
Opinion statement
Cervical cancer (CC) is most often caused by the human papillomavirus (HPV). In principle, these ties to the virus should make HPV tumors a relatively easy target for clearance by the immune system. However, these HPV-associated tumors have evolved strategies to escape immune attack. Checkpoint inhibition immunotherapy, which has had remarkable success in cancer treatment, has the potential to overcome the immune escape in CC by harnessing the patient’s own immune system and priming it to recognize and kill tumors. Recent work involving PD-1/PD-L1 inhibitors in CC lends credence to this belief, as pembrolizumab has shown evidence of clinical efficacy and consequently been granted accelerated approval by the FDA. That being said, the oncologic outcomes following monotherapy with these biologics have mostly been modest and variable, and this can be attributed to alternative resistance mechanisms to tumor response. The use of therapies that stimulate immune responses via checkpoint-independent activation will therefore augment release of T cell inhibition by checkpoint inhibitors for stronger and more sustained clinical responses. Such a combinatorial approach holds promise for weak- or non-responders to checkpoint therapies as supported by evidence from various, recent pre-clinical, and preliminary clinical studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Tremendous improvements have been made in strategies designed to prevent development of CC. Prophylactic vaccines and various population-based screening strategies now offer significant protection against development of invasive disease [1,2,3]. However, CC remains a significant clinical challenge and the burden of the disease is high. Implementation of some of preventative measures has been met with serious challenges. Uptake rates of the vaccine has been suboptimal in resource-poor settings as well as developed markets such as the US [4,5,6]. Even if 100% coverage could be achieved, given that the vaccine is most suitable for young women pre-infection, there remain approximately 3 generations of women who are ineligible for vaccination. Currently, CC ranks the 4th commonest cancer by incidence and 4th largest contributor to female cancer-related deaths [7].
To a large extent, the fatality rate of CC is explained by the paucity of effective and targeted therapies. Unlike early-stage CC, which can be treated with surgery, the later stages of CC are typically treated with some combination of chemotherapy and radiation. Unfortunately, the disease often recurs or persists. For the patients in this persistence, recurrence, or metastasis setting, a cocktail of cisplatin or paclitaxel with bevacizumab (Avastin™) is the standard of care [8]. There are no other standard treatment options beyond this regimen if it fails. Several cytotoxic and targeted chemotherapy clinical trials addressing second-line therapies have been initiated, though only modest gains in survival or intolerable toxicities have been realized thus far [8, 9]. CC patients in this population therefore represent a serious and unmet clinical need.
The recent advent of immunotherapy, particularly immune checkpoint inhibition, in the treatment of cancer could help to fill this void. Immune checkpoint blockade (ICB) has shifted the paradigm in the treatment of both solid and hematological cancers. Currently, the FDA has approved checkpoint inhibitors for the treatment of a variety of disparate cancers in both a first- and second-line therapy setting. Moreover, unprecedentedly long durations of response have been observed, even in heavily previously treated patients presenting with relapsed and metastatic disease [10, 11]. Such success stories have inspired the current flurry of activity in search of immune checkpoint inhibitors for the treatment of cancers without effective first- or second-line therapies. With respect to cervical cancer, the hope for finding an effective immune checkpoint inhibitor has received a lift with the recent FDA priority review of pembrolizumab (Keytruda™) in 2018 [12]. This means that patients who experience recurrence now have pembrolizumab as an available option. Despite the priority review, the majority of patients did not respond to pembrolizumab, and further clinical validation is required. Finding answers to overcome this challenge posed by non-responders will likely involve untangling the complex immune biology of CC. In the immediate future, however, combinatorial therapies present a tantalizing alternative to standard of care. Studies reported in the literature offer ample preliminary supporting evidence to clinicians and investigators to rationally design effective combinations using checkpoint inhibitors so as to extend clinical benefit to those patients currently not responding [13,14,15,16].
In this review, we highlight and rationalize how immune response is compromised during development and progression of CC and how certain therapies can maximize the response of CC patients to checkpoint inhibitors.
Immune escape and CC development: from infection to malignancy
Evidence shows that following an infection of the mucosa by HPV, the host’s immune system is often able to eliminate the virus. About 90% of all HPV infections are acute and subclinical, resolving spontaneously within the first 2 years. In cases where the infection manifests clinically, pre-cancerous lesions (cervical intraepithelial neoplasias) are observed in the cervix. In majority of these patients, both the innate and adaptive immunity will eradicate the lesions by deploying macrophages, Th1 and CD8 T cells, directed against HPV-encoded proteins [17,18,19,20].
Nonetheless, lesions sometimes fail to regress in immune compromised patients and in cases where the virus successfully evades the immune surveillance. To maintain a low profile and avoid immune attack, the virus keeps expression of viral antigenic proteins low and away from sentinel immune cells. The life cycle also does not involve a viremic phase, and host cells are not lysed when new virions are shed [19, 21]. Additionally, the HPV oncoproteins E6 and E7 are known to antagonize innate immunity viral sensors such as Toll-like receptors (TLRs) and their downstream targets IRFs, NFκB, and type I interferons (IFNs), that facilitate the secretion of pro-inflammatory cytokines (IL-1β, TNFα, IL-6, and IL-8) and chemokines (CCL20) [17, 22,23,24]. Without chemokines and pro-inflammatory cytokines, keratinocytes are unable to recruit and activate resident immune cells such as Langerhans, stromal dendritic cells (DCs), and macrophages. Together with the down-regulation of major histocompatibility complexes (MHC I and MHC II) by HPV [17, 21, 25], the virus impairs antigen acquisition, presentation, and the provision of a stimulus for humoral- and cytotoxic T cell-mediated immunity to create an environment of immunological ignorance.
Immunological ignorance allows the virus to persist and establish chronic infection. The more the virus lingers during chronic infection, the higher the probability of stochastic events of HPV DNA integrating into the host genome. These events are enhanced by cellular stress due to viral proteins or inflammation from factors such as co-infections and smoking. Indeed, in our lab we have shown that an isoform of E6 down-regulates expression of the anti-oxidant enzymes and elevates levels of reactive oxygen species (ROS) in HPV-positive cells. The ROS levels cause significant DNA damage and induce the integration of plasmid and episomal HPV DNA into cervical keratinocytes [22, 26,27,28,29]. Following successful integration, certain viral genes are often lost, including E2. Loss of E2 releases control of expression of the oncoproteins, E6 and E7, and the overexpression of E6 and E7 that ensues will undermine the functions of telomeres, tumor suppressors, apoptosis machinery, and genomic maintenance. This leads to transformation and sustained proliferation of infected cells [30,31,32,33].
Although integration of HPV DNA is considered a hallmark of cervical cancer, the progression to malignancy still requires additional events. One of these important changes occurs in the tumor micro-environment (TME), and reciprocal interactions between keratinocytes and stromal cells enable transformed cells to escape anti-tumor immune defenses [34]. Studies show that IL-6 is one of the initiators and among its pleitropic functions, it exerts influence on stromal monocytes and myeloid cells and activates STAT3 [35,36,37,38,39,40,41,42]. STAT3 activation increases the production of CCL2, a chemokine that promotes inflammation through attraction of additional myelomonocytic infiltrates. CCL2 also brings about architectural changes that promote progression towards neoplasia via production of MMP-9 [22, 23, 39, 40, 43, 44]. IL-1β is another key pro-inflammatory cytokine whose levels become elevated. Studies show that IL-1β activates cancer-associated fibroblasts (CAFs) into inflammatory cells that secrete a host of carcinogenesis-promoting factors including CCL20 and Cox-2. In turn, more monocytes, macrophages, neutrophils, and Th17 cells are recruited into the stroma. It is from these cells that tumor-associated macrophages (TAMs), tumor-associated neutrophils (TANs), and myeloid-derived suppressor cells (MDSCs) are derived [45,46,47,48] .
TAMs and MDSCs promote the down-regulation of IFN-γ and IL-12 and up-regulate several inhibitory cytokines and skew the Th1–Th2 balance towards Th2 cells. TAMs will also drive expression of proteolytic enzymes and angiogenic factors such as VEGF [22, 23, 34, 43, 49,50,51,52]. Another equally important tolerogenic mediator is stromal dendritic cells (DCs). E6 and E7 tend to reduce expression of the chemokine receptor, CCR7 [44]. This prevents migration of DCs to the lymph nodes for antigen presentation and traps them in the stroma. The immature stromal DCs will add to the growing number of suppressive factors by producing IDO, TGF-β, IL-10, and Foxp3+ Tregs [18, 22, 23]. Due to all of these changes, the general clinical observation is that as the immunosuppressive milieu strengthens, the CIN grade also increases in parallel [18, 22]. Even though research is still building on this topic, it is becoming clear that tolerogenicity noted in CINs is not simply an inconsequential phenotype meant to help disarm antigen presentation and generation of peripheral Th1 and CD8 cytotoxic anti-tumor responses. Rather, it is a necessary step that helps to create a protective and immune privileged niche for invasive cervical carcinoma to fully develop.
Immune checkpoints and immune checkpoint blockade
The various soluble mediators, suppressive metabolites, and pro-tumor immune cells discussed above represent the many strategies used by CC to drive its progression. In addition to these immune suppressors, immune checkpoints are an additional modulator that also depress response to tumors. With the advent of immunotherapy however, these various factors also represent opportunities for targeting by immuno-therapeutic agents. Blockade of checkpoints in particular harnesses the body’s own immune system to fight tumors, and this has clinically revolutionized the treatment of many cancers [10, 11].
Immune checkpoints are physiological negative regulators of the immune system that help to maintain immune homeostasis and self-tolerance. They achieve this by arresting the activation and proliferation of T cells in both lymphoid and peripheral tissues. CTLA-4 is the main checkpoint in lymph nodes that inhibits T cells after successful priming and initial activation. PD-1 is the peripheral compartment checkpoint that regulates activation of effector T cells. PD-1’s primary ligand is known as PD-L1 and is expressed on various somatic cells including APCs. When the PD-L1 ligand binds to PD-1, an inhibitory signaling follows and arrests further activation of effector T cells [10, 11, 53].
Cancers can co-opt this mechanism by up-regulating the expression of these immune checkpoints on various cells in the TME, thereby disabling T cells that recognize the antigens they display [10, 11, 54]. Immune checkpoints therefore allow cancers to neutralize any anti-tumor response mounted against them despite expression of immunogenic antigens. Eventually, chronic stimulation of TCRs by antigens found on surviving tumor cells will diminish the effector function of tumor-reactive T cells and lead to their exhaustion. As such, the use of antibodies that antagonize immune checkpoints should reverse this T cell dysfunction. In current clinical practice, the blockade of immune checkpoints not only re-boots anti-tumor immunity, but it also results in durable tumor regression and remission in some patients [10, 11].
Biological basis for ICB in cancer: is ICB likely to be effective in treating CC?
Generally, the factors that predict the response of tumors to ICB in various malignancies include the mutational load, immune profile, and status of immune checkpoint expression. High mutational load is associated with more neo-antigens, greater TCR diversity, and robust effector function of T cells. Accordingly, cancers associated with chronic mutagens or deficiency in DNA mismatch repair (dMMR) tend to display a higher clinical response to ICB [55,56,57]. The immune profile is also important because T cells are ultimately the entities that engage the tumor cells and mediate the effects of checkpoint inhibitors. High responses to ICB have hence been observed in tumors with an immune-inflamed phenotype. “Inflamed” tumors are usually infiltrated with tumor antigen-specific CD8 T cells, pro-inflammatory cytokines, Th1 (T-bet+) cells, and Th1-type chemokines. Non-inflamed tumors, on the other hand, have no or low levels of CD8 T cells [58, 59]. Ultimately, the expressions of the PD-1, CTLA-4, and PD-L1 molecules are also essential because these immune checkpoint molecules generally serve as a surrogate for the pre-existence of activated tumor-reactive T cells. Granted, some patients negative for PD-L1 expression still respond to anti-PD1/PDL1 therapies and vice versa, calling the predictive reliability of its expression into question, but PD-L1 is considered a reasonable biomarker to use to clinically stratify patients [55, 57, 60].
The Genetic landscape and mutational load in CC
The molecular features of CC tumors are consistent with cancers that respond to ICB. CC is a virally induced malignancy that constitutively expresses two viral oncoproteins, E6 and E7, that have no human homologs. Because of their viral origins, E6 and E7 have long been regarded as primarily responsible for the immunogenicity of HPV tumors. Indeed, this notion has been confirmed by various E6/E7 vaccines that have entered clinical trials, as well as the promising anti-tumor activity of autologous E6/E7-directed T cells reported in recent adoptive cell transfer (ACT) patient studies [61, 62].
The immunogenicity of CC also stems from non-viral sources, such as somatic mutations in the genome of host cells. E6 and E7 are notorious for driving APOBEC-signature mutations and copy number alterations, as well as impairing the DNA damage repair response [63,64,65,66]. Genetic alterations resulting from such aberrations are likely to result in neo-antigens that have sufficient immune reactivity. Some studies have already identified specific neo-epitopes in cervical cancer tumors, including those emanating from oncogenic driver genes [67•]. The other non-viral source of immunogenicity are tumor-expressed testis associated (TTA) genes. Some cancers, including cervical cancer, express these non-self germline genes [62]. Specifically, significant expression of the NY-ESO-1 and MAGE-A family genes, TAAs with a long history of antigenicity, was detected in nearly 50% of recurrent CC tumors [68]. The importance of these neo-epitopes and TTAs in conferring T cell reactivity to CC tumors was elegantly demonstrated by a recent study published in science. Antigens of HPV origin were compared against epitopes resulting from somatic mutations as well as TTAs derived from ACT patients in remission. Interestingly, non-HPV specific TILs from these patients demonstrated higher TCR clonality and functional avidity than those directed against HPV epitopes [69••]. Even though the sample size in this study was small, this finding reveals that there potentially exist multiple strong epitopes for use in immunotherapies other than HPV proteins. In fact, studies that have looked at the total number of mutations in CC in comparison with other cancers have found the data to be quite promising. CC has a fairly diverse and high number of neo-antigens, and its median number of mutations clusters together with cancers that already have been approved for ICB therapy, such as head and neck and bladder cancers [61, 63, 69••, 70,71,72]. From a standpoint of mutations and antigens alone, therefore, CC appears to be well suited for ICB-based clinical interventions.
Tumor-reactive T cell recruitment in CC
Immunologists and clinicians have long observed that the accumulations of TILs generally portend good patient outcomes [13, 73, 74]. This correlation is not unique to standard therapies, however, as pre-existing tumor-specific TILs are also the active agents following treatment with immune checkpoint inhibitors. As such, their recruitment into the TME is an essential determinant of the response of tumors to ICB. In CC patients, active, polyclonal TILs are evident in both primary tumors and lymph nodes, and their presence correlates with outcomes [75]. For instance, CC tumors that have high levels of CD8+ T cells with tumor-specific immune responses display a lower frequency of metastasis to lymph nodes [76]. Interestingly, TILs within the tumor epithelial cells are better predictors of good prognosis in CC patients after radiotherapy and chemotherapy than are TILs in the stroma [77]. Furthermore, these parenchymal TILs tend to be co-expressed with activation markers of tumor-lytic T cells. The evidence for the presence of T cells that infiltrate CC in response to tumor-specific antigens is also apparent in ACT studies. For example, in CC patients undergoing ACT therapy, the positive relationship between tumor-specific TILs and outcomes has held, in that only patients displaying T cell infiltrates specific for HPV proteins in the resected tumors experienced an objective clinical response [62]. That said, the fact that CC still advances and metastasizes in patients presenting these effector, tumor-specific TILs points to the existence of powerful negative regulators in the TME such as the immune checkpoints.
Expression of immune checkpoints in CC
Even though TILs express activation markers, co-inhibitory checkpoints that exhaust T cells and attenuate their responses are also expressed in CC [78,79,80]. Inhibitory immune receptors such as CTLA4, PD-1, and TIGIT (T cell immunoreceptor with Ig and ITIM domains) are up-regulated on CD4 and CD8 T cells of tumors. In particular, PD-1 and CTLA-4 can be found on CD103+ CD8 T cells embedded in the tumor parenchyma. Not only are these CD103+ CD8 T cells tumor-reactive, but they are also associated with good prognoses, making their inactivation particularly problematic [77]. An important research study by Stevanovic et al. provided even more compelling evidence for PD-1 expression in CC and its connection to tumor specificity. This study found that the PD1+ T cell compartment is also the niche where high avidity tumor-lytic T cells generated from both somatic mutations and viral epitopes reside [69••]. Other findings similarly show fairly high expression of PD-1 ranging from 46.97 to 60.82% [81, 82]. Moreover, there is evidence for existence of alternate immune checkpoints such as TIM-3 and LAG-3 on memory anti-tumor CD8 T cells [83].
Another critical component of the immune checkpoint system that plays an important role in CC is PD-L1. The expression of PD-L1 in CC is elevated on tumor cells as well as on TAMs and DCs [79•]. In about 67% of CC specimens, a copy gain mutation in the 9p24.1 locus for PD-L1 was found [84]. For reference, classical Hodgkin lymphoma (CHL) is another cancer with similarly high PD-L1 expression due to a copy gain mutation in the same locus. In CHL, virtually all patients carry this mutation, which confers a remarkable sensitivity to PD1 blockade. Specifically, the average clinical objective response rate (ORR) to PD-1 blockade in CHL is about 80%, the highest ORR to ICB measured in all cancers to date [10]. Whether the immune phenotype in CC with the copy gain mutation is similar to that seen in CHL is not yet clear.
In addition to the high expression of PD-L1 due to genetic amplification, PD-L1 expression in CC can also increase due to adaptive resistance. PD-L1, similarly to PD-1, has been found to co-localize with TIL densities. One study has found that about 60% of TILs and tumor cells expressed PD-1 and PD-L1, respectively [81, 85, 86]. Another study has found that even though normal epithelial cells express no PD-L1, about 95% of CIN and 80% of CC tissues express PD-L1 [87]. Such high expression patterns support earlier findings that local immunity is impaired early on and that impairment is used as a means of immune escape. Based on this evidence, therefore, inhibition of immune checkpoints is predicted to restore protective immunity.
Other HPV-associated cancers: HNSCC as a benchmark?
A reasonable precedent for assessing the potential of ICB in CC is head and neck squamous cell carcinoma (HNSCC), as both CC and HNSCC are often caused by HPV. HNSCC has had two ICB agents, pembrolizumab and nivolumab, approved for use by the FDA in 2016 [10]. Importantly, HPV+ HNSCC displayed a better clinical response to ICB than did HPV− HNSCC. In addition, HNSCC and CC cluster in the same group in terms of total and median number of mutations and predicted response to ICB. Furthermore, it is not only the number of mutations that these two cancers share; the patterns and characteristics of the mutations found in these two cancers are also similar [70, 71]. It is a reasonable prediction, therefore, that the successful inhibition of immune checkpoints in HNSCC could inform similar endeavors in CC. Nonetheless, caution should be practiced in extrapolating these findings. For example, there are differences in the anatomical location and natural history of infection when comparing HPV+ HNSCC with HPV+ CC. In HNSCC, the HPV infects the oropharyngeal epithelium in the tonsils, and tonsils are a lymphoid-rich organ. On the other hand, in CC, the HPV infects the mucosal epithelia of the cervix, which is relatively shielded from the immune surveillance provided by lymphatic tissue. Also, some studies have shown that the local HPV-specific T cell response, Th1 levels, and IFN-γ production are relatively better in HPV+ HNSCC [88,89,90]. Together, the molecular and immune landscape in CC suggests feasibility and potential therapeutic benefit of employing ICB in the treatment of CC.
Clinical experience regarding the use of single checkpoint inhibitors in CC
The clinical success of ICB in malignant diseases was initially tested in cancers such as melanoma, NSCLC, and RCC over the past 5 years [10], while gynecological cancers such as CC have been considered only more recently. However, there have been a number of clinical trials in recent years exploring the effectiveness of single checkpoint inhibitors in CC, with the number growing every year [8, 91]. For perspective on the progress made so far, a few key studies involving the antibodies ipilimumab, nivolumab, and pembrolizumab are discussed below.
Ipilimumab
The Chicago N01 Consortia study as reported by Lheureux et al. was one of the key studies that first investigated CTLA-4 inhibitors in CC. Heavily pretreated patients were administered the CTLA-4 antibody, ipilimumab. From the standpoint of clinical benefit, the performance of ipilimumab was below par. The objective response, PFS, and OS were low and not close to the targeted end points. However, ipilimumab demonstrated safety and manageable toxicities in CC patients. Today, more clinical trials involving ipilimumab or other CTLA-4 antibodies are being conducted using this study as a reference [8, 92].
Nivolumab
Nivolumab, which targets the PD-1 immune checkpoint, is another monoclonal antibody that has been studied in CC patients quite extensively. Check-358 is one of the key early studies that assessed the efficacy and safety of nivolumab in CC [8, 92]. The study included 19 patients who had disease in the R/M setting and prior systemic therapies. The overall response rate was 26.3%, one of the highest yet found in CC ICB studies. Nivolumab also showed low toxicity and signs of a durable response in some patients. The major conclusion reached in this study was that nivolumab has some encouraging activity and a good safety profile. Future confirmatory nivolumab studies were thus warranted for validation, and one such study is the NRGGY002 trial [8]. Unfortunately, even though safety was confirmed, the findings presented at the ASCO annual meeting in 2018 concluded that the anti-tumor activity of nivolumab in CC is low. Therefore, definitive answers regarding the clinical efficacy of nivolumab in CC are not yet available.
Pembrolizumab
Considering the challenges that ipilimumab and nivolumab have faced, pembrolizumab, another anti-PD1 antibody, may represent the most promising single ICB therapy tested in CC thus far. In 2016, results from the KEYNOTE-028 phase 1b study in 24 patients who had advanced CC disease and prior systemic treatment showed that pembrolizumab is well tolerated with relatively good responses. The median OS was approximately 11 months in these patients, with an ORR of about 17% [8, 9]. These findings were followed up by the KEYNOTE-158 study in a larger patient population. Results of the KEYNOTE-158 study were presented at the 2018 ASCO annual meeting. In 91% of the patients studied, the duration of response was at least 6 months, and the OS was 9.4 months. Despite the relatively low ORR of 14.3%, the major conclusion was that pembrolizumab had a durable anti-tumor activity and a safe profile. Based on this, the FDA accelerated the approval of pembrolizumab, with a final approval given in June 2018, making it the first approved checkpoint inhibitor for the treatment of CC. All patients who responded were positive for PD-L1, and none without PD-L1 responded [8, 9, 92, 93]. Even though the 14% RR of pembrolizumab may not have been sufficient for other cancers, its approval underscores the complete lack of viable, alternative second-line therapies in CC. Now patients in the relapse setting can use this antibody, giving them a second chance at survival.
Combinations in ICB: the quest for increasing ICB response in patients
Although the studies described above regarding single immune checkpoint therapy in CC are encouraging, it is clear that there is still much work to be done before the great potential of ICB, as seen in some other malignancies, can be fully realized. Ipilimumab and nivolumab, albeit safe in CC patients, have exhibited low anti-tumor activity. Pembrolizumab is the only antibody approved thus far, but its approval is still partial; full approval is contingent on future clinical studies that substantiate the initial clinical benefit observed. In addition, the accelerated approval was based on a study that showed 14% ORR, meaning only a select few patients are likely to benefit from pembrolizumab.
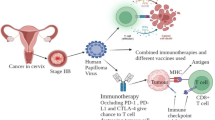
In this context, it should be kept in mind that cervical cancer meets most criteria regarding ICB suitability, from its robust PD1/PDL1 expression and a T cell-inflamed phenotype to the mutational load and immunogenicity of HPV+ tumors. So how then are the impressive ICB responses observed in other cancers not seen in CC? Clearly, there are gaps in our understanding of the immune biology in CC as it pertains to ICB and the strategies to boost the paltry 14% ORR to maybe 25–30% or more. More light thus needs to be shed on the immune landscape of CC, and greater research efforts are needed in identifying biomarkers other than PD-L1 and ascertaining their robustness in predicting who benefits. Additional studies are needed on inflammatory gene signatures and the pathways to target in combination therapy as well as the identity of mutations, viral peptide, and non-viral epitope pools in CC. Furthermore, immunosuppressive mediators such as TAMs, Tregs, MDSCs, IDO, and alternate checkpoints and their impact on disease progression and the efficacy of ICB need to be elucidated [94, 95]. CC tumors are known to down-regulate expression of MHC complexes for antigen presentation. Also, a recent comprehensive multi-omic study showed that CC is more heterogeneous and molecularly complex than previously thought, which might suggest that the effectiveness and durability of ICB depend on how patients are stratified for treatment [64]. All these features are potential escape mechanisms contributing to the attenuation of ICB efficacy (Fig. 1). While research efforts will continue to unravel the immune context of CC and these mechanisms, combination of therapies represent an alternative and viable route forward in the immediate future. Exciting findings are being reported regarding ICB combinations in other cancers [96, 97], and the data presented in the literature supports the anticipation that certain agents and modalities in combination with ICB may have synergistic effects and extend clinical benefit to many non-responders in CC. These modalities include cytotoxic chemotherapy, targeted small molecules, radiation therapy, and vaccines.

Combination therapies to potentiate the efficacy of ICB monotherapy. Various putative mechanisms of resistance (innate or acquired) to ICB may arise due to tumor heterogeneity, lack of sufficiently reactive neo-antigens, inability of T cells to infiltrate tumors, impairment of antigen presentation, exhaustion of T cells and/or increased expression of immunosuppressive metabolic mediators, immune suppressive cells and alternate immune checkpoints. ICB as a monotherapy may help to re-activate anti-tumor T cell immunity and ameliorate exhaustion, while chemo, targeted, and radiation-mediated therapy may promote antigenicity, improve IFN signaling and intra-tumor infiltration, and increase expression of PD-1/PD-L1. Combinations of ICB with any of these modalities will potentially enhance function of DCs and antigen presentation, reduce depletion of immunosuppressive milieu, and enhance diversity and robustness of tumor reactivity of CD4 and CD8 T cells as well as potential systemic immunity, together leading to significantly improved immunological tumor control. + represents stimulating TCR-MHC interactions and – represent inhibitory PD1/PDL1 or B7/CTLA-4 interactions on T cells.
Chemotherapy and radiation: basis for synergy with ICB
Single immune checkpoint inhibitors in CC have failed to objectively shrink tumors or prolong survival in the majority of patients. Such observations in these patients may be attributed to (i) their tumors being immunologically cold, lacking TILs and antigens and (ii) their tumors having diminished effector T cell activity due to checkpoint-independent immunosuppression. Can chemotherapy or radiotherapy overcome these challenges?
This may indeed be the case. An accumulating body of evidence in various cancers shows that both chemotherapy (CT) and radiotherapy (RT) can have immune-stimulatory effects. With regard to cold tumors, RT and CT can induce immunogenic cell death (ICD) that increases the formation of cancer-specific peptides or DNA and exposure of damage-associated molecular patterns (DAMPs) in immune cell poor tumors [14, 98,99,100,101]. The available tumor antigens and DAMPs will augment antigen presentation, T cell priming, and activation either directly or as adjuvants and boost T cell infiltration.
PD1/PDL1-independent immunological barriers such as inhibitory immune cells, suppressive cytokines, and metabolites can also limit the efficacy of ICB. Nevertheless, certain chemotherapies and radiation regimens have been shown to deplete immune regulatory factors (MDSCs, Tregs, IDO), increase expression of class I/II MHCs, and enhance function of DCs for T cell priming. This also makes the TME more hospitable, freeing up PD1+/PDL1+ CTLs and poising them for ICB-mediated re-activation. Combination therapies therefore could potentiate the efficacy of ICB by acting as spark plugs that alter the tumor immune landscape and make it more supportive of ICB therapy (Fig. 1).
In many cancers such as melanoma, NSCLC, and breast and bladder cancers, combinations of ICB with cytotoxic chemotherapies, targeted small molecules, monoclonal antibodies, and radiation therapy are already being tested in clinical trials with promising results [73, 97, 99, 101]. In particular, the FDA has already approved the combination of chemotherapy (pemetrexed and cisplatin/carboplatin) with pembrolizumab for first-line treatment of metastatic NSCLC [102]. Another great case in point is the recent FDA approval of atezolizumab in combination with nanoparticle albumin-bound (nab)-paclitaxel in triple negative breast cancer (TNBC). As expected, the combination therapy was better than either therapy alone, with overall response jumping from 10% in the monotherapy trial to about 40% in the combination trial [103]. Even though chemotherapy might exacerbate toxicity of ICB, these examples of success of chemotherapy cooperating with ICB to broaden clinical response should encourage the testing of combinations in cancers such as cervical cancer where modest activity and response rates are being observed with monotherapy.
Chemotherapy: pre-clinical models and preliminary clinical evidence
Combinatorial studies of ICB with cytotoxic chemotherapy in HPV-associated cancers are still in their infancy relative to cancers such as NSCLC. However, a number of pre-clinical studies provide a rationale for conducting such trials in the clinic. Research findings reported by Spanos et al. almost a decade ago provided the first clear link between chemo-sensitivity and tumor immunogenicity in HPV-associated tumors. The study found that HNSCC tumors in mice were sensitive to cisplatin only when the immune system of the tumor-bearing host was competent [104]. The authors concluded that the sensitization to cisplatin was primarily due to the induction of the immune response in immune-competent mice and thus challenged the previous understanding that the effectiveness of chemotherapy stems autonomously from its cytotoxic or cytostatic effects on tumor cells. This seminal study has since been corroborated by more recent studies. For example, the Schmitt group has found that not only does cisplatin induce ICD and up-regulate PD-L1, but it also enhances antigen presentation and T cell tumor reactivity. When the group combined anti-PDL1 therapy and cisplatin subsequently, synergistic anti-tumor effects were observed [105]. In line with this study, PD-L1 overexpression following cisplatin has also been observed clinically in HNSCC patients [106].
Importantly, similar findings have been found in the context of CC. In mice, the treatment of tumors with cisplatin was associated with an increase in PD-1, implying recent in vivo T cell activation. A cisplatin plus anti-PD1 cocktail was associated with better tumor regression and reconstitution of tumor-active T cells [107]. In patients with CC, cisplatin was also shown to support immune cell activation and infiltration of the tumors by T cells. Consistently,
the activation of T cells was also associated with the development of potentially deleterious adaptive resistance (PD-L1 up-regulation), providing another reason for combining ICB with chemotherapy [82]. Another standard cytotoxic regimen that has been investigated for immune-modulatory effects in the clinic is carboplatin plus paclitaxel. The immune cell subpopulation that was most affected by this chemotherapy doublet was myeloid suppressor cells. Their abrogation was followed by enhancement in DC function, T cell reactivity, and prolonged patient survival [108]. The take home from their findings was that relief of tumor-lytic T cells from myeloid-driven immunosuppression may be a critical step needed before activity of these cells can be fully restored by immunotherapies. This is line with the evidence that shows that tumor-directed TILs in CC tumors are often accompanied by MDSCs, Tregs, and a variety of suppressive cytokines [79].
As summarized above, a fairly extensive collection of pre-clinical information in the literature supports the concept of ICB in combination with cytotoxic chemotherapy. Combinatorial studies of ICB with targeted chemotherapy therapy, on the other hand, are still few in number and limited in scope. Nonetheless, the potential in some studies is evident. For example, treatment of cervical cancer cells in vitro with Smac mimetics has shown evidence of necroptosis and up-regulation of markers for ICD (HMGB1, CRT, ATP) [109]. Clinical trials for CC involving Smac mimetics in combination with checkpoint inhibitors are also already underway. Another recent study explored potential novel targeted therapy effectors for combination with immune checkpoint blockade using gene expression analysis of recurrent CC tissues. An evaluation of cancer immune genes and cancer-specific molecular pathways yielded new promising targets in the DNA damage repair pathway (e.g., PARP) and epithelial-mesenchymal transition-related genes (e.g., STAT3) [110]. These targets could be considered in future pre-clinical and clinical trial immune checkpoint combination studies. Table 1 lists ongoing ICB combinatorial clinical trials in CC.
Radiation therapy: pre-clinical models and preliminary clinical evidence
Pre-clinical and retrospective clinical evidence in HNSCC and CC involving ICB and RT combinations is consistent with that observed using chemotherapy. HNSCC preclinical models show that PD-L1 is up-regulated on tumor and myeloid cells following RT. When checkpoint blockade is applied, T cell infiltration into the tumor lesions with concomitant enhancement of tumor control and prolonged mouse survival are observed [111•]. The same phenotypes and outcomes are also seen clinically in HPV+ HNSCC patients who are treated with radiotherapy [112, 113].
Rodent studies modeling CC also confirm the immuno-modulatory role of RT. In the Wistar rat model, treatment with RT led to overexpression of PD-L1, as seen in other cancers [114]. In a TC-1 mouse model, RT failed to clear tumors in immune-compromised mice, again showing the interplay between host immunity and RT [104]. In addition, combinations involving antigen challenge were shown to increase the robustness of tumor-targeted T cell generation by radiotherapy in mice [77]. While these pre-clinical reports are promising, there remain hurdles in integrating RT with ICB clinically, particularly with regard to determining the optimum radiation dosage, fractions to use, and the order of combination with immunotherapy. Low radiation may not be sufficiently immunogenic, while high radiation can destroy the very immune cells that mediate ICB effects. In addition, it is not yet definitive as to whether fractionated or conventional radiation is more effective [99, 115]. More research therefore is needed to fine-tune the conditions for optimized regimens. To that end, some studies in CC are already looking into how RT affects the frequency of immune cells and their activation in the clinic. In a clinical study by Dorta-Estremera et al., patients treated with fractionated radiation initially experienced a transient decline in CD4 and CD8 T cells. Within 2 weeks, however, a proliferative phenotype of these cells rebounded, together with increased TCR diversity and enhanced local antigen presentation [116]. Similar preliminary findings of improved efficacy in the combination setting were found in another clinical study [117]. These data bolster the idea that RT and ICB together may yield more effective disease control. Ongoing combination clinical trials are shown in Table 1, and results from the early phases of some these combination clinical trials are already showing encouraging signs of tolerability, feasibility, and effectiveness of chemoradiation-immunotherapy combinations [12, 118].
Therapeutic vaccines: basis for synergy with ICB
Suboptimal T cell priming, activation, and expansion are some of the early events involved in disabling protective tumor immunity. Lack of antigens or their acquisition can be the primary cause for the below par lymphocyte activation seen in CC. Vaccination can help overcome this hurdle by artificially generating and magnifying the low population of TA-specific T cells and increase their reactivity upon tumor challenge. In this way, vaccines help to improve the presentation process of tumor antigens and subsequent activation of T cells [61, 62]. However, despite observations that therapeutic vaccines against CC are capable of inducing immunogenicity, most have failed to show meaningful clinical anti-tumor activity as a single therapy in patients. Studies in vitro as well as preclinical models suggest that the clinical potential of vaccines may be restricted by tumor-mediated immune suppression, which includes immune checkpoints that deactivate the CTLs triggered by the vaccines [17, 61, 62]. Combining vaccines and checkpoint inhibitors, given their non-redundancy in anti-tumor response, along with the possibility that ICB could overcome limitations to vaccine efficacy, carries significant clinical potential.
Although discussion is needed as to which group of patients should be targeted by vaccine/ICB combinations, patients that make up the late-stage CC population are potential beneficiaries. Studies have shown that in contrast to early-stage CC, which abounds with tumor-reactive TILs, aggressive late-stage CC is poorly infiltrated. A study by Komedeu et al. concluded that the patients in this group may require additional interventions such as vaccines to pre-condition their tumors with T cells and thus make them candidates for a high ICB response [77]. In addition to this group, patients who express PD-L1 as a result of tumor intrinsic signaling as opposed to an immunogenic response may also benefit tremendously from vaccines. Overall, the prospects for clinical success for all patients who will end up using CC therapeutic vaccines are improving. The recent findings that dominant tumor-reactive T cell populations are actually directed against some previously unidentified germline antigens and neo-epitopes and not necessarily against HPV onco-proteins are expected to transform the field [67, 69••]. Based on this newly available broader panel of strong and non-tolerant epitopes, we should, in theory, be able to design better vaccines, monovalent or polyvalent, that will increase the amplitude and duration of immune response. For now, most therapeutic vaccines are still based on various peptide or full protein forms of E6 and E7.
Therapeutic vaccines: pre-clinical models and preliminary clinical evidence
Just as with chemotherapy and radiation therapy, vaccination improves the efficacy of immune checkpoint inhibitors compared with either therapy alone. In one mouse study, immunization against E6/E7 genes using viral vectors resulted in infiltration of activated CD8+ CTLs into tumors followed by immunosuppression due to PD-L1 up-regulation. When PD-1 blockade was applied, potent tumor regression was observed [119]. Similar observations were made when a DC-based vaccination strategy against E7 was used followed by the treatment of the tumors with a PD-L1 inhibitor [120]. Inhibition of checkpoints and activation of co-stimulators in combination with an E6/E7 peptide vaccine has also been found to generate E7-specific CD8 T cells and Th CD4 cells [121]. Another DNA vaccine with potential for clinical success in CC is the live-attenuated Listeria monocytogenes-based vaccine. An Lm-LOO-E7 vaccine designed to secrete the E7 protein in combination with a PD1 inhibitor was associated with improved antigen presentation, TILs frequency, and significant tumor inhibition. Moreover, the combination also depleted the inhibitory regulatory cells (MDSCs and Tregs) and resulted in complete remissions in some mice [122]. These observations have been recently corroborated by an Lm-LOO-E6 vaccine combined with a PDL1 inhibitor in SiHa and TL-1 subcutaneous mouse models [123]. Another study that combined 4-1BB agonist or CTLA-4 blockade with E6/E7 peptide vaccine in an orthotopic syngeneic mouse model also observed depletion of immunosuppressive cells in addition to curative efficacy [124•]. More importantly, data from a recent phase II trial appears to validate these pre-clinical studies as a combination of an HPV-peptide vaccine with nivolumab yielded an ORR of 33%, which is about double on what we have generally seen with anti-PD1 monotherapies [125••]. This emerging clinical evidence has buoyed the hope that this combinatorial approach will work in patients and will encourage more studies in the future. Table 1 shows other vaccines currently being tested in combination with ICB.
Dual checkpoint inhibition
Dual immune checkpoint inhibitors are also a logical combination that may increase ICB efficacy in cervical cancer. CTLA-4 antibodies trigger T cell priming and clearance of inhibitory regulatory cells. PD-1/PDL1 inhibitors, on the other hand, enhance activation of peripheral T cells and help counteract adaptive resistance. In HPV-associated cancers, no pre-clinical data is yet available to back the use of these checkpoints together. However, the mechanistic non-redundancy of these two types of checkpoint inhibitors, their individual overexpression and demonstrated tolerability in CC, may result in additive or synergistic effects in combination. Proof that PD-1 and CTLA-4 inhibitors can be more efficacious together has already been provided by dual inhibition studies in melanoma. The combination of nivolumab (anti-PD1) plus ipilimumab (anti-CTLA4) increased the response rate by 2-fold from about 30 to 60%. Not only was response improved, but patient survival was also higher in the dual inhibition group. The approval of the two inhibitors by the FDA has opened the door for dual ICB in many other cancers [94]. The trade off, however, that comes with this increased activity is higher toxicity. Only when the response is high enough and there are no alternatives can the toxicity be justified [10, 126]. See Table 1 for similar dual checkpoint inhibitor studies for CC in clinical trials.
Conclusions
ICB has shown a marked therapeutic success in cancers such as NSLC and CHL. In cancers such as CC and other gynecological cancers, similar prospects in the future are likely, noting that research on efficacy is still in its infancy. ICB monotherapy, particularly pembrolizumab, has offered a ray of hope for some CC patients with recurrent or metastatic disease. For the majority of patients who did not respond, efforts are already underway to try to convert the non-responders into responders. A number of additional factors in the TME outside of the PD1:PDL1 axis have been suggested to contribute to ICB escape in non-responders. Therefore, composite biomarkers and a multi-modal approach that increases tumor immunogenicity, T cell priming, and T cell intratumoral infiltration might help to circumvent intrinsic or acquired resistance to ICB. Past and ongoing clinical studies we highlighted in this review will help to inform further investigations, better design of future clinical trials, and reveal strategies on how to effectively translate or integrate ICB with other treatments for better cure rates. Even though challenges lie ahead, the accelerated approval of pembrolizumab, the follow-up studies involving combinations thereof, and other studies aimed at personalized care and better predictive biomarkers could improve the utility of ICB in the clinical management of CC in upcoming years.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Eskander RN, Tewari KS. Beyond angiogenesis blockade: targeted therapy for advanced cervical cancer. J Gynecol Oncol. 2014;25(3):249–59.
Lowy DR, Schiller JT. Prophylactic human papillomavirus vaccines. J Clin Invest. 2006;116(5):1167–73.
Stanley M. Prophylactic HPV vaccines. Drugs Today (Barc). 2007;43(10):737–44.
Brotherton JM. Human papillomavirus vaccination: where are we now? J Paediatr Child Health. 2014;50(12):959–65.
Niccolai LM, Hansen CE. Practice- and community-based interventions to increase human papillomavirus vaccine coverage: a systematic review. JAMA Pediatr. 2015;169(7):686–92.
Bruni L, Diaz M, Barrionuevo-Rosas L, Herrero R, Bray F, Bosch FX, et al. Global estimates of human papillomavirus vaccination coverage by region and income level: a pooled analysis. Lancet Glob Health. 2016;4(7):e453–63.
Torre LA, Islami F, Siegel RL, Ward EM, Jemal A. Global cancer in women: burden and trends. Cancer Epidemiol Biomarkers Prev. 2017;26(4):444–57.
Minion LE, Tewari KS. Cervical cancer - State of the science: from angiogenesis blockade to checkpoint inhibition. Gynecol Oncol. 2018;148(3):609–21.
Borcoman E, Le Tourneau C. Pembrolizumab in cervical cancer: latest evidence and clinical usefulness. Ther Adv Med Oncol. 2017;9(6):431–9.
Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359(6382):1350–5.
Smyth MJ, Ngiow SF, Ribas A, Teng MW. Combination cancer immunotherapies tailored to the tumor microenvironment. Nat Rev. Clin Oncol. 2016;13(3):143–58.
Liu YL, Zamarin D. Combination immune checkpoint blockade strategies to maximize immune response in gynecological cancers. Curr Oncol Rep. 2018;20(12):94.
Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell. 2015;28(6):690–714.
Gong J, Le TQ, Massarelli E, Hendifar AE, Tuli R. Radiation therapy and PD-1/PD-L1 blockade: the clinical development of an evolving anticancer combination. J Immunother Cancer. 2018;6(1):46.
Gotwals P, Cameron S, Cipolletta D, Cremasco V, Crystal A, Hewes B, et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat Rev. Cancer. 2017;17(5):286–301.
Hughes PE, Caenepeel S, Wu LC. Targeted therapy and checkpoint immunotherapy combinations for the treatment of cancer. Trends Immunol. 2016;37(7):462–76.
Bashaw AA, Leggatt GR, Chandra J, Tuong ZK, Frazer IH. Modulation of antigen presenting cell functions during chronic HPV infection. Papillomavirus Res. 2017;4:58–65.
Kobayashi A, Weinberg V, Darragh T, Smith-McCune K. Evolving immunosuppressive microenvironment during human cervical carcinogenesis. Mucosal Immunol. 2008;1(5):412–20.
Stanley MA. Epithelial cell responses to infection with human papillomavirus. Clin Microbiol Rev. 2012;25(2):215–22.
Welters MJ, de Jong A, van den Eeden SJ, van der Hulst JM, Kwappenberg KM, Hassane S, et al. Frequent display of human papillomavirus type 16 E6-specific memory t-Helper cells in the healthy population as witness of previous viral encounter. Cancer Res. 2003;63(3):636–41.
Grabowska AK, Riemer AB. The invisible enemy - how human papillomaviruses avoid recognition and clearance by the host immune system. Open Virol J. 2012;6:249–56.
Smola S. Immunopathogenesis of HPV-associated cancers and prospects for immunotherapy. Viruses. 2017;9(9).
Smola S, Trimble C, Stern PL. Human papillomavirus-driven immune deviation: challenge and novel opportunity for immunotherapy. Ther Adv Vaccines. 2017;5(3):69–82.
Yang X, Cheng Y, Li C. The role of TLRs in cervical cancer with HPV infection: a review. Signal Transduct Target Ther. 2017;2:17055.
Vermeulen CF, Jordanova ES, Zomerdijk-Nooijen YA, ter Haar NT, Peters AA, Fleuren GJ. Frequent HLA class I loss is an early event in cervical carcinogenesis. Hum Immunol. 2005;66(11):1167–73.
Chen Wongworawat Y, Filippova M, Williams VM, Filippov V, Duerksen-Hughes PJ. Chronic oxidative stress increases the integration frequency of foreign DNA and human papillomavirus 16 in human keratinocytes. Am J Cancer Res. 2016;6(4):764–80.
Chen Y, Williams V, Filippova M, Filippov V, Duerksen-Hughes P. Viral carcinogenesis: factors inducing DNA damage and virus integration. Cancers (Basel). 2014;6(4):2155–86.
Fernandes JV, TA DEMF, JC DEA, Cobucci RN, MG DEC, Andrade VS, et al. Link between chronic inflammation and human papillomavirus-induced carcinogenesis (Review). Oncol Lett. 2015;9(3):1015–26.
Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev. Cancer. 2010;10(8):550–60.
Williams VM, Filippova M, Filippov V, Payne KJ, Duerksen-Hughes P. Human papillomavirus type 16 E6* induces oxidative stress and DNA damage. J Virol. 2014;88(12):6751–61.
Williams VM, Filippova M, Soto U, Duerksen-Hughes PJ. HPV-DNA integration and carcinogenesis: putative roles for inflammation and oxidative stress. Future Virol. 2011;6(1):45–57.
Kessis TD, Connolly DC, Hedrick L, Cho KR. Expression of HPV16 E6 or E7 increases integration of foreign DNA. Oncogene. 1996;13(2):427–31.
Roden RBS, Stern PL. Opportunities and challenges for human papillomavirus vaccination in cancer. Nat Rev. Cancer. 2018;18(4):240–54.
Mazibrada J, Ritta M, Mondini M, De Andrea M, Azzimonti B, Borgogna C, et al. Interaction between inflammation and angiogenesis during different stages of cervical carcinogenesis. Gynecol Oncol. 2008;108(1):112–20.
Tjiong MY, van der Vange N, ten Kate FJ, Tjong AHSP, ter Schegget J, Burger MP, et al. Increased IL-6 and IL-8 levels in cervicovaginal secretions of patients with cervical cancer. Gynecol Oncol. 1999;73(2):285–91.
Iglesias M, Plowman GD, Woodworth CD. Interleukin-6 and interleukin-6 soluble receptor regulate proliferation of normal, human papillomavirus-immortalized, and carcinoma-derived cervical cells in vitro. Am J Pathol. 1995;146(4):944–52.
Artaza-Irigaray C, Molina-Pineda A, Aguilar-Lemarroy A, Ortiz-Lazareno P, Limon-Toledo LP, Pereira-Suarez AL, et al. E6/E7 and E6(*) From HPV16 and HPV18 upregulate IL-6 expression independently of p53 in keratinocytes. Front Immunol. 2019;10:1676.
Barros MR Jr, de Oliveira THA, de Melo CML, Venuti A, de Freitas AC. Viral modulation of TLRs and cytokines and the related immunotherapies for HPV-associated cancers. J Immunol Res. 2018;2018:2912671.
Hess S, Smola H, Sandaradura De Silva U, Hadaschik D, Kube D, Baldus SE, et al. Loss of IL-6 receptor expression in cervical carcinoma cells inhibits autocrine IL-6 stimulation: abrogation of constitutive monocyte chemoattractant protein-1 production. J Immunol. 2000;165(4):1939–48.
Ren C, Cheng X, Lu B, Yang G. Activation of interleukin-6/signal transducer and activator of transcription 3 by human papillomavirus early proteins 6 induces fibroblast senescence to promote cervical tumourigenesis through autocrine and paracrine pathways in tumor microenvironment. Eur J Cancer. 2013;49(18):3889–99.
Morgan EL, Macdonald A. Autocrine STAT3 activation in HPV positive cervical cancer through a virus-driven Rac1-NFkappaB-IL-6 signaling axis. PLoS Pathog. 2019;15(6):e1007835.
Wei LH, Kuo ML, Chen CA, Chou CH, Lai KB, Lee CN, et al. Interleukin-6 promotes cervical tumor growth by VEGF-dependent angiogenesis via a STAT3 pathway. Oncogene. 2003;22(10):1517–27.
Morgan EL, Wasson CW, Hanson L, Kealy D, Pentland I, McGuire V, et al. STAT3 activation by E6 is essential for the differentiation-dependent HPV18 life cycle. PLoS Pathog. 2018;14(4):e1006975.
Pahne-Zeppenfeld J, Schroer N, Walch-Ruckheim B, Oldak M, Gorter A, Hegde S, et al. Cervical cancer cell-derived interleukin-6 impairs CCR7-dependent migration of MMP-9-expressing dendritic cells. Int J Cancer. 2014;134(9):2061–73.
Spurgeon ME, den Boon JA, Horswill M, Barthakur S, Forouzan O, Rader JS, et al. Human papillomavirus oncogenes reprogram the cervical cancer microenvironment independently of and synergistically with estrogen. Proc Natl Acad Sci U S A. 2017;114(43):E9076–E85.
Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell. 2010;17(2):135–47.
Dudas J, Fullar A, Bitsche M, Schartinger V, Kovalszky I, Sprinzl GM, et al. Tumor-produced, active interleukin-1beta regulates gene expression in carcinoma-associated fibroblasts. Exp Cell Res. 2011;317(15):2222–9.
Barros MR Jr, de Melo CML, Barros M, de Cassia Pereira de Lima R, de Freitas AC, Venuti A. Activities of stromal and immune cells in HPV-related cancers. J Exp Clin Cancer Res. 2018;37(1):137.
Bais AG, Beckmann I, Lindemans J, Ewing PC, Meijer CJ, Snijders PJ, et al. A shift to a peripheral Th2-type cytokine pattern during the carcinogenesis of cervical cancer becomes manifest in CIN III lesions. J Clin Pathol. 2005;58(10):1096–100.
Gosmann C, Mattarollo SR, Bridge JA, Frazer IH, Blumenthal A. IL-17 suppresses immune effector functions in human papillomavirus-associated epithelial hyperplasia. J Immunol. 2014;193(5):2248–57.
Jacobs N, Giannini SL, Doyen J, Baptista A, Moutschen M, Boniver J, et al. Inverse modulation of IL-10 and IL-12 in the blood of women with preneoplastic lesions of the uterine cervix. Clin Exp Immunol. 1998;111(1):219–24.
Kanodia S, Fahey LM, Kast WM. Mechanisms used by human papillomaviruses to escape the host immune response. Curr Cancer Drug Targets. 2007;7(1):79–89.
Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10.
Xu-Monette ZY, Zhang M, Li J, Young KH. PD-1/PD-L1 blockade: have we found the key to unleash the antitumor immune response? Front Immunol. 2017;8:1597.
Maleki Vareki S, Garrigos C, Duran I. Biomarkers of response to PD-1/PD-L1 inhibition. Crit Rev. Oncol Hematol. 2017;116:116–24.
Mirzaei R, Sarkar S, Yong VW. T Cell exhaustion in glioblastoma: intricacies of immune checkpoints. Trends Immunol. 2017;38(2):104–15.
Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8(328):328rv4.
Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–30.
Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying cancers based on T cell infiltration and PD-L1. Cancer Res. 2015;75(11):2139–45.
Gibney GT, Weiner LM, Atkins MB. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016;17(12):e542–e51.
Vici P, Pizzuti L, Mariani L, Zampa G, Santini D, Di Lauro L, et al. Targeting immune response with therapeutic vaccines in premalignant lesions and cervical cancer: hope or reality from clinical studies. Expert Rev. Vaccines. 2016;15(10):1327–36.
Zsiros E, Tsuji T, Odunsi K. Adoptive T cell therapy is a promising salvage approach for advanced or recurrent metastatic cervical cancer. J Clin Oncol. 2015;33(14):1521–2.
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–21.
Cancer Genome Atlas Research N, Albert Einstein College of M, Analytical Biological S, Barretos Cancer H, Baylor College of M, Beckman Research Institute of City of H, et al. Integrated genomic and molecular characterization of cervical cancer. Nature. 2017;543(7645):378–84.
Litwin TR, Clarke MA, Dean M, Wentzensen N. Somatic host cell alterations in HPV carcinogenesis. Viruses. 2017;9(8).
Vieira VC, Leonard B, White EA, Starrett GJ, Temiz NA, Lorenz LD, et al. Human papillomavirus E6 triggers upregulation of the antiviral and cancer genomic DNA deaminase APOBEC3B. MBio. 2014;5(6).
• Qin Y, Ekmekcioglu S, Forget MA, Szekvolgyi L, Hwu P, Grimm EA, et al. Cervical cancer neoantigen landscape and immune activity is associated with human papillomavirus master regulators. Front Immunol. 2017;8:689. Identified novel non-viral antigens that will re-strategize immunotherapy targeting or vaccine design.
Napoletano C, Bellati F, Tarquini E, Tomao F, Taurino F, Spagnoli G, et al. MAGE-A and NY-ESO-1 expression in cervical cancer: prognostic factors and effects of chemotherapy. Am J Obstet Gynecol. 2008;198(1):99 e1–7.
•• Stevanovic S, Pasetto A, Helman SR, Gartner JJ, Prickett TD, Howie B, et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science. 2017;356(6334):200–5. Significantly revealed that neo- and germline antigens were the potent and dominant immunogens in anti-tumor response of CC patients that underwent complete regression.
Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9(1):34.
Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214–8.
Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med. 2017;377(25):2500–1.
Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumors: impact on clinical outcome. Nat Rev. Cancer. 2012;12(4):298–306.
Tung NM, Winer EP. Tumor-infiltrating lymphocytes and response to platinum in triple-negative breast cancer. J Clin Oncol. 2015;33(9):969–71.
de Vos van Steenwijk PJ, Heusinkveld M, Ramwadhdoebe TH, Lowik MJ, van der Hulst JM, Goedemans R, et al. An unexpectedly large polyclonal repertoire of HPV-specific T cells is poised for action in patients with cervical cancer. Cancer Res. 2010;70(7):2707–17.
Piersma SJ, Jordanova ES, van Poelgeest MI, Kwappenberg KM, van der Hulst JM, Drijfhout JW, et al. High number of intraepithelial CD8+ tumor-infiltrating lymphocytes is associated with the absence of lymph node metastases in patients with large early-stage cervical cancer. Cancer Res. 2007;67(1):354–61.
Komdeur FL, Prins TM, van de Wall S, Plat A, Wisman GBA, Hollema H, et al. CD103+ tumor-infiltrating lymphocytes are tumor-reactive intraepithelial CD8+ T cells associated with prognostic benefit and therapy response in cervical cancer. Oncoimmunology. 2017;6(9):e1338230.
Chang H, Hong JH, Lee JK, Cho HW, Ouh YT, Min KJ, et al. Programmed death-1 (PD-1) expression in cervical intraepithelial neoplasia and its relationship with recurrence after conization. J Gynecol Oncol. 2018;29(3):e27.
• Heeren AM, Koster BD, Samuels S, Ferns DM, Chondronasiou D, Kenter GG, et al. High and interrelated rates of PD-L1 + CD14+ antigen-presenting cells and regulatory T cells mark the microenvironment of metastatic lymph nodes from patients with cervical cancer. Cancer Immunol Res. 2015;3(1):48–58. Findings in this study suggest that maximizing checkpoint therapy efficacy may require counteracting the actions of immunosuppressive effectors cells and mediators.
Heeren AM, Punt S, Bleeker MC, Gaarenstroom KN, van der Velden J, Kenter GG, et al. Prognostic effect of different PD-L1 expression patterns in squamous cell carcinoma and adenocarcinoma of the cervix. Mod Pathol. 2016;29(7):753–63.
Feng YC, Ji WL, Yue N, Huang YC, Ma XM. The relationship between the PD-1/PD-L1 pathway and DNA mismatch repair in cervical cancer and its clinical significance. Cancer Manag Res. 2018;10:105–13.
Meng Y, Liang H, Hu J, Liu S, Hao X, Wong MSK, et al. PD-L1 expression correlates with tumor infiltrating lymphocytes and response to neoadjuvant chemotherapy in cervical cancer. J Cancer. 2018;9(16):2938–45.
Heeren AM, Rotman J, Stam AGM, Pocorni N, Gassama AA, Samuels S, et al. Efficacy of PD-1 blockade in cervical cancer is related to a CD8(+)FoxP3(+)CD25(+) T cell subset with operational effector functions despite high immune checkpoint levels. J Immunother Cancer. 2019;7(1):43.
Howitt BE, Sun HH, Roemer MG, Kelley A, Chapuy B, Aviki E, et al. Genetic basis for PD-L1 expression in squamous cell carcinomas of the cervix and vulva. JAMA Oncol. 2016;2(4):518–22.
Liu C, Lu J, Tian H, Du W, Zhao L, Feng J, et al. Increased expression of PDL1 by the human papillomavirus 16 E7 oncoprotein inhibits anticancer immunity. Mol Med Rep. 2017;15(3):1063–70.
Son CH, Fleming GF, Moroney JW. Potential role of radiation therapy in augmenting the activity of immunotherapy for gynecologic cancers. Cancer Manag Res. 2017;9:553–63.
Mezache L, Paniccia B, Nyinawabera A, Nuovo GJ. Enhanced expression of PD L1 in cervical intraepithelial neoplasia and cervical cancers. Mod Pathol. 2015;28(12):1594–602.
Andersen AS, Koldjaer Solling AS, Ovesen T, Rusan M. The interplay between HPV and host immunity in head and neck squamous cell carcinoma. Int J Cancer. 2014;134(12):2755–63.
Lyford-Pike S, Peng S, Young GD, Taube JM, Westra WH, Akpeng B, et al. Evidence for a role of the PD-1:PD-L1 pathway in immune resistance of HPV-associated head and neck squamous cell carcinoma. Cancer Res. 2013;73(6):1733–41.
Ock CY, Keam B, Kim S, Lee JS, Kim M, Kim TM, et al. Pan-cancer immunogenomic perspective on the tumor microenvironment based on PD-L1 and CD8 T cell infiltration. Clin Cancer Res. 2016;22(9):2261–70.
Saglam O, Conejo-Garcia J. PD-1/PD-L1 immune checkpoint inhibitors in advanced cervical cancer. Integr Cancer Sci Ther. 2018;5(2).
De Felice F, Marchetti C, Palaia I, Ostuni R, Muzii L, Tombolini V, et al. Immune check-point in cervical cancer. Crit Rev. Oncol Hematol. 2018;129:40–3.
Ramanathan P, Dhandapani H, Jayakumar H, Seetharaman A, Thangarajan R. Immunotherapy for cervical cancer: Can it do another lung cancer? Curr Probl Cancer. 2018;42(2):148–60.
Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. 2018;118(1):9–16.
Heeren AM, de Boer E, Bleeker MC, Musters RJ, Buist MR, Kenter GG, et al. Nodal metastasis in cervical cancer occurs in clearly delineated fields of immune suppression in the pelvic lymph catchment area. Oncotarget. 2015;6(32):32484–93.
Emens LA, Middleton G. The interplay of immunotherapy and chemotherapy: harnessing potential synergies. Cancer Immunol Res. 2015;3(5):436–43.
Kersten K, Salvagno C, de Visser KE. Exploiting the immunomodulatory properties of chemotherapeutic drugs to improve the success of cancer immunotherapy. Front Immunol. 2015;6:516.
Dosset M, Vargas TR, Lagrange A, Boidot R, Vegran F, Roussey A, et al. PD-1/PD-L1 pathway: an adaptive immune resistance mechanism to immunogenic chemotherapy in colorectal cancer. Oncoimmunology. 2018;7(6):e1433981.
Ko EC, Formenti SC. Radiotherapy and checkpoint inhibitors: a winning new combination? Ther Adv Med Oncol. 2018;10:1758835918768240.
Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev. Immunol. 2013;31:51–72.
Binder DC, Fu YX, Weichselbaum RR. Radiotherapy and immune checkpoint blockade: potential interactions and future directions. Trends Mol Med. 2015;21(8):463–5.
Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. 2018;378(22):2078–92.
Esteva FJ, Hubbard-Lucey VM, Tang J, Pusztai L. Immunotherapy and targeted therapy combinations in metastatic breast cancer. Lancet Oncol. 2019;20(3):e175–e86.
Spanos WC, Nowicki P, Lee DW, Hoover A, Hostager B, Gupta A, et al. Immune response during therapy with cisplatin or radiation for human papillomavirus-related head and neck cancer. Arch Otolaryngol Head Neck Surg. 2009;135(11):1137–46.
• Tran L, Allen CT, Xiao R, Moore E, Davis R, Park SJ, et al. Cisplatin alters antitumor immunity and synergizes with PD-1/PD-L1 inhibition in head and neck squamous cell carcinoma. Cancer Immunol Res. 2017;5(12):1141–51. Demonstrated standard chemotherapy modulates anti-tumor response and potentiates checkpoint inhibitors in vivo.
Ock CY, Kim S, Keam B, Kim S, Ahn YO, Chung EJ, et al. Changes in programmed death-ligand 1 expression during cisplatin treatment in patients with head and neck squamous cell carcinoma. Oncotarget. 2017;8(58):97920–7.
van Meir H, Nout RA, Welters MJ, Loof NM, de Kam ML, van Ham JJ, et al. Impact of (chemo)radiotherapy on immune cell composition and function in cervical cancer patients. Oncoimmunology. 2017;6(2):e1267095.
Welters MJ, van der Sluis TC, van Meir H, Loof NM, van Ham VJ, van Duikeren S, et al. Vaccination during myeloid cell depletion by cancer chemotherapy fosters robust T cell responses. Sci Transl Med. 2016;8(334):334ra52.
Yang H, Ma Y, Chen G, Zhou H, Yamazaki T, Klein C, et al. Contribution of RIP3 and MLKL to immunogenic cell death signaling in cancer chemotherapy. Oncoimmunology. 2016;5(6):e1149673.
Roszik J, Ring KL, Wani KM, Lazar AJ, Yemelyanova AV, Soliman PT, et al. Gene expression analysis identifies novel targets for cervical cancer therapy. Front Immunol. 2018;9:2102.
• Oweida A, Lennon S, Calame D, Korpela S, Bhatia S, Sharma J, et al. Ionizing radiation sensitizes tumors to PD-L1 immune checkpoint blockade in orthotopic murine head and neck squamous cell carcinoma. Oncoimmunology. 2017;6(10):e1356153. Showed that radiation therapy synergizes therapeutically with anti-PD-L1 agents in vivo.
Nagasaka M, Zaki M, Kim H, Raza SN, Yoo G, Lin HS, et al. PD1/PD-L1 inhibition as a potential radiosensitizer in head and neck squamous cell carcinoma: a case report. J Immunother Cancer. 2016;4:83.
Ou D, Adam J, Garberis I, Blanchard P, Nguyen F, Levy A, et al. Clinical relevance of tumor infiltrating lymphocytes, PD-L1 expression and correlation with HPV/p16 in head and neck cancer treated with bio- or chemo-radiotherapy. Oncoimmunology. 2017;6(9):e1341030.
Zhang SA, Niyazi HE, Hong W, Tuluwengjiang GL, Zhang L, Zhang Y, et al. Effect of EBI3 on radiation-induced immunosuppression of cervical cancer HeLa cells by regulating Treg cells through PD-1/PD-L1 pathway. Tumor Biol. 2017;39(3):1010428317692237.
Wang Y, Deng W, Li N, Neri S, Sharma A, Jiang W, et al. Combining immunotherapy and radiotherapy for cancer treatment: current challenges and future directions. Front Pharmacol. 2018;9:185.
Dorta-Estremera S, Colbert LE, Nookala SS, Yanamandra AV, Yang G, Delgado A, et al. Kinetics of intratumoral immune cell activation during chemoradiation for cervical cancer. Int J Radiat Oncol Biol Phys. 2018;102(3):593–600.
Sharabi A, Kim SS, Kato S, Sanders PD, Patel SP, Sanghvi P, et al. Exceptional response to nivolumab and stereotactic body radiation therapy (SBRT) in neuroendocrine cervical carcinoma with high tumor mutational burden: management considerations from the center for personalized cancer therapy at UC San Diego Moores Cancer Center. Oncologist. 2017;22(6):631–7.
•• Mayadev JS, Enserro D, Lin YG, Da Silva DM, Lankes HA, Aghajanian C, et al. Sequential ipilimumab after chemoradiotherapy in curative-intent treatment of patients with node-positive cervical cancer. JAMA Oncol. 2019. One of the first studies to demonstrate feasibility, safety and efficacy of combining standard therapy with ICB in patients.
Rice AE, Latchman YE, Balint JP, Lee JH, Gabitzsch ES, Jones FR. An HPV-E6/E7 immunotherapy plus PD-1 checkpoint inhibition results in tumor regression and reduction in PD-L1 expression. Cancer Gene Ther. 2015;22(9):454–62.
Liu Z, Zhou H, Wang W, Fu YX, Zhu M. A novel dendritic cell targeting HPV16 E7 synthetic vaccine in combination with PD-L1 blockade elicits therapeutic antitumor immunity in mice. Oncoimmunology. 2016;5(6):e1147641.
Bartkowiak T, Singh S, Yang G, Galvan G, Haria D, Ai M, et al. Unique potential of 4-1BB agonist antibody to promote durable regression of HPV+ tumors when combined with an E6/E7 peptide vaccine. Proc Natl Acad Sci U S A. 2015;112(38):E5290–9.
Mkrtichyan M, Chong N, Abu Eid R, Wallecha A, Singh R, Rothman J, et al. Anti-PD-1 antibody significantly increases therapeutic efficacy of Listeria monocytogenes (Lm)-LLO immunotherapy. J Immunother Cancer. 2013;1:15.
Lin PL, Cheng YM, Wu DW, Huang YJ, Lin HC, Chen CY, et al. A combination of anti-PD-L1 mAb plus Lm-LLO-E6 vaccine efficiently suppresses tumor growth and metastasis in HPV-infected cancers. Cancer Med. 2017;6(9):2052–62.
• Dorta-Estremera S, Chin RL, Sierra G, Nicholas C, Yanamandra AV, Nookala SMK, et al. Mucosal HPV E6/E7 peptide vaccination in combination with immune checkpoint modulation induces regression of HPV(+) oral cancers. Cancer Res. 2018;78(18):5327–39. Demonstrated in an orthotopic model that vaccines can cooperate effectively with ICB and result in curative efficacy of HPV+ tumors.
•• Massarelli E, William W, Johnson F, Kies M, Ferrarotto R, Guo M, et al. Combining immune checkpoint blockade and tumor-specific vaccine for patients with incurable human papillomavirus 16-related cancer: a phase 2 clinical trial. JAMA Oncol. 2019;5(1):67–73. A significant phase II study that demonstrated the potential and clinical utility of therapeutic vaccines in combination with ICB in patients.
Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377(14):1345–56.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Lennox Chitsike declares that there is no conflict of interest. Penelope Duerksen-Hughes declares that there is no conflict of interest.
Human and animal rights and informed consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Gynecologic Cancers
Rights and permissions
About this article

Cite this article
Chitsike, L., Duerksen-Hughes, P. The Potential of Immune Checkpoint Blockade in Cervical Cancer: Can Combinatorial Regimens Maximize Response? A Review of the Literature. Curr. Treat. Options in Oncol. 21, 95 (2020). https://doi.org/10.1007/s11864-020-00790-4
Accepted:
Published:
DOI: https://doi.org/10.1007/s11864-020-00790-4