- Invariant natural killer T cell
- Mucosal-associated invariant T cell