
Opinion statement
Hormone receptor positive (HR+), human epidermal growth factor receptor 2 negative (HER-2-) metastatic breast cancer (MBC) is the most common subtype of breast cancer. Due to therapeutic advances with molecularly targeted therapies, the prognosis for patients with metastatic disease has improved significantly. The advent of CDK4/6 inhibitors (CDK4/6i) has changed the treatment paradigm for patients with HR+HER2-MBC. CDK4/6i allowed for marked improvement in overall survival, delaying the time to chemotherapy initiation, and improved quality of life for our patients. Efforts are now focused on the best approach(es) for patients after progression on CDK4/6i. Can we further harness the benefit of CDK4/6i in novel combinations at the time of progression? Should we continue CDK4/6i or proceed other novel agents or endocrine therapies? As we advance our treatment strategies for HR+HER2-MBC, there is no longer a one-size-fits-all model, but instead a multifaceted and personalized approach lending to improved outcomes for our patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction: CDK4/6 inhibition in metastatic hormone receptor positive breast cancer
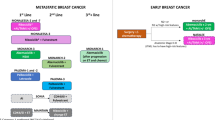
Due to therapeutic advances with molecularly targeted therapies, the prognosis for patients with HR + HER2-MBC has improved significantly, due in large part to the approval of CDK4/6i [1]. The landmark, phase III trials leading to the FDA approval of CDK4/6i demonstrated a striking OS benefit of 60 months or more in postmenopausal women and men [2••, 3, 674••, 5••, 8••]. The trials showed a similar improvement in PFS from 14 to 25–30 months when used in combination with ET, rather than ET alone [9,10,11,12,13, 14]. Likewise, CDK4/6i demonstrated PFS improvement in the second line with fulvestrant in patients who progressed on an AI alone or had metastatic relapse on adjuvant AI within 12 months or more [12, 14,15,16, 17] (Table 1).
In premenopausal women, the efficacy of CDK4/6i has been studied in a randomized fashion in the front-line setting in the MONALEESA-7 study where ribociclib was used in combination with goserelin and tamoxifen or an AI. The improvement in PFS seen in the ribociclib arm was similar to that seen in postmenopausal women in MONALEESA-2; therefore, ribociclib is the CDK4/6i of choice in the first line for premenopausal women [29]. In the second line setting, palbociclib and fulvestrant are approved irrespective of menopausal status, based on PALOMA-3 which included premenopausal women who received goserelin [30]. Abemaciclib can also be used in premenopausal patients in the second line setting with fulvestrant based on data with 72 pre/perimenopausal patients in combination with a GnRH agonist with a significantly improved PFS and overall response rate (ORR) with a generally tolerable safety profile [3].
In patients with brain metastasis or leptomeningeal involvement, clinicians often preferentially turn to abemaciclib as the CDK4/6i of choice based on data from Tolaney et al., in which abemaciclib was found to have an intracranial clinical benefit rate of 24% in patients with heavily pretreated HR + MBC. Abemaciclib was proven to cross the blood brain barrier, achieving therapeutic concentrations in brain tissue [31]. Both palbociclib and ribociclib may have CNS activity; however, these agents have not been specifically studied in this population.
Even in patients with aggressive, life-threatening disease, the practice of giving chemotherapy rather than CDK4/6i in the first line, is quickly changing with results of the phase II RIGHT Choice trial. For patients treated with ribociclib plus ET in visceral crisis, a 1-year improvement in PFS was seen when compared with combination chemotherapy (24 months ribociclib vs. 12.3 months chemotherapy). Additionally, ribociclib with ET had a similar median time to treatment response as compared to combination chemotherapy (4.9 months ribociclib vs. 3.2 months chemotherapy), moving CDK4/6i to the forefront as the preferred, front-line therapy, even for this patient population with aggressive disease [32•].
Preclinical and clinical efforts are ongoing to better understand resistance mechanisms to CDK4/6i. Investigators are evaluating novel strategies with targeted combinations to inhibit crosstalk between pathways of resistance. Approaches include combining CDK4/6i with SERDs, FGFR3, ERK, AKT, CDK2, and mTOR/PI3K inhibitors, with the goal of improving outcomes for our patients on CDK4/6i.
The great debate: overall survival differences between CDK4/6 inhibitors in the frontline
Despite different designs for the landmark trials evaluating CDK4/6i with ET, the hazard ratios for PFS are strikingly similar (HR 0.54–0.58) (Table 1). As OS data for the randomized trials matured, however, there are apparent differences among the studies, causing clinicians to question which CDK4/6i is the best first line choice for postmenopausal women or men with HR + HER2-MBC.
The randomized, phase III studies involving ribociclib and abemaciclib for use in the frontline have demonstrated an OS benefit compared to ET alone (MONALEESA-2, MONALEESA-3, MONALEESA-7, and MONARCH-3). Palbociclib, alternatively, did not demonstrate an OS benefit in PALOMA-2 where the mOS was numerically longer in the experimental arm (53.9 months with palbociclib and 51.2 months with placebo), but not statistically significant overall (HR 0.956 [95% CI, 0.777–1.777] p = 0.3378) [6].
The difference in OS data in PALOMA-2 could be attributed to a number of factors. Follow-up was not available on notable proportion of patients (21% in placebo arm versus 13% in the palbociclib arm). Due to the regulatory approval of palbociclib during the conduct of PALOMA-2, patients likely withdrew consent to receive commercial CDK4/6i outside of the trial. A post hoc sensitivity analysis, which excluded the patients lost to follow-up, resulted in a mOS of 51.6 months for palbociclib versus 44.6 months for placebo (HR 0.869 [95% CI, 0.706–1.069] [6].
As these studies did not directly compare ribociclib and palbociclib, the results from each trial could truly reflect a difference in efficacy. Many clinicians offer ribociclib as the frontline CDK4/6i of choice, given the mature, statistically significant OS data. Despite abemaciclib demonstrating a consistent OS benefit in randomized phase III trials, ribociclib currently remains the preferred CDK4/6i due to less gastrointestinal issues compared to abemaciclib. Further head-to-head comparison trials are needed to answer the debate as to which CDK4/6i is best in the frontline. This study design will be pursued in the HARMONIA trial, which will randomize patients to receive either palbociclib or ribociclib and address the question of which CDK4/6i is best in the frontline in HER2-enriched patients (NCT05207709).
Continuing a CDK 4/6 inhibitor at the time of progression
Identifying best treatment options following progression on CDK4/6i remains an unmet medical need. One strategy is sequential CDK4/6i after progression. Two phase II randomized trials have been reported evaluating this approach: PACE (evaluating continuation of palbociclib with a change in ET backbone and/or with immunotherapy) and MAINTAIN (changing the CDK4/6i and ET backbone) (Table 1).
The PACE trial is a multicenter, phase II study, in which patients who progressed on prior CDK 4/6i were randomized 1:2:1 to fulvestrant plus palbociclib, fulvestrant alone, fulvestrant plus palbociclib, and avelumab. Palbociclib was the most prevalent CDK4/6i used in the frontline (90.9%), with a minority of patients receiving ribociclib (4.5%) and abemaciclib (4.1%). After a median follow-up of 23.6 months, mPFS was 4.6 months in the fulvestrant + palbociclib arm, 4.8 months in the fulvestrant arm, and 8.1 months in the triplet arm with avelumab. The OS was longer in the avelumab arm than in the other two arms (24.6 vs. 27.5 vs. 42.5 months, respectively). The PACE trial was powered to evaluate the treatment effect of palbociclib with fulvestrant; therefore, data from the avelumab arm is exploratory in nature and has not led to a change in the standard of care. The continued use of palbociclib after progression with adjustment of the ET backbone did not provide a clinical benefit and is therefore not recommended as a treatment strategy at this time [24•].
The phase II MAINTAIN trial evaluated the efficacy of fulvestrant or exemestane + / − ribociclib in patients who had progressed on prior CDK 4/6i (84% palbociclib, 11% ribociclib, 2% abemaciclib). The fulvestrant or exemestane + ribociclib arm had a statistically significant improvement in PFS at 5.33 months versus 2.76 months in the placebo arm (HR = 0.56 (95% CI: 0.37–0.83), p = 0.004). At 6 months, 42% in the ribociclib arm versus 24% on the placebo arm had not progressed. While at 12 months, 25% in the ribociclib arm versus 7% on the placebo arm had not progressed [25•]. These data are now mentioned in NCCN guidelines. It is important to reiterate that MAINTAIN was not a registration trial and did not lead to a change in the label; however, this offers initial evidence that patients benefit from treatment with ribociclib in combination with a switch in their ET backbone after progression on prior CDK4/6i and ET. The hazard ratio for patients who received prior ribociclib was similar to those who received prior palbociclib; however, only 14/199 evaluable patients received prior ribociclib.
The postMONARCH phase III, global, randomized study aims to evaluate whether continuation of abemaciclib with a switch in the ET after progression on a prior CDK4/6i may provide benefit. Eligible patients are randomized 1:1 to receive abemaciclib or placebo plus fulvestrant. Patients who experience a metastatic recurrence on or after treatment with CDK4/6i in the adjuvant setting are also eligible, making this the first study to evaluate this cohort of patients. Completion of accrual is anticipated in summer 2023. We are hopeful the results will guide the optimal therapy following metastatic relapse now that CDK4/6i are deployed in the adjuvant setting and yield another treatment option for our patients [26] (Table 1).
Mechanisms of resistance to CDK4/6 inhibitors
Due to the numerous resistance mechanisms, even the best responders have decreased efficacy of CDK4/6i overtime; however, patients who develop acquired resistance still demonstrate durable clinical benefit typically exceeding 6 months. Many driver events are implicated in resistance to CDK4/6i impacting both intrinsic and acquired resistance from changes in RB1, AURKA, CCNE1/2, CDK2, AKT/mTOR, RAS/MAPK, FGFR1/2, and ERRB2 (Table 2). We will briefly highlight the biologic mechanisms proposed to cause resistance to CDK4/6i and the ongoing efforts to address these pathways of resistance and improve clinical outcomes for patients.
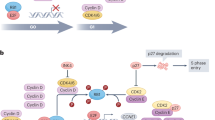
Loss of retinoblastoma protein (Rb1)
The tumor suppressor protein Rb1 is a key checkpoint in the cell cycle. Constitutive activation of the cell cycle can occur when there are mutations in Rb via activation of E2F and cycle-CDK2 axis. With loss of Rb1, there is no longer dependence on CDK4/6. In PALOMA-3, ctDNA was enriched with Rb1 mutations in patients who progressed on palbociclib (1/127 versus 0/68 in placebo arm) [33]. Clinical and preclinical data show Rb1 loss can cause de novo resistance to CDK4/6i. Loss of function mutations in Rb1 found in CDK4/6 naive tumors portend a worse prognosis with decreased PFS [34, 35]. Breast cancer cell lines with acquired resistance to CDK4/6i with Rb1 loss had enhanced sensitivity to the novel AURKA inhibitor (Aurora Kinase A), a serine/threonine kinase that contributes to the regulation of cell cycle progression [36]. A phase Ib trial with erbumine (AURKA inhibitor) after progression on a CDK4/6i is underway. In this early phase trial, a patient with MBC treated with erbumine derived a clinical benefit for 11 months after progression on prior CDK4/6i [36]. Future studies are needed to evaluate if AURKA inhibitors with CDK4/6i could prevent the development of acquired resistance to CDK4/6i.
CCNE1/2 and CDK2 amplification
Cyclin E-CDK2 phosphorylates Rb1, allowing release of E2F, causing progression of the cell cycle of G1 to S phase. CCNE1 encodes cyclin E and, when overexpressed, causes resistance to CDK4/6i [35, 37]. Some CDK4/6i-resistant cells in preclinical studies lose dependence on cyclin D1-CDK4/6 signaling and use the MAPK-AKT signaling cascade or bypass through the PI3K/AKT/mTOR pathway. In preclinical studies, the CDK2 inhibitor roscovitine allowed evasion of resistance to CDK4/6i in CCNE1-amplified cells [35]. There is an ongoing phase I/II trial evaluating the CDK2 inhibitor, PF-07104091, with and without palbociclib and letrozole to evaluate if inhibition of cyclin E-CDK2 may be a successful strategy to overcome resistance (NCT04553233).
Notably, when CHK1 regulates CDK2 activity in breast cancer cells, the cells do not tolerate activity in S-phase [38]. In applying this knowledge, CCNE2-amplified breast cancer cells were sensitive to CHK1 inhibition with prexasertib [36], proposing CHK1 inhibition as another way to combat resistance to CDK4/6i. Prexasertib is currently being evaluated in combination with chemotherapy in several advanced cancers, including breast cancer (NCT02124148).
FGFR1/2 activation
Fibroblast growth factor receptor (FGFR) signaling pathways are key players in cell differentiation and survival [39] and are responsible for resistance to CDK4/6i and ET [40, 41]. FGFR1 activates the RAS/MEK/ERK and PI3K/AKT pathways in endocrine-resistant breast cancer [39]. In preclinical models, FGFR1 activation causes resistance to palbociclib with fulvestrant [42]. Furthermore, FGFR1 signaling via FGFR2 promotes endocrine resistance and CDK4/6i resistance [39]. Through ctDNA analysis from PALOMA-3, acquired FGFR2 mutations or amplification events were present in 4/195 patients at the time of disease progression and were associated with worse PFS [33, 43]. Similarly, in analysis of ctDNA from the MONALEESA-2 study, patients with FGFR1 amplification events had inferior PFS (10.61 months vs. 24.84 months p = 0.075). The higher the level of FGFR1 expression, the shorter the PFS [44].
Researchers have found FGFR1/2 amplified breast cancer cells with resistance to CDK4/6i can be re-sensitized to CDK4/6i through treatment with FGFR inhibitors such as lucitanib and erdafitinib [44, 45]. A phase Ib trial, with erdafitinib, an FGFR inhibitor, is being given with palbociclib and fulvestrant in patients with FGFR-amplified tumors that progressed on prior CDK4/6i (NCT03238196). The correlative objectives of this study will be important in determining the therapeutic predictive role of FGFR1-4, CCND1-2, CDK4, and CDK6 amplifications, Rb1 and ESR1 mutations on clinical outcomes. FGFR1 amplification levels will also be evaluated as an early surrogate of response. In parallel, a phase II trial is recruiting patients to investigate activity of TAS-120, an FGFR inhibitor currently approved for the treatment of cholangiocarcinoma, with fulvestrant in patients treated with a prior CDK4/6i (NCT04024436).
Another possible strategy to target FGFR1/2 upregulation is through downstream targets via the MAPK or AKT/mTOR pathways, as key players in these pathways are overexpressed in FGFR1/2 amplified cells [45]. To this end, FGFR1/2 amplified cells have been sensitive to treatment with meiotic chromosome-axis-associated kinase (MEK) inhibitors and SH2 containing protein tyrosine phosphatase-2 (SHP2) inhibitors [45]. A randomized phase II trial evaluated the efficacy of selumetinib, a MEK inhibitor, with fulvestrant in patients with HR + MBC who progressed on prior AI therapy. This trial stopped accrual early as the experimental arm did not reach the pre-specified disease control rate (DCR). DCR was 23% with selumetinib + fulvestrant and 50% with placebo (n = 46). The addition of selumetinib to fulvestrant may have deteriorated the efficacy of ET in some patients [46]. Alternatively, a first-in-human phase I study of JAB-3312, an SHP inhibitor, is underway in patients with MBC (NCT04045496) and may potentially prove a more effective method to prevent the upregulation of FGFR1/2 and in turn CDK4/6i resistance.
RAS/MAPK and AKT/mTOR activation
Activating mutations in RAS oncogenes are identified in tumor biopsy specimens from patients resistant to CDK4/6i. Efforts to overcome these mechanisms of resistance are ongoing through the evaluation of the ERK1/2 inhibitor, LY321499 in phase I study with and without abemaciclib in HR + MBC (NCT02857270).
AKT inhibition has proven an effective method to treat HR + HER2- tumors with resistance to ET through the promising phase III CAPItello-291 trial with capivasertib. Capivasertib plus fulvestrant doubled the PFS compared to placebo with fulvestrant in patients who had progressed on prior AI [47]. The TAKTIC trial evaluates the additional efficacy of combining AKT inhibition with CDK4/6i in patients who have progressed on prior CDK4/6i. The AKT1 inhibitor, ipatasertib, was given in the phase Ib/II setting with an AI, fulvestrant, or triplet combination (fulvestrant + ipatasertib + palbociclib). Eighty-four percent of patients enrolled were treated with a prior CDK4/6i. The clinical benefit rate (CBR), defined as the percentage of patients who achieved a partial response or stable disease for 6 months or more, was 48% across the study with a 57% benefit in those who were on triplet therapy. The triplet cohort had a mPFS of 5.5 months and mOS of 24.5 months (n = 77) [48] and was generally well tolerated overall, suggesting the addition of AKT inhibition to a CDK4/6i may be helpful to prolong the efficacy of continued CDK4/6i in the second line.
ERBB2 activation
Beyond ERBB2’s known involvement in oncogenic signaling causing endocrine resistance, preclinical work has determined ERBB2 activation mutations bestow resistance to CDK4/6i [49, 50]. Wander et al. found ERBB2 mutations in 5 of 41 patients with resistance to CDK4/6i. ERBB2 mutant cell lines activate downstream signaling pathways such as MAPK and AKT/mTOR, further highlighting the importance of crosstalk between multiple mechanisms of resistance to CDK4/6i [36].
In preclinical models, cells with ERBB2 activating mutations were sensitive to HER2 kinase inhibition with neratinib [50]. These data prompted the phase II SUMMIT trial which evaluated the combination of fulvestrant + neratinib + trastuzumab in patients with prior exposure to CDK4/6i with metastatic HER2 mutant breast cancer. In patients receiving triple therapy (n = 45), objective response rate was 38%, with a CBR of 21 months, median duration of response of 14.4 months, and mPFS of 8.2 months [51] (NCT01953926).
Novel combinations carrying CDK4/6 inhibitors into the future
Novel combination studies are being investigated to seek creative ways to prolong the clinical benefit of CDK4/6i. Ongoing innovative combinations with SERDs and mTOR/PI3K inhibitors are key areas of interest.
CDK4/6 inhibitors plus SERD
Important progress has been made in the investigation of novel ER degraders and antagonists to address the challenge of endocrine resistance [52]. Orally bioavailable SERDs, which bind the estrogen receptor causing degradation and downregulation, have shown promising activity, especially in patients with ESR1 mutations. In January 2023, the FDA approved the first oral SERD, elacestrant, for use by postmenopausal women and men with ESR1 mutated tumors, who have progressed on at least one prior ET, including a CDK4/6i [53]. Data from the EMERALD trial revealed the duration of prior treatment with CDK4/6i impacted the PFS benefit of elacestrant, regardless of ESR1 status. Specifically, for those with ESR1 mutations, 6, 12, and 18 months of prior CDK4/6i treatment, correlated with a PFS of 4.14, 8.61, and 8.61 months, respectively [54]. Based on the extended PFS benefit, elacestrant is being explored with other targeted agents (alpelisib, everolimus, CDK4/6i) with ET in the phase Ib/II ELEVATE trial in patients who have received prior AI and CDK4/6i. The design of the ELEVATE trial will also shed light on the utility of continued CDK4/6i after progression. While one of the study arms utilizing CDK4/6i in combination allows for prior CDK4/6i exposure, the other CDK4/6i combination arm will enroll patients treated only with ET, not prior CDK4/6i (Table 1) [27].
The next-generation oral SERD, camizestrant, also improved PFS when compared with fulvestrant in the phase II SERENA-2 trial in patients who progressed on at least one ET. Randomization is stratified so 50% of patients had prior CDK4/6i exposure. Camizestrant improved outcomes in patients with ESR1 mutated tumors, with a PFS benefit of 9.2 months (150 mg) vs. 2.2 months with fulvestrant [55•]. Early results led to the phase III SERENA-6 trial assessing the combination of camizestrant with palbociclib or abemaciclib vs. AI plus CDK4/6i in patients with ESR1 mutations. In this trial, ctDNA is checked every 8–12 weeks, while patients are treated with CDK4/6i and AI. If an ESR1 mutation develops, patients are randomized to continue their current therapy or switch to camizestrant with CDK4/6i. This trial will answer several important questions: whether combining an oral SERD with CDK4/6i at the first indication of endocrine resistance will improve outcomes and whether ctDNA can tell us how well therapy is working in real time [28]. By addressing these unmet needs, SERENA-6 was granted fast track designation by the FDA. SERENA-4 is also investigating camizestrant plus palbociclib for use in the first line setting [56].
Imlunestrant, another next generation oral SERD, is being studied in combination with abemaciclib with or without ET in the EMBER phase I study. No safety signals or dose-limiting toxicities were identified in phase I. The majority of treatment-related adverse events (TrAEs) included grade 1 nausea (33.3%), fatigue (27.5%), and diarrhea (23.2%) [57], which are similar to the TrAEs seen with abemaciclib alone. EMBER-3, is a phase III, randomized study comparing imlunestrant versus exemestane or fulvestrant in patients who progressed on prior AI or CDK4/6i. One arm will include imlunestrant plus abemaciclib with the primary endpoint of PFS. Key secondary endpoints are OS and ORR [58] (Table 1).
CDK4/6 plus mTOR/PI3K inhibitor
Further trials are underway to investigate triplet therapy with CDK4/6i, ET, and a third additional therapeutic, targeting the mTOR or PI3K pathways. The TRINITI-1 phase I/II trial explored the triplet combination of ribociclib, everolimus, and exemestane in patients who progressed on prior CDK4/6i. At week 24, the CBR was 41.1% (95% CI 31.1–51.6%), which met the primary endpoint; however, due to drug-drug interactions with everolimus and ribociclib, this triplet combination is not moving forward into development [59].
A separate, multicenter, phase Ib study evaluating a different triplet combination with ribociclib, fulvestrant, and a PI3K inhibitor (buparlisib or alpelisib) stopped enrollment early due to unexpected toxicity, further demonstrating the difficulty of establishing safe and tolerable triplet therapy regimens in this setting [60].
A larger, phase II/III trial is recruiting 400 participants to study the efficacy and safety of palbociclib, fulvestrant, and inavolisib, a PI3K alpha-inhibitor, versus palbociclib, fulvestrant, and placebo in patients who progressed during treatment or within 12 months of completing adjuvant ET and have not received systemic therapy for metastatic disease (NCT04191499). Data from this trial will highlight whether bringing a PI3K inhibitor forward in combination with CDK4/6i earlier in the metastatic setting will be important for our patients with PI3KCA mutations.
A first-in-class pan-PI3K/mTOR inhibitor, gedatolisib, is being investigated in combination with palbociclib and ET. Gedatolisib has unique pharmacokinetic properties different from other currently approved therapies that target PI3K or mTOR alone [61]. In this phase Ib trial, arm A received gedatolisib plus palbociclib/letrozole first line; arm B received gedatolisib plus palbociclib/fulvestrant (CKD 4/6i naïve); arm C & D had previous treatment with CKD4/6i and received gedatolisib plus palbocilib/fulvestrant. In arm C, gedatolisib was dosed weekly, and in arm D, gedatolisib was dosed 3-weeks-on and 1-week-off. The most common grade 3 or 4 adverse events included stomatitis, neutropenia, and fatigue. Overall, the favorable safety profile and antitumor activity supports the use of the 3-weeks-on, 1-week-off dosing schedule with triplet therapy.
Based on these results, the phase III VIKTORIA-1 trial is further evaluating the efficacy of gedatolisib plus fulvestrant with or without palbociclib vs. standard of care following progression on CDK4/6i (NCT05501886). The primary outcome is OS, and key secondary outcomes include ORR, and duration of response in PIK3CA wild type and PIK3CA mutated breast cancers. Treatment arms are randomized according to PIK3CA mutational status. The trial began enrolling patients in fall 2022 with expected primary completion in 2024.
These studies exemplify the devotion to significant efforts aimed at overcoming resistance and evasion to ET, further employing the profound effect of CDK 4/6i for our patients with HR + HER2-MBC. Results from these trials will inform clinical decisions regarding CDK4/6i in later-line treatments and better elucidate a strategy for utilizing continued CDK4/6i after disease progression in the first line. In summary, these novel combination strategies will continue to shape our treatment paradigm for later-line treatment options for HR + HER2-MBC.
Conclusions
CDK4/6i as the recommended, standard, first-line treatment for patients with HR + HER2-MBC continues to make a meaningful impact in the lives of our patients. Ribociclib has become the CDK4/6i of choice in the first line due to OS data and more tolerable side effect profile, even in patients with visceral crisis. Efforts focused on the best approaches for patients after progression on CDK4/6i have shown benefit when switching from palbociclib to ribociclib with a new ET backbone such as exemestane or fulvestrant. Triplet regimens with SERDs and targeted therapies with ET and/or CDK4/6i are underway in hopes to delay resistance to CDK4/6i. Many questions remain. After progression on CDK4/6i, should we use a single agent or combination? Should the combination involve another CDK4/6i? How do we best sequence various agents or combinations? Ultimately, multifaceted treatment options involving CDK4/6i will exist, allowing us to take a personalized approach to the treatment of patients with HR + HER2-MBC.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
McAndrew NP, Finn RS. Clinical review on the management of hormone receptor–positive metastatic breast cancer. JCO Oncology Practice. 2022;18(5):319–27.
•• Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Hart L, et al. Overall survival with ribociclib plus letrozole in advanced breast cancer. N Engl J Med. 2022;386(10):942–50. Final OS analysis of the randomized, double-blind, placebo-controlled, phase III MONALEESA-2 trial with ribociclib plus letrozole demonstrating OS benefit in the front line.
Neven PFP, Chia S, Jerusalem G, Im S, Petrakova K, Bianchi G, Martin Jimenez M, Nusch A, Sonke GS, de la Cruz merino L, Zarate JP, Wang Y, Chakravatty A, Wang C, Slamon D. editor Updated overall survival (OS) results from the first-line population in the phase III MONALEESA-3 trial of postmenopausal patients with HR+HER2- advanced breast cancer treated with ribociclib + fulvestrant. ESMO Breast Cancer Congres; 2022.
•• M.P. Goetz MT, J. Huober, J. Sohn, O. Tredan, I.H. Park, M. Campone, S.C. Chen, L.M. Manso Sanchez, S. Paluch-Shimon, G. van Hal, A. Shahir, H. Iwata, S. Johnston, editor MONARCH 3: interim overall survival (OS) results of abemaciclib plus a nonsteroidal aromatase inhibitor (NSAI) in patients (pts) with HR+, HER2- advanced breast cancer (ABC). ESMO Congress; 2022. Second interim OS analysis from the randomized, double-blind, placebo-controlled, phase III MONARCH-3 trial with abemaciclib plus AI demonstrating OS benefit in front line.
•• Lu Y-S, Im S-A, Colleoni M, Franke F, Bardia A, Cardoso F, et al. Updated overall survival of ribociclib plus endocrine therapy versus endocrine therapy alone in pre- and perimenopausal patients with HR+/HER2− advanced breast cancer in MONALEESA-7: a phase III randomized clinical trial. Clin Cancer Res. 2022;28(5):851–9. (Exploratory OS analysis with extended follow-up (median 53.5 months) for the randomized, double-blind, placebo-controlled, phase III MONALEESA-7 trial showing consistent OS benefit with ribociclib plus ET in peri/premenopausal patients in the front line.)
Finn RS, Boer K, Bondarenko I, Patel R, Pinter T, Schmidt M, et al. Overall survival results from the randomized phase 2 study of palbociclib in combination with letrozole versus letrozole alone for first-line treatment of ER+/HER2- advanced breast cancer (PALOMA-1, TRIO-18). Breast Cancer Res Treat. 2020;183(2):419–28.
Llombart-Cussac A, Pérez-García JM, Bellet M, Dalenc F, Gil-Gil M, Ruíz-Borrego M, et al. Fulvestrant-palbociclib vs letrozole-palbociclib as initial therapy for endocrine-sensitive, hormone receptor–positive, ERBB2-negative advanced breast cancer: a randomized clinical trial. JAMA Oncol. 2021;7(12):1791–9.
•• Finn RS, Rugo HS, Dieras VC, Harbeck N, Im S-A, Gelmon KA, et al. Overall survival (OS) with first-line palbociclib plus letrozole (PAL+LET) versus placebo plus letrozole (PBO+LET) in women with estrogen receptor–positive/human epidermal growth factor receptor 2–negative advanced breast cancer (ER+/HER2− ABC): Analyses from PALOMA-2. J Clin Oncol. 2022;40(17_suppl):LBA1003-LBA. Planned OS analysis for the randomized, double-blind, placebo-contolled, phase III PALOMA-2 trial with palbociclib and letrozole, where palbo + letrozole had numerically longer OS compared to placebo + letrozole, but the results were not statistically significant.
Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375(18):1738–48.
Finn RS, Martin M, Rugo HS, Jones S, Im SA, Gelmon K, et al. Palbociclib and letrozole in advanced breast cancer. N Engl J Med. 2016;375(20):1925–36.
Slamon DJ, Neven P, Chia S, Fasching PA, De Laurentiis M, Im SA, et al. Phase III Randomized study of ribociclib and fulvestrant in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: MONALEESA-3. J Clin Oncol. 2018;36(24):2465–72.
Sledge GW Jr, Toi M, Neven P, Sohn J, Inoue K, Pivot X, et al. MONARCH 2: Abemaciclib in combination with fulvestrant in women with HR+/HER2- advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol. 2017;35(25):2875–84.
Tripathy D, Im SA, Colleoni M, Franke F, Bardia A, Harbeck N, et al. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): a randomised phase 3 trial. Lancet Oncol. 2018;19(7):904–15.
Turner NC, Ro J, André F, Loi S, Verma S, Iwata H, et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med. 2015;373(3):209–19.
Cristofanilli M, Rugo HS, Im SA, Slamon DJ, Harbeck N, Bondarenko I, et al. Overall survival with palbociclib and fulvestrant in women with HR+/HER2- ABC: updated exploratory analyses of PALOMA-3, a double-blind, phase III randomized study. Clin Cancer Res. 2022;28(16):3433–42.
Cristofanilli M, Turner NC, Bondarenko I, Ro J, Im S-A, Masuda N, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016;17(4):425–39.
Sledge GW Jr, Toi M, Neven P, Sohn J, Inoue K, Pivot X, et al. The effect of abemaciclib plus fulvestrant on overall survival in hormone receptor–positive, ERBB2-negative breast cancer that progressed on endocrine therapy—MONARCH 2: a randomized clinical trial. JAMA Oncol. 2020;6(1):116–24.
Hortobagyi GN, Stemmer SM, Burris HA, Yap Y-S, Sonke GS, Hart L, et al. Overall survival with ribociclib plus letrozole in advanced breast cancer. N Engl J Med. 2022;386(10):942–50.
Slamon DJ, Neven P, Chia S, Jerusalem G, De Laurentiis M, Im S, et al. Ribociclib plus fulvestrant for postmenopausal women with hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer in the phase III randomized MONALEESA-3 trial: updated overall survival. Ann Oncol. 2021;32(8):1015–24.
Slamon DJ, Neven P, Chia S, Fasching PA, De Laurentiis M, Im S-A, et al. Overall survival with ribociclib plus fulvestrant in advanced breast cancer. N Engl J Med. 2019;382(6):514–24.
Lu YS, Im SA, Colleoni M, Franke F, Bardia A, Cardoso F, et al. Updated overall survival of ribociclib plus endocrine therapy versus endocrine therapy alone in pre- and perimenopausal patients with HR+/HER2- advanced breast cancer in MONALEESA-7: a phase III randomized clinical trial. Clin Cancer Res. 2022;28(5):851–9.
Sledge Jr GW, Toi M, Neven P, Sohn JH, Inoue K, Pivot X, Okera M, Masuda N, Kaufman PA, Koh H, Grischke E-M, Conte P, Andre V, Bian Y, Shahir A, val Hal G, Llombart-Cussac A. Final overall survival analysis of Monarch 2 : a phase 3 trial of abemaciclib plus fulvestrant in patients with hormone receptor-positive, HER2-negative advanced breast cancer. San Antonio Breast Cancer Conference; 2022 December 8.
Turner NC, Slamon DJ, Ro J, Bondarenko I, Im S-A, Masuda N, et al. Overall survival with palbociclib and fulvestrant in advanced breast cancer. N Engl J Med. 2018;379(20):1926–36.
• Mayer EL, Ren Y, Wagle N, Mahtani R, Ma CX, DeMichele A, Cristofanili M, Meisel J, Miller KD, Jolly T, Riley E, Qamar R, Sharma P, Reid S, Sinclair N, Faggen M, Block C, Ko N, Patridge AH, Chen WY, DeMeo MK, Attaya V, Okopoebo A, Liu Y, Gauthier E, Bustein H, Regan MM, Tolaney SM. Palbociclib after CDK4/6i and endocrine therapy (PACE): a randomized phase II study of fulvestrant, palbociclib, and avelumab for endocrine pre-treated ER+/HER2- metastatic breast cancer. San Antonio Breast Cancer Conference; 2022 December 8; San Antonio, TX. First randomized phase II study demonstrating combination of palbociclib with fulvestrant beyond progression on prior CDK4/6i did not significantly improve PFS compaed to using fulvestrant alone.
• Kalinsky K, Chiuzan C, Singh M, Trivedi M, Novik Y, Tiersten A, Raptis G, Baer L, Young Oh S, Zelnak AB, Wisinski KB, Andreopoulou E, Gradishar WJ, Stringer-Reasor E, Reid SA, O'Dea A, O'Regan R, Crew KD, Hershman DL. A randomized, phase II trial of fulvestrant or exemestane with or without ribociclib after progression on anti-estrogen therapy plus cyclin-dependent kinase 4/6 inhibition (CDK 4/6i) in patients (pts) with unresectable or hormone receptor–positive (HR+), HER2-negative metastatic breast cancer (MBC): MAINTAIN trial. ASCO Annual Meeting; 2022; Chicago, IL. First randomized, placebo-controlled trial demonstrating significant PFS benefit when switching ET in combination with ribociclib after progression on prior CDK4/6i.
Kalinsky K, Layman RM, Kaufman PA, Graff SL, Bianchini G, Martin M, et al. postMONARCH: a phase 3 study of abemaciclib plus fulvestrant versus placebo plus fulvestrant in patients with HR+, HER2-, metastatic breast cancer following progression on a CDK4 & 6 inhibitor and endocrine therapy. J Clin Oncol. 2022;40(16_suppl):TPS1117-TPS.
Rugo H, Bardia A, Cortés J, Curigliano G, Hamilton E, Hurvitz S, et al. Abstract OT2–01–03: ELEVATE: a phase 1b/2, open-label, umbrella study evaluating elacestrant in various combinations in women and men with metastatic breast cancer (mBC). Cancer Res. 2023;83(5_Supplement):OT2-01-3-OT2--3.
Bidard F-C, Kalinsky K, Cristofanilli M, Bianchini G, Chia SK, Janni W, et al. Abstract OT2-11-05: SERENA-6: A Phase III study to assess the efficacy and safety of AZD9833 (camizestrant) compared with aromatase inhibitors when given in combination with palbociclib or abemaciclib in patients with HR+/HER2- metastatic breast cancer with detectable ESR1m who have not experienced disease progression on first-line therapy. Cancer Res. 2022;82(4_Supplement):OT2-11-05-OT2-11–05.
Im S-A, Lu Y-S, Bardia A, Harbeck N, Colleoni M, Franke F, et al. Overall survival with ribociclib plus endocrine therapy in breast cancer. N Engl J Med. 2019;381(4):307–16.
Loibl S, Turner NC, Ro J, Cristofanilli M, Iwata H, Im SA, et al. Palbociclib combined with fulvestrant in premenopausal women with advanced breast cancer and prior progression on endocrine therapy: PALOMA-3 results. Oncologist. 2017;22(9):1028–38.
Tolaney SM, Sahebjam S, Le Rhun E, Bachelot T, Kabos P, Awada A, et al. A phase II study of abemaciclib in patients with brain metastases secondary to hormone receptor-positive breast cancer. Clin Cancer Res. 2020;26(20):5310–9.
• Lu YS, Bin Mohd Mahidin El, Azim H, Eralp Y, Yap YS, Im SA, Rihani J, Bowels J, Delgar Alfaro T, Wu J, Gao M, Slimane K. Primary results from the randomized phase II right choice trial of premenopausal patients with aggressive HR+/HER2− advanced breast cancer treated with ribociclib + endocrine therapy vs physician’s choice combination chemotherapy. San Antonio Breast Conference; 2022. The first prospective study comparing a CDK4/6i + ET with combination chemotherapy, demonstrating PFS superiority of ribociclib + ET rather than combination chemotherapy in patients with aggressive clinical features or rapidly progressing or highly symptomatic disease, favoring CDK4/6i as the firstline, standard-of-care for patients with visceral crisis.
O’Leary B, Cutts RJ, Liu Y, Hrebien S, Huang X, Fenwick K, et al. The genetic landscape and clonal evolution of breast cancer resistance to palbociclib plus fulvestrant in the PALOMA-3 trial. Cancer Discov. 2018;8(11):1390–403.
Li Z, Razavi P, Li Q, Toy W, Liu B, Ping C, et al. Loss of the FAT1 tumor suppressor promotes resistance to CDK4/6 inhibitors via the hippo pathway. Cancer Cell. 2018;34(6):893-905.e8.
Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I, et al. Early Adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. 2016;76(8):2301–13.
Wander SA, Cohen O, Gong X, Johnson GN, Buendia-Buendia JE, Lloyd MR, et al. The genomic landscape of intrinsic and acquired resistance to cyclin-dependent kinase 4/6 inhibitors in patients with hormone receptor-positive metastatic breast cancer. Cancer Discov. 2020;10(8):1174–93.
Taylor-Harding B, Aspuria PJ, Agadjanian H, Cheon DJ, Mizuno T, Greenberg D, et al. Cyclin E1 and RTK/RAS signaling drive CDK inhibitor resistance via activation of E2F and ETS. Oncotarget. 2015;6(2):696–714.
Sakurikar N, Thompson R, Montano R, Eastman A. A subset of cancer cell lines is acutely sensitive to the Chk1 inhibitor MK-8776 as monotherapy due to CDK2 activation in S phase. Oncotarget. 2016;7(2):1380.
Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Can Res. 2010;70(5):2085–94.
Brooks AN, Kilgour E, Smith PD. Molecular pathways: fibroblast growth factor signaling: a new therapeutic opportunity in cancer. Clin Cancer Res. 2012;18(7):1855–62.
Mao P, Kusiel J, Cohen O, Wagle N. Abstract PD4–01: The role of FGF/FGFR axis in resistance to SERDs and CDK4/6 inhibitors in ER+ breast cancer. Cancer Res. 2018;78(4_Supplement):PD4-01-PD4-.
Formisano L, Lu Y, Jansen VM, Bauer JA, Hanker AB, Sanders ME, et al. gain-of-function kinase library screen identifies FGFR1 amplification as a mechanism of resistance to antiestrogens and CDK4/6 inhibitors in ER+ breast cancer. Cancer Res. 2017;77(13_Supplement):1008.
O’Leary B, Cutts RJ, Huang X, Hrebien S, Liu Y, André F, et al. Circulating tumor DNA Markers for early progression on fulvestrant with or without palbociclib in ER+ advanced breast cancer. J Natl Cancer Inst. 2021;113(3):309–17.
Formisano L, Lu Y, Servetto A, Hanker AB, Jansen VM, Bauer JA, et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat Commun. 2019;10(1):1373.
Mao P, Cohen O, Kowalski KJ, Kusiel JG, Buendia-Buendia JE, Cuoco MS, et al. Acquired FGFR and FGF alterations confer resistance to estrogen receptor (ER) targeted therapy in ER+ metastatic breast cancer FGFR/FGF alterations confer resistance to endocrine therapy. Clin Cancer Res. 2020;26(22):5974–89.
Zaman K, Winterhalder R, Mamot C, Hasler-Strub U, Rochlitz C, Mueller A, et al. Fulvestrant with or without selumetinib, a MEK 1/2 inhibitor, in breast cancer progressing after aromatase inhibitor therapy: a multicentre randomised placebo-controlled double-blind phase II trial, SAKK 21/08. Eur J Cancer. 2015;51(10):1212–20.
Turner N, Oliveira M, Howell SJ, Dalenc F, Cortés J, Gomez H, et al. Abstract GS3–04: GS3–04 Capivasertib and fulvestrant for patients with aromatase inhibitor-resistant hormone receptor-positive/human epidermal growth factor receptor 2-negative advanced breast cancer: results from the Phase III CAPItello-291 trial. Cancer Res. 2023;83(5_Supplement):GS3-04-GS3-.
Wander SA, Keenan JC, Niemierko A, Juric D, Spring LM, Supko J, Vidula N, Isakoff SJ, Ryan L, Padden S, Fisher E, Newton A, Moy B, Ellisen LW, Micalizzi DS, Bardia A. editor Combination therapy with the AKT inhibitor, ipatasertib, endocrine therapy, and a CDK4/6 inhibitor for hormone receptor positive (HR+)/HER2 negative metastatic beast cancer: results from the phase I TAKTIC trial. SABCS; 2022 December 8, 2022; San Antonio.
Nayar U, Cohen O, Kapstad C, Cuoco MS, Waks AG, Wander SA, et al. Acquired HER2 mutations in ER+ metastatic breast cancer confer resistance to estrogen receptor–directed therapies. Nat Genet. 2019;51(2):207–16.
Medford AJ, Dubash TD, Juric D, Spring L, Niemierko A, Vidula N, et al. Blood-based monitoring identifies acquired and targetable driver HER2 mutations in endocrine-resistant metastatic breast cancer. NPJ Precis Oncol. 2019;3(1):18.
Jhaveri KL, Goldman JW, Hurvitz SA, Guerrero-Zotano A, Unni N, Brufsky A, et al. Neratinib plus fulvestrant plus trastzuzumab (N+F+T) for hormone receptor-positive (HR+), HER2-negative, HER2-mutant metastatic breast cancer (MBC): outcomes and biomarker analysis from the SUMMIT trial. J Clin Oncol. 2022;40(16_suppl):1028-.
Hamilton EP, Patel MR, Armstrong AC, Baird RD, Jhaveri K, Hoch M, et al. A first-in-human study of the new oral selective estrogen receptor degrader AZD9496 for ER(+)/HER2(-) advanced breast cancer. Clin Cancer Res. 2018;24(15):3510–8.
FDA approves elacestrant for ER-positive, HER2-negative, ESR1-mutated advanced or metastatic breast cancer FDA.gov2023 [updated 1/27/2023. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-elacestrant-er-positive-her2-negative-esr1-mutated-advanced-or-metastatic-breast-cancer. Accessed 4/5/2023.
Bardia A, Bidard F-C, Neven P, Streich G, Montero AJ, Forget F, et al. Abstract GS3–01: GS3–01 EMERALD phase 3 trial of elacestrant versus standard of care endocrine therapy in patients with ER+/HER2- metastatic breast cancer: Updated results by duration of prior CDK4/6i in metastatic setting. Cancer Res. 2023;83(5_Supplement):GS3-01-GS3-.
• Oliveira M, Pominchuck D, Nowecki Z, Hamilton E, Kulyaba Y, Andabekov T, et al. Abstract GS3–02: GS3–02 Camizestrant, a next generation oral SERD vs fulvestrant in post-menopausal women with advanced ER-positive HER2-negative breast cancer: Results of the randomized, multi-dose Phase 2 SERENA-2 trial. Cancer Res. 2023;83(5_Supplement):GS3–02-GS3-. Randomized, phase II SERENA-2 trial demonstrates next-generation oral SERD, camizestrant, offers meaningful PFS benefit when compared to faslodex in post-menopausal patients who progressed on prior ET, prompting further studies with CDK4/6i and camizestrant in combination in SERENA-4 and SERENA-6.
André F, Im S-A, Neven P, Baird RD, Ettl J, Goetz MP, et al. Abstract OT2-11-06: SERENA-4: A Phase III comparison of AZD9833 (camizestrant) plus palbociclib, versus anastrozole plus palbociclib, for patients with ER-positive/HER2-negative advanced breast cancer who have not previously received systemic treatment for advanced disease. Cancer Res. 2022;82(4_Supplement):OT2-11-06-OT2-11–06.
Lloyd MR, Spring LM, Bardia A, Wander SA. Mechanisms of resistance to CDK4/6 blockade in advanced hormone receptor-positive, HER2-negative breast cancer and emerging therapeutic opportunities. Clin Cancer Res. 2022;28(5):821–30.
Jhaveri K, Harbeck N, Aftimos P, Kim S-B, Pivot X, Saura C, et al. Abstract OT2-11-01: EMBER-3: A randomized phase 3 study of LY3484356, a novel, oral selective estrogen receptor degrader vs investigator’s choice of endocrine therapy of either fulvestrant or exemestane, in patients with estrogen receptor-positive, human epidermal growth factor receptor 2-negative, locally advanced or metastatic breast cancer previously treated with endocrine-based therapy. Cancer Res. 2022;82(4_Supplement):OT2-11-01-OT2-11–01.
Bardia A, Hurvitz SA, DeMichele A, Clark AS, Zelnak A, Yardley DA, et al. Phase I/II trial of exemestane, ribociclib, and everolimus in women with HR(+)/HER2(-) advanced breast cancer after progression on CDK4/6 inhibitors (TRINITI-1). Clin Cancer Res. 2021;27(15):4177–85.
Tolaney SM, Im YH, Calvo E, Lu YS, Hamilton E, Forero-Torres A, Bachelot T, Maur M, Fasolo A, Tiedt R, Nardi L, Stammberger U, Abdelhady AM, Ruan S, Lee SC. Phase Ib study of ribociclib plus fulvestrant and ribociclib plus fulvestrant plus PI3K inhibitor (alpelisib or buparlisib) for HR+ advanced breast cancer. Clin Cancer Res. 2021;27(2):418–28.
Layman R, Wesolowski R, Han H, Specht JM, Stringer-Reasor EM, Dees EC, et al. Abstract PD13–02: phase Ib expansion study of gedatolisib in combination with palbociclib and endocrine therapy in women with ER+ metastatic breast cancer. Cancer Research. 2022;82(4_Supplement):PD13-02-PD13-02.
Hortobagyi GN, Stemmer SM, Burris HA, Yap Y-S, Sonke GS, Paluch-Shimon S, et al. Ribociclib as first-line therapy for HR-positive, Advanced Breast Cancer. N Engl J Med. 2016;375(18):1738–48.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Elizabeth Sakach declares she has no conflict of interest. Merve Keskinkilic declares she has no conflict of interest. Sarah Wood declares she has no conflict of interest. Madison Canning declares she has no conflict of interest. Kevin Kalinsky reports advising/consulting for Genentech/Roche, Immunomedics, Seattle Genetics, Oncosec, 4D pharma, Daicchi Saknyo, Puma Biotechnology, Mersna, Menarini Silicon Biosystems, Myovant Sciences, Takeda. His spouse is currently an employee at EQRX.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human and animals subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sakach, E., Keskinkilic, M., Wood, S. et al. CDK4/6 Inhibition in the Metastatic Setting: Where Are We Headed?. Curr. Treat. Options in Oncol. 24, 1103–1119 (2023). https://doi.org/10.1007/s11864-023-01109-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11864-023-01109-9