
Abstract
Sheath blight disease (ShB) severely affects rice production; however, the details of defense against ShB remain unclear. To understand the rice defense mechanism against ShB, an RNA sequencing analysis was performed using Rhizoctonia solani inoculated rice leaves after 48 h of inoculation. Among them, 3417 genes were upregulated and 2532 were downregulated when compared with the control group (> twofold or < 1/2). In addition, the differentially expressed genes were classified via Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and MapMan analyses. Fifty-nine GO terms and seven KEGG pathways were significantly enriched. A MapMan analysis demonstrated that the phytohormone and metabolic pathways were significantly altered. Interestingly, the expression levels of 359 transcription factors, including WRKY, MYB, and NAC family members, as well as 239 transporter genes, including ABC, MFS, and SWEET, were significantly changed in response to R. solani AG1-IA inoculation. Additionally, OsWRKY53 and OsAKT1 negatively regulate the defense response in rice against R. solani via gain of function study for OsWRKY53 and loss of function study for OsAKT1, respectively. Furthermore, several differentially expressed genes contain R. solani-responsive cis acting regulatory elements in their promoter regions. Taken together, our analyses provide valuable information for the additional study of the defense mechanisms against ShB, and the candidate genes identified in this study will be useful resource for future breeding to enhance resistance against ShB.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rice (Oryza sativa L.) is a globally important staple crop and feeds 50% of the global human population (Wilson and Talbot 2009; Fahad et al. 2014). As the population grows, rice production needs to be increased; however, plant diseases threaten rice yields. Sheath blight (ShB), one of the three major diseases of rice, infects rice in a wide area and results in particularly severe yield losses (Lee and Rush 1983). It causes lesions on the rice sheath, leaf, and panicle, resulting in withered leaves and sheaths, as well as a decreased seed setting rate, and can reduce the yield by more than 50% in severe cases (Marchetti and Bollich 1991). Currently, treatment with fungicides is the main approach to protecting rice from ShB (Singh et al. 2019). However, fungicides are harmful to the environment, and the long-term use of fungicides could increase the risk of fungicide resistance. Therefore, alternative methods of control have been attempted, such as breeding disease-resistant varieties.
Breeding crops with a durable resistance to pathogens is an ideal strategy to manage plant diseases; however, disease resistance breeding against ShB has lagged far behind, and the primary reason is a lack of donors that exhibit proper resistance (Bonman et al. 1992). Resistance to ShB is considered to be a quantitative trait that is controlled by multiple genes, and some of these quantitative trait loci have been mapped and functionally characterized (Li et al. 1995; Richa et al. 2016, 2017). Extensive studies have analyzed the molecular basis of resistance to ShB. PR (pathogenesis-related) genes are major contributors to a plant’s defense against pathogens, and the overexpression of OsOSM1, which belongs to the PR5 family, could enhance resistance to ShB (Xue et al. 2016). The overexpression of OsACS2 promotes rice’s defense against rice blast fungus and ShB (Helliwell et al. 2013). Recent research has shown that the F-box gene ZmFBL41 and its corresponding homolog in rice, OsFBX61, are negative regulators of resistance to ShB. ZmFBL41 targets ZmCAD, which is responsible for lignin biosynthesis and 26S proteasome-mediated degradation; additionally, recent results have indicated that the content of lignin affects resistance to ShB (Li et al. 2019). We previously identified that brassinosteroids (BRs) biosynthesis gene D2 and receptor gene BRI1 negatively regulate rice resistance, whereas the ethylene signaling genes EIN2 and EIL1 positively regulate rice resistance to ShB. The BR signaling transcription factor RAVL1 activates key BR- and ethylene-signaling pathways to modulate rice’s defense against ShB (Yuan et al. 2018). OsWRKY4, OsWRKY13, OsWRKY30, and OsWRKY80 have been reported to positively regulate resistance to ShB (Peng et al. 2012, 2016; Wang et al. 2015; John Lilly and Subramanian 2019), and more recently, we identified that sugar will eventually be exported by sugar transporter 11 (SWEET11), which negatively regulates rice’s resistance to ShB (Gao et al. 2018). Although much progress has been made, knowledge about how rice plants defend themselves against ShB is still fragmented and limited.
Plants rapidly reprogram their transcription profile to respond to external stimuli. Monitoring transcriptome changes provides insights into understanding how plants manage under adverse conditions (Matsumura et al. 2003). Numerous reports illustrate that RNA-Seq research plays an important role in understanding several aspects of pathogen–plant interactions. A set of time-series transcriptome analyses has revealed that functional effector-triggered immunity (ETI) responses rely on an appropriate time point for transcriptional reprogramming upon challenge with virulent or avirulent strains of Pseudomonas syringae (Mine et al. 2018). Transcriptome analysis has provided useful information for understanding the resistance mechanism of Brassica napus to clubroot disease (Mei et al. 2019) and was performed to investigate the regulatory mechanism of Pi9 and Pi21, two R (resistance) genes that control blast fungus (Zhang et al. 2016; Jain et al. 2017). Differences in the molecular basis between the interaction of resistant and susceptible cultivars or species with the pathogens (Zhang et al. 2017a, b; Zhang et al. 2017a, b) suggest that RNA-Seq is an ideal tool to dissect the resistance mechanism of host plants (Venu et al. 2007).
In this study, we performed an RNA-Seq assay using rice leaves with or without R. solani AG1-IA inoculation for 48 h. A total of 5949 differentially expressed genes depict the transcription landscape of rice’s response to R. solani infection, which were then further classified via Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and MapMan analyses. In addition, differentially expressed transcription factors and transporters were collected and analyzed. Our results provide new insights into rice’s defense mechanism against R. solani and may contribute to resistance breeding.
Materials and methods
Plant growth and inoculation
Wild-type (WT) rice (Oryza sativa L. Japonica. cv. Dongjin and Longjing 11), OsWRKY53 overexpression plants, and Oswrky53 and Osakt1 mutants were used in this study. One-month-old plants were used for R. solani inoculation by means of the method described in earlier literature (Yuan et al. 2018). All plants were grown under greenhouse conditions (temperature 23–30°C; relative humidity 80%; and 12 h light: 12 h dark) at Shenyang Agricultural University, China, and propagated by selfing. In brief, a 10-cm-long piece was cut from the second youngest leaf of the main tiller and placed on moistened filter paper in a Petri dish (diameter, 36 cm; height, 2.5 cm). Each replicate comprised six leaves, and four replicates per line were used, in a completely randomized design. Colonized potato dextrose agar (PDA) blocks (diameter, 7 mm) were excised using a circular cutter and placed on the abaxial surface of each leaf piece. Leaves were incubated in a chamber with continuous light, at 25°C for 72 h. The filter paper was kept moist with sterile water. After 72 h, the length and width of the lesions within each leaf piece was measured using Image J Fiji software and the percentage of leaf covered with lesions was calculated (Yuan et al. 2018).
RNA extraction and qRT-PCR assay
Rice leaves were inoculated with R. solani for 0, 24, 48, and 72 h, and then the leaves were collected for RNA extraction. Total RNA was isolated from 1-month-old seedlings using the RNAiso Plus reagent (Takara, Dalian, China) and then treated with RQ1 Rnase-free Dnase (Promega, Madison, WI, USA) to remove genomic DNA contamination. Complementary DNA was synthesized using the Reverse Transcription kit (Takara), and all these experiments were conducted according to the manufacturer’s instructions. qRT-PCR assays were performed using the BIO-RAD CFX96 real-time PCR system (Bio-Rad, Hercules, CA, USA) with ChamQ Universal SYBR qPCR Master Mix (Vazyme, Nanjing, China) by using one of the RNAs used in the RNA-sequencing. The gene expression levels were normalized against Ubiquitin as previously described (Moon et al. 2019). The primers used in the qRT-PCR assays are listed in Supporting Information Table S5.
mRNA sequencing and data analysis
Three biological replicates were performed for the RNA-Seq analysis. The total RNA of each sample was extracted using the TRIzol reagent (Invitrogen, CA, USA) before it was quantified and qualified using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA), NanoDrop (Thermo Fisher Scientific Inc., Waltham, MA, USA), and a 1.0% agarose gel. One microgram of total RNA with a RIN value above seven was used for the following library preparation. Next-generation sequencing library preparations were constructed according to the manufacturer’s instructions (NEBNext® Ultra™ RNA Library Prep Kit for Illumina®, San Diego, CA, USA).
The poly(A) mRNA isolation was performed using a NEBNext Poly(A) mRNA Magnetic Isolation Module (New England Biolabs, Ipswich, MA, USA). mRNA fragmentation and priming were performed using a NEBNext First Strand Synthesis Reaction Buffer and NEBNext Random Primers. First-strand cDNA was synthesized using ProtoScript II Reverse Transcriptase, and the second-strand cDNA was synthesized using a Second Strand Synthesis Enzyme Mix. Purified double-strand cDNA by AxyPrep Mag PCR Clean-up (Corning, Corning, NY, USA) was then treated with the End Prep Enzyme Mix to repair both ends and add a dA-tailing in one reaction, followed by a T-A ligation to add adaptors to both ends. Size selection of the adaptor-ligated DNA was performed using an AxyPrep Mag PCR Clean-up kit (Corning), and fragments of ~ 360 bp (with an approximate insert size of 300 bp) were recovered. Each sample was then amplified by PCR for 11 cycles using P5 and P7 primers, with both primers carrying sequences that could anneal with the flow cell to perform bridge PCR and with the P7 primer carrying a six-base index allowing multiplexing. The PCR products were cleaned using AxyPerp Mag PCR Clean-up (Axygen), validated using an Agilent 2100 Bioanalyzer (Agilent Technologies), and quantified by a Qubit 2.0 Fluorometer (Invitrogen).
Next, libraries with different indices were multiplexed and loaded on an Illumina Hi-Seq instrument according to the manufacturer’s instructions (Illumina). Sequencing was performed using a 2 × 150 bp paired-end configuration; image analysis and base calling were conducted using Hi-Seq Control Software (HCS) + OLB + GAPipeline-1.6 (Illumina) on a Hi-Seq instrument. Quality control, mapping, expression analysis, differential expression analysis, GO and KEGG enrichment analysis, novel transcripts prediction, alternative splicing, differential exon usage, principal component analysis, and protein–protein interaction analyses were performed as following. Hypergeometric distribution method was used to enrich the GO category, and calculate the Hyper P value. Enrichment of GO members among differentially expressed genes (DEGs) was performed with the GO enrichment function in rice oligonucleotide array database (https://ricephylogenomics-khu.org/ROAD_old/analysis/go_enrichment.shtml, temporary available for updating). After retrieving the enrichment data, we applied the criteria query number > 2, hyper P value < 0.05, and fold enrichment value (query number/query expected number) > 2 and visualized with ggplot2 R package. The KEGG pathway enrichment was performed to find out the significant pathway of the DEGs according to KEGG pathway. ClusterProfiler R package was used to select the significant pathway, and the threshold of significance was defined by P value of < 0.05 (Trapnell et al. 2009; Cole et al. 2010; Love et al. 2014; Rao et al. 2014; Damian et al. 2015; Cao et al, 2012; Yu et al, 2012). The sequencing data analysis was processed and analyzed with GENEWIZ (Suzhou, China).
MapMan analysis
The experiment was performed as reported elsewhere. Put simply, a table of RNA-Seq results, including gene ID and fold change data, was imported to generate a schematic diagram that shows enriched pathways or processes (Thimm et al. 2010; Hwang et al. 2018).
Identification of differentially expressed TFs and transporters
Utilizing the rice TF database (https://ricephylogenomics.ucdavis.edu/tf/index.shtml) and the rice transporter database (https://ricephylogenomics.ucdavis.edu/transporter/) (Dardick et al. 2007; Jung et al. 2010), the genes listed in these databases were searched for in our RNA-Seq data. Isolated differentially expressed TFs and transporters were classified according to their families. The nomenclature and locus number of the rice SWEET genes refer to a previous publication (Chen et al. 2010).
Results
Transcriptome analysis of R. solani-infected rice leaves
ShB is a severe disease that can affect rice production. However, the molecular mechanism of how rice defends itself against ShB remains unclear. To analyze rice’s defense mechanism against ShB, an RNA-Seq analysis was performed. Before performing the sequencing, the optimized time point for the response of rice to R. solani AG1-IA was tested. One-month-old rice plants inoculated with R. solani AG1-IA and the expression of the two PR genes PBZ1 and PR1b were examined 0, 24, 48, and 72 h postinoculation (hpi). The results showed that OsPBZ1 and OsPR1b exhibited the highest expression at 48 hpi (Fig. 1). Therefore, 48 hpi was chosen for the RNA-Seq analysis.

Expression of OsPBZ1 and OsPR1b in response to inoculation with Rhizoctonia solani AG1-IA. (a) One-month-old seedlings were inoculated with R. solani AG1-IA, and the leaves were sampled at 0, 24, 48, and 72 h post inoculation (hpi). R. solani infection-mediated expression patterns of OsPBZ1 (b) and OsPR1b (c) were analyzed by quantitative real-time PCR (qRT-PCR). Data are the means ± standard error (SE) of three repeated experiments. Significant differences between different time points compared with 0 hpi are shown (*P < 0.05, **P < 0.01, ***P < 0.001)
RNA sequencing data were established by 150 bp pair-end sequencing and 2.86 × 108 reads of three biological replicates. The Q20 and Q30 values were 98% and 95%, respectively, indicating that the RNA-Seq quality is high enough for further evaluation. On average, 95% of the reads were mapped to the Nipponbare reference genome (Os-Nipponbare-Reference-IRGSP-1.0; GenBank assembly accession: GCA_001433935.1). A total of 92.73%, 3.86%, and 3.41% of these reads were mapped to the exon, intergenic, and intron regions, respectively. Ht-Seq software was used to perform the gene expression assay based on a calculation of the FPKM (fragments per kilobases per million reads) values of the genes (Mortazavi et al. 2008). To test reproducibility of these data, we calculated correlation coefficient values between replicates. As a result, we found that the average correlation coefficient value between replicates of control was 0.965 and that of the R. solani AG1-IA inoculation was 0.703. Although samples inoculated with R. solani AG1-IA were less consistent than those of control, previous study treated with pathogens such as Xanthomonas oryzae pv. oryzae and X. oryzae pv. oryzicola retains similar trends and results in meaningful data from DEGs (Figure S1; Seo et al. 2008). Next, the volcano and heat map diagrams display the differentially expressed genes (DEGs) from RNA sequencing (Fig. 2a). Genes whose expression variation is more than twofold between the control and inoculated leaves (Log2FC = > 1, P ≤ 0.05) were considered as DEGs, and 5949 genes were significantly changed, including 3417 upregulated and 2532 downregulated genes (Fig. 2b, Table S1).

Volcano and heat map diagrams of RNA sequencing data. (a) Distribution of differentially expressed genes (DEGs). Red and blue spots represent up- and downregulated DEGs, respectively. (b) Hierarchical clustering map exhibiting DEGs with or without R. solani inoculation. The numbers 1–3 above the map indicate the three replicates of RNA sequencing. Up- and downregulated genes are marked on the right side of the map. The color key image indicates the up- and downregulation of DEGs
Verification of DEGs by quantitative real-time PCR (qRT-PCR)
To validate the RNA sequencing results, seven upregulated and two downregulated genes were randomly chosen and further confirmed by qRT-PCR. The qRT-PCR results indicate that seven genes (OsSWEET2a, OsSWEET14, OsWRKY108, OsERF096, OsNAC3, OsPR1b, and LOC_Os07g36560) were induced, whereas OsMST1 and LOC_Os03g24860 were repressed by inoculation with R. solani at 48 hpi (Fig. 3), suggesting that the RNA-Seq and qRT-PCR results are consistent.

Validation of DEGs by qRT-PCR analysis. Seven upregulated genes (OsSWEET2a, OsSWEET14, OsWRKY108, OsERF096, OsNAC3, OsPR1b, and LOC_Os07g36560) and two downregulated genes (OsMST1 and LOC_Os03g24860) were evaluated by qRT-PCR. The gray and black bars indicate the qRT-PCR and RNA-Seq results, respectively
GO, KEGG, and MapMan analyses of DEGs
To classify the DEGs, GO and KEGG enrichment assays were performed. To identify core information concerning the response, we set the value of fold enrichment = > 5 as the threshold of GO analysis. The DEGs were divided into 59 GO terms; the most enriched GO terms of upregulated genes were sensory perception of chemical stimulus, the carbohydrate biosynthetic process, response to other organisms, and the glucose catabolic process, and the most enriched downregulated genes were protein import, the fructose 2,6-bisphosphate metabolic process, sensory perception, and megasporogenesis (Fig. 4a). The DEGs were classified into different biochemical or signaling transduction pathways through the KEGG analysis. The results show that the most enriched pathways are carbon metabolism, photosynthesis, and biosynthesis of amino acids among the seven KEGG pathways (Fig. 4b). The results of GO and KEGG analyses suggest that carbon metabolism- and photosynthesis-associated genes represent the most enriched DEGs.

GO and KEGG analyses of DEGs. (a) DEGs were classified into 59 GO terms, including response to other organisms, the glucose catabolic process, protein import, and sensory perception. The number of DEGs belonging to each category was represented by red, pink, and blue ovals. (b) DEGs were divided into seven KEGG pathways, photosynthesis, carbon metabolism, and biosynthesis of amino acids. The different sizes of the red ovals indicate the number of DEGs in each pathway
To obtain additional insights into the DEGs, a MapMan analysis was performed. MapMan data indicated that biotic stress, metabolism, hormones, and the regulation of transcription, as well as transporters, were all enriched. In the biotic stress overview, phytohormones, abiotic stress, redox state, secondary metabolites, signaling, proteolysis, and cell wall biosynthesis-related were enriched. Among them, proteolysis, signaling, redox state, cell wall, and secondary metabolic-associated genes were the most enriched (Fig. 5). In the cell function overview, the regulation of transcription, regulation, redox state, enzyme families, and transporters were the most enriched functions, suggesting that DEGs with these functions are major participants in rice’s response to R. solani infection (Figure S2. In the regulation overview, the transcription factor was the most enriched term (Figure S3). The transporter overview showed that 20 types of transporters, including ATP-binding cassette (ABC) transporters, nitrate and ammonium transporters, and amino acid transporters, were classified. The ABC transporters had the highest numbers, and the potassium, sugar, and amino acid transporters were significantly enriched (Figure S4). In addition, previous work has reported that the disruption of lignin biosynthesis in rice significantly impairs resistance to ShB (Li et al. 2019); therefore, we conducted a MapMan analysis concerning secondary metabolism. In the secondary metabolism overview, phenylpropanoids, which are structural components of the cell wall, had the highest numbers; lignin and lignans were also shown to be significantly enriched (Figure S5). The DEGs in the biosynthesis pathways of the phenylpropanoids and lignins were further inspected, and a MapMan analysis showed that the genes responsible for almost every step of phenylpropanoid and lignin biosynthesis were upregulated (Figure S6, Table 1). These results suggest that the reinforcement of cell walls, especially regarding the biosynthesis of lignins, is a major part of rice’s response to R. solani infection.

MapMan analysis of biotic stress-related DEGs. The biotic stress overview showed that phytohormones, abiotic stress, redox state, secondary metabolites, and cell wall-related genes were enriched. Abscisic acid, defense, redox-related state, transcription factors, cell wall, heat shock, and secondary metabolic-associated genes were highly enriched. Boxes with green and red colors indicate down and upregulated genes by R. solani, respectively
Identification of differentially expressed transcription factors and transporters
Transcription factors (TFs), such as the WRKY family, transporters, and the SWEET genes, are well known for their function in a plant’s defense against disease. Therefore, the differentially expressed TFs and transporters from RNA-Seq data were analyzed and classified. The rice TF database (https://ricephylogenomics.ucdavis.edu/tf/index.shtml) and the rice transporter database (https://ricephylogenomics.ucdavis.edu/transporter/) were used to isolate the TFs and transporters that expressed a differential response to R. solani inoculation (Dardick et al. 2007; Jung et al. 2010). In general, 359 differentially expressed TFs with 45 different types were identified. Among these TFs, WRKY, AP2-EREBP, bHLH, MYB, and TIFY were the most enriched TF families, and showed 40, 39, 31, 28, and 11 members in each type, respectively (Fig. 6a, Table S2). Further, a qRT-PCR analysis was performed to verify the expression patterns of the TFs in response to R. solani. The qRT-PCR results indicated that three WRKY members (OsWKRY28, OsWRKY32, and OsWRKY53) and three TIFY family genes (OsJAZ5, OsJAZ6, and OsJAZ9) were induced by infection with R. solani. OsWRKY28 exhibited the highest induction rate (approximately 20-fold) of the three WRKYs, and OsJAZ6 exhibited the highest expression (approximately 20-fold) among the JAZ genes at 48 hpi (Fig. 6b–f).

Isolation of differentially expressed transcription factors (TFs) and qRT-PCR verification. (a) Among the DEGs, differentially expressed TFs were collected. A total of 40 types of TFs, including WRKY, MYB, AUX/IAA, and C2H2 zinc finger families, were enriched. The R. solani-dependent expression of OsWKRY28 (b), OsWRKY32 (c), OsWRKY53 (d), OsJAZ5 (e), OsJAZ6 (f), and OsJAZ9 (g) was analyzed via qRT-PCR. Data are the means ± SE of three repeated experiments. Significant differences between different time points compared with 0 hpi are shown (*P < 0.05, **P < 0.01, ***P < 0.001)
In addition, 239 differentially expressed transporters with 53 different types were isolated. As the recently characterized SWEET sugar transporters (Chen et al. 2010) were not included in the Rice Transporter Database, we manually added SWEET to the list of transporters. Among the differentially expressed transporters, ABC, AAAP, POT, and F-ATPase were the most enriched superfamilies, accounting for 22, 21, 19, and 18 members, respectively (Fig. 7a). A qRT-PCR analysis was performed to verify the RNA-Seq data. The results showed that OsAKT1, OsSWEET2b, and a monosaccharide transporter (OsMST4) were significantly induced, whereas OsMST8 was suppressed by R. solani infection (Fig. 7b–e). In addition, OsSWEET2a and OsSWEET14 were induced, whereas OsMST1 was repressed by R. solani inoculation (Fig. 3), and the expression patterns of the seven transporters were similar to the results observed after the RNA-Seq analysis.

Isolation of differentially expressed transporters and qRT-PCR verification. (a) Transporters that belong to 53 types of superfamilies, including ABC, AAAP, POT, and F-ATPase, were enriched. The expression levels of OsAKT1 (b), OsSWEET2b (c), OsMST4 (d), and OsMST8 (e) were verified by a qRT-PCR analysis. Data are the means ± SE of three repeated experiments. Significant differences between different time points compared with 0 hpi are shown (**P < 0.01, ***P < 0.001, ****P < 0.0001, n.s. not significant)
OsWRKY53 and OsAKT1 regulate rice’s resistance to ShB
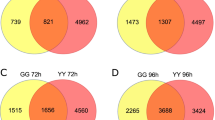
As 40 WRKY genes differentially responded to R. solani infection, a genetic study was performed to evaluate their functions in rice’s defense against ShB. OsWRKY53 was reported to be involved in BR signaling (Tian et al. 2017) and was significantly induced by R. solani infection. However, its function in rice’s defense mechanisms is unclear. The R. solani AG1-IA detachment assay showed that the Oswrky53 genome editing-created mutant was less susceptible, whereas the OsWRKY53 overexpressor (OE) was more susceptible, to ShB compared with the wild-type control Longjing11 (Fig. 8a). The size of the lesion area was 35.63% of the total surface area of Longjing11 leaves, 9.97% of Oswrky53 leaves, and 77.15% of OsWRKY53 OE leaves (Fig. 8b).

Evaluation of OsWRKY53 and OsAKT1 mutants in response to sheath blight disease compared with the wild-type control (WT). (a) Response of the Oswrky53 and OsWRKY53 overexpressor (OE) to R. solani AG1-IA compared with the WT plants. The leaves at 24, 48, 72, and 96 dpi were photographed. (b) Percentage of leaf area covered with lesions in the Oswrky53 and OsWRKY53 OE compared with the WT plants after 72 hpi. (c) Response of Osakt1 to R. solani AG1-IA compared with the WT plants. The leaves at 24, 48, 72, and 96 dpi were photographed. (d) Percentage of leaf area covered with lesions in Osakt1 compared with the WT plants after 72 hpi. Data are the means ± SE (n > 15). Significant differences between mutants compared with WT are shown (***P < 0.001, ****P < 0.0001)
Interestingly, the potassium transporter OsAKT1 was significantly induced by R. solani, and the rice blast fungus Magnaporthe grisea secreted the effector protein AvrPiz-t, which targets OsAKT1 to inhibit the interaction between CIPK23 and OsAKT1 and partially block the potassium influx to increase its virulence (Shi et al. 2018). However, the function of OsAKT1 in rice’s defense against ShB has not been examined. The results of inoculation with R. solani AG1-IA showed that the Osakt1 mutant (Li et al. 2014) was less susceptible to ShB compared with its corresponding wild-type Dongjin (DJ) (Fig. 8c). The lesion area was 33.69% of the total surface area of DJ leaves and 25.43% of Osakt1 leaves (Fig. 8d). These results suggest that both OsWRKY53 and OsAKT1 negatively regulate rice’s defense against ShB.
Identification of R. solani-responsive cis-acting regulatory elements (CREs) in DEGs’ promoters
A previous study identified that a promoter of R. solani-induced genes commonly harbors four types of CRE sequences: GCTGA, TATAT, GTTGA, and TATTT (Li et al. 2017a, b; Yang et al. 2017). To test whether the upregulated genes also carry CRE motifs, the 1.5 kb of promoter sequences from 31 OsWRKYs, four SWEETs, and five MSTs were searched. The results indicate that all the genes that were analyzed harbor putative CREs. Among them, OsWRKY75 contains the highest number of CREs with 36 in its promoter region, and OsWRKY70 contains the lowest number of putative CREs with only one (Table 2). Among the 40 gene promoters examined, 53 GCTGA, 186 TATAT, 61 GTTGA, and 169 TATTT CRE sequences were identified, indicating that the numbers of TATAT and TATTT motifs are higher than those of GCTGA and GTTGA.
Discussion
ShB is a major rice disease that severely affects yield production; however, defense mechanisms against this disease remain unknown (Marchetti and Bollich 1991). In this study, we performed RNA-Seq-based transcriptome analyses to dissect the molecular mechanism of how rice defends itself against ShB. Several previously reported genes that contribute resistance to ShB, such as OsACS2 (Helliwell et al. 2013), 14-3GF14f (Karmakar et al. 2019), OsPAL4 (Tonnessen et al. 2015), OsPR4b (Zhu et al. 2006), OsMYB4 (Pooja et al. 2015), and OsASR2 (Li et al. 2018), are present in our DEG dataset. These results confirm the effectiveness of our RNA-Seq experiment. Under this condition, a total of 5949 genes were differentially and significantly expressed: 3417 upregulated genes and 2532 downregulated genes. These genes were further enriched into 59 GO terms, including a response to other organisms, the glucose catabolic process, protein import, and sensory perception. In addition, a KEGG analysis showed that the DEGs were classified into photosynthesis, carbon metabolism, and the biosynthesis of amino acids. Furthermore, several TFs and transporters were significantly changed. The promoter sequence analysis of WRKYs, SWEETs, and MSTs indicated that a number of DEGs harbor R. solani-responsive CREs in their promoters. However, there is no correlation between the number of CREs and induction fold.
Phytohormone-related genes were significantly changed
A MapMan analysis using DEGs indicated that phytohormones, particularly the auxin, BR, abscisic acid (ABA)-, and jasmonic acid (JA)-related genes, changed significantly. Our previous study identified that BR signaling negatively regulates rice’s defense against ShB (Yuan et al. 2018), and exogenously treated auxin promotes rice’s defense against ShB, and also identified that the auxin polar transporter OsPIN1a positively regulates rice’s defense against ShB (Sun et al. 2019). JA regulates plant defenses against necrotrophic fungi (Glazebrook 2005), and the JAZ family of proteins are key regulators of JA signaling. In our results, the expression of 11 JAZ genes was significantly altered by R. solani inoculation. A number of ABA-related genes were also enriched. Some research has demonstrated that ABA is involved in pathogen–plant interactions as a regulator of immunity (Audenaert et al. 2002; Ulferts et al. 2015; Lievens et al. 2017). These results suggest that infection by R. solani rapidly influences net phytohormone signaling, which might be important in rice’s defense against ShB. Of note, additional experiments would be valuable to investigate the detailed role of phytohormone signaling in rice’s defense against ShB.
Cell wall- and redox state-related genes were significantly changed
Plant cell wall provide mechanical support and defense against the invasion of pathogens (Mirabet et al. 2011; Hamann 2012). R. solani penetrates host cells using hyphae, requiring enzymes that can be secreted. A previous study found that the R. solani AG1-IA genome contains an expanded set of genes encoding cell wall-degrading enzyme, such as those encoding pectinase, xylanase, and laccase, suggesting that cell wall degradation is also an important process for infection by R. solani AG1-IA (Zheng et al. 2013). Secondary cell wall deposition, a burst of reactive oxygen species (ROS), and the expression of PR genes are important parts of plant immunity. Secondary cell wall deposition will reinforce this barrier to prevent further invasion by pathogens or block the infected cell to stop the spread of pathogens. In this context, lignin, a component of secondary cell walls, was focused on for further inspection because of its resistance to physical disruption and enzymatic degradation. A recent breakthrough exploring the molecular basis of ShB resistance showed that lignin positively regulates resistance to ShB in both maize and rice (Li et al. 2019). A MapMan analysis illustrated that cell wall-related genes, especially the biosynthesis genes of lignin, are significantly enriched in DEGs, suggesting that alteration of the cell wall is a common strategy in the defense against pathogens. In addition, another significantly enriched MapMan term is redox state-related genes. The redox state tightly correlates with ROS production and salicylic acid signaling (Mou et al. 2003; Noctor et al. 2018). ROS plays an important role in plant immunity, because they are signal molecules that induce programmed cell death to protect the host from pathogen infection (Alvarez et al. 1998). These results suggest that the cell wall- and redox state-related genes might play an important role in rice’s defense against ShB.
R. solani infection significantly alters carbon and nitrogen metabolism
Nitrogen and carbon sources are necessary for living organisms and need to be obtained from the host plants by pathogens. A KEGG analysis indicated that photosynthesis, and pyruvate metabolism- and amino acid metabolism-related genes were significantly enriched. In addition, a MapMan overview showed that several genes related to starch, sucrose, TCA, and amino acid metabolism were significantly changed. Comparative proteomic and metabolomic analyses illustrated that altering the energy and primary metabolism contributes to rice’s resistance to ShB (Karmakar et al. 2019). These results suggest that plants might rapidly reprogram their carbon and nitrogen metabolisms to provide energy and metabolic sources for defense when they are infected by pathogens.
TFs might play a role in rice’s defense against ShB
In RNA-Seq data, 359 TFs among the 5949 genes, comprising 6.03%, were identified to be significantly altered by R. solani infection. These TFs include 40 WRKY, 39 AP2-EREBP, 28 MYB, 11 TIFY, and several indeterminate domain (IDD) family genes. WRKYs are well known for their pivotal roles in plant responses to stress conditions. Several studies have illustrated that OsWRKY4, OsWRKY13, OsWRKY30, and OsWRKY80 positively regulate rice’s resistance to ShB (Peng et al. 2012, 2016; Wang et al. 2015; John Lilly and Subramanian 2019). In this study, we identified that OsWRKY53 was induced by R. solani; a further genetic study showed that OsWRKY53 overexpression in the plant rendered it more susceptible to ShB, whereas Oswrky53 rendered it less susceptible. OsWRKY53 was previously reported to positively regulate BR signaling downstream of the BR receptor OsBRI1 (Tian et al. 2017). Our previous study showed that BR signaling negatively regulates rice’s defense against ShB (Yuan et al. 2018), implying that OsWRKY53 might activate BR signaling to suppress rice’s defense against ShB. A few IDD family genes, including OsIDD14/LPA1, were changed in the RNA-Seq data. Our recent result has shown that LPA1 activates the expression of OsPIN1a to promote rice’s defense against ShB (Sun et al. 2019), and the RNA-Seq data further suggest that LPA1 activation on R. solani infection might be important in promoting rice’s defense. In addition, OsWRKY4, OsWRKY13, OsWRKY30, and OsWRKY80 regulate the resistance of rice to ShB (Peng et al. 2012, 2016; Wang et al. 2015; John Lilly and Subramanian 2019). These results suggest that TFs might play a key role in rice’s defense against ShB, and this will be interesting to examine in additional experiments.
Diverse types of transporters are involved in rice’s defense against ShB
The cell membrane is a selective-permeable physical barrier that separates cell components from the external environment. To communicate with other cells or respond to external stimuli, plant cells use membrane-anchored proteins such as receptors, transporters, and enzymes. In plant–pathogen interaction systems, transporters play a crucial role in plant defense against invading microbes. In total, 239 transporters were isolated from 5949 DEGs, including ABC, AAAP, AMT, and SWEET family members. In Arabidopsis, the pleiotropic drug resistance transporters PEN3 and PDR12 mediate a camalexin export that induces resistance to Botrytis cinerea (He et al. 2019). In wheat, a broad-spectrum resistance gene, Lr34, was reported to be an ABC transporter, which transports ABA (Krattinger et al. 2019). In addition, the expression levels of sugar, nitrate, ammonium, amino acids, and peptide transporters were obviously changed. The SWEET family members, sugar transporters that have been recently identified, play a key role in phloem loading, seed filling, nectar secretion, and feeding pathogens (Chen et al. 2010, 2012; Lin et al. 2014; Yang et al. 2018). In rice, several SWEET members are transcriptionally activated by TAL effectors secreted by Xanthomonas oryzae pv. oryzase (Xoo), the causal agent of bacterial leaf blight, to increase susceptibility (Yang et al. 2006; Romer et al. 2010; Streubel et al. 2013). OsSWEET11 is a type of SWEET sugar transporter that negatively regulates rice’s defense against ShB (Gao et al. 2018). In RNA-Seq data, we identified that OsSWEET2a and OsSWEET2b were both dramatically induced by R. solani, suggesting that other SWEET members besides OsSWEET11 might also play a role in rice and R. solani interactions. In Arabidopsis, AMT1; 1 changes the basal defense, inducing resistance to Pseudomonas syringae and Plectosphaerella cucumerina (Pastor et al. 2014), and TaAMTs was induced in wheat by a Puccinia graminis f. sp. tritici infection (Li et al. 2017a, b). Our RNA-Seq data and qRT-PCR analysis showed that the rice potassium influx transporter OsAKT1 transcriptionally activated a response to R. solani infection. OsAKT1 facilitates potassium absorption, and a high level of potassium concentration in planta confers resistance to rice blast disease, while potassium inhibits the growth of M. grisea on media (Shi et al. 2018). In response to R. solani, Osakt1 has improved rice resistance to this pathogen. These data indicate that potassium may differentially regulate the resistance of rice to different types of fungal pathogens and strongly suggest that several transporters might be involved in the regulation of rice’s defense against ShB. In addition, OsWRKY53 and OsAKT1 are induced by R. solani infection, but these two induced DEGs negatively regulate rice defense to ShB, implying that R. solani might has a mechanism to transcriptionally activate negative regulators to suppress rice defense during infection. The detailed mechanism might need further analysis for the clarity.
Supplemental Table 1. The list of DEGs by R. solani infection.
Supplemental Table 2. The list of differentially expressed transcription factors.
Supplemental Table 3. The list of differentially expressed transporters.
Supplemental Table 4. List of MapMan analysis belonging to six Binnames.
Supplemental Table 5. Primer sequences used in this study.
Supplemental Fig. 1. Correlation coefficient analysis between replicates of control and R. solani AG1-IA inoculation.
Supplemental Fig. 2. MapMan analysis of cell function related DEGs.
Supplemental Fig. 3. MapMan analysis of regulation processes related DEGs.
Supplemental Fig. 4. MapMan analysis of transporter related DEGs.
Supplemental Fig. 5. MapMan analysis of secondary metabolism related DEGs.
Supplemental Fig. 6. MapMan analysis of lignin biosynthesis related DEGs.
References
Alvarez ME, Pennell RI, Meijer P-J, Ishikawa A, Dixon RA, Lamb C (1998) Reactive oxygen intermediates mediate a systemic signal network in the establishment of plant immunity. Cell 92:773–784
Audenaert K, De Meyer GB, Hofte MM (2002) Abscisic acid determines basal susceptibility of tomato to Botrytis cinerea and suppresses salicylic acid-dependent signaling mechanisms. Plant Physiol 128(2):491–501
Bonman JM, Khush GS, Nelson RJ (1992) Breeding rice for resistance to pests. Annu Rev Phytopathol 30:507–528
Cao P, Jung K, Choi D, Hwang D, Zhu J, Ronald PC (2012) The rice oligonucleotide array database: an atlas of rice gene expression. Rice 5(1):17
Chen LQ, Hou BH, Lalonde S, Takanaga H, Hartung ML, Qu XQ, Guo WJ, Kim JG, Underwood W, Chaudhuri B, Chermak D, Antony G, White FF, Somerville SC, Mudgett MB, Frommer WB (2010) Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 468(7323):527–532
Chen LQ, Qu XQ, Hou BH, Sosso D, Osorio S, Fernie AR, Frommer WB (2012) Sucrose efflux mediated by SWEET proteins as a key step for phloem transport. Science 335(6065):207–211
Cole T, Williams BA, Geo P, Ali M, Gordon K, Baren MJ, Van SSL, Wold BJ, Lior P (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28(5):511–515
Damian S, Andrea F, Stefan W, Kristoffer F, Davide H, Jaime HC, Milan S, Alexander R, Alberto S, Tsafou KP (2015) STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43:4–47
Dardick C, Chen J, Richter T, Ouyang S, Ronald P (2007) The rice kinase database. A phylogenomic database for the rice kinome. Plant Physiol 143(2):579–586
Fahad S, Nie L, Khan FA, Chen Y, Hussain S, Wu C, Xiong D, Jing W, Saud S, Khan FA, Li Y, Wu W, Khan F, Hassan S, Manan A, Jan A, Huang J (2014) Disease resistance in rice and the role of molecular breeding in protecting rice crops against diseases. Biotechnol Lett 36(7):1407–1420
Gao Y, Zhang C, Han X, Wang ZY, Ma L, Yuan P, Wu JN, Zhu XF, Liu JM, Li DP, Hu YB, Xuan YH (2018) Inhibition of OsSWEET11 function in mesophyll cells improves resistance of rice to sheath blight disease. Mol Plant Pathol 19(9):2149–2161
Glazebrook J (2005) Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu Rev Phytopathol 43:205–227
Hamann T (2012) Plant cell wall integrity maintenance as an essential component of biotic stress response mechanisms. Front Plant Sci 3:77
He Y, Xu J, Wang X, He X, Wang Y, Zhou J, Zhang S, Meng X (2019) The Arabidopsis pleiotropic drug resistance transporters PEN3 and PDR12 mediate camalexin secretion for resistance to Botrytis cinerea. Plant Cell 31(9):2206–2222
Helliwell EE, Wang Q, Yang Y (2013) Transgenic rice with inducible ethylene production exhibits broad-spectrum disease resistance to the fungal pathogens Magnaporthe oryzae and Rhizoctonia solani. Plant Biotechnol J 11(1):33–42
Hwang S, Kim Y, Sohn S, Choi D (2018) Gene expression profiling provides insight into the escape behavior of deepwater rice during submergence. J Plant Biol 61(6):374–382
Jain P, Singh PK, Kapoor R, Khanna A, Solanke AU, Krishnan SG, Singh AK, Sharma V, Sharma TR (2017) Understanding host-pathogen interactions with expression profiling of NILs carrying rice-blast resistance Pi9 gene. Front Plant Sci 8:93
John Lilly J, Subramanian B (2019) Gene network mediated by WRKY13 to regulate resistance against sheath infecting fungi in rice (Oryza sativa L.). Plant Sci 280:269–282
Jung KH, Cao P, Seo YS, Dardick C, Ronald PC (2010) The Rice Kinase Phylogenomics Database: a guide for systematic analysis of the rice kinase super-family. Trends Plant Sci 15(11):595–599
Karmakar S, Datta K, Molla KA, Gayen D, Das K, Sarkar SN, Datta SK (2019) Proteo-metabolomic investigation of transgenic rice unravels metabolic alterations and accumulation of novel proteins potentially involved in defence against Rhizoctonia solani. Sci Rep 9(1):10461
Kim E, Kim Y, Hong W, Lee C, Jeon J, Jung K (2019) Genome-wide analysis of root hair preferred RBOH genes suggests that three RBOH genes are associated with Auxin-mediated root hair development in rice. J Plant Biol 62(3):229–238
Krattinger SG, Kang J, Bräunlich S, Boni R, Chauhan H, Selter LL, Robinson MD, Schmid MW, Wiederhold E, Hensel G, Kumlehn J, Sucher J, Martinoia E, Keller B (2019) Abscisic acid is a substrate of the ABC transporter encoded by the durable wheat disease resistance gene Lr34. New Phytol 223(2):853–866
Lee FN, Rush MC (1983) Rice sheath blight a major rice disease. Plant Dis 67(7):829–832
Li J, Long Y, Qi GN, Li J, Xu ZJ, Wu WH, Wang Y (2014) The Os-AKT1 channel is critical for K+ uptake in rice roots and is modulated by the rice CBL1-CIPK23 complex. Plant Cell 26(8):3387–3402
Li N, Chen J, Yang F, Wei S, Kong L, Ding X, Chu Z (2017a) Identification of two novel Rhizoctonia solani-inducible cis-acting elements in the promoter of the maize gene, GRMZM2G315431. Sci Rep 7:42059
Li N, Lin B, Wang H, Li X, Yang F, Ding X, Yan J, Chu Z (2019) Natural variation in ZmFBL41 confers banded leaf and sheath blight resistance in maize. Nat Genet 51(10):1540–1548
Li N, Wei S, Chen J, Yang F, Kong L, Chen C, Ding X, Chu Z (2018) OsASR2 regulates the expression of a defence-related gene, Os2H16, by targeting the GT-1 cis-element. Plant Biotechnol J 16(3):771–783
Li T, Liao K, Xu X, Gao Y, Wang Z, Zhu X, Jia B, Xuan Y (2017b) Wheat ammonium transporter (AMT) gene family: diversity and possible role in host-pathogen interaction with stem rust. Front Plant Sci 8:1637
Li Z, Pinson SRM, Marchetti MA, Stansel JW, Park WDJT (1995) Characterization of quantitative trait loci (QTLs) in cultivated rice contributing to field resistance to sheath blight (Rhizoctonia solani). Theor Appl Genet 91(2):382–388
Lievens L, Pollier J, Goossens A, Beyaert R, Staal J (2017) Abscisic acid as pathogen effector and immune regulator. Front Plant Sci 8:587
Lin IW, Sosso D, Chen LQ, Gase K, Kim SG, Kessler D, Klinkenberg PM, Gorder MK, Hou BH, Qu XQ, Carter CJ, Baldwin IT, Frommer WB (2014) Nectar secretion requires sucrose phosphate synthases and the sugar transporter SWEET9. Nature 508(7497):546–549
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15(12):550
Marchetti MA, Bollich CN (1991) Quantification of the relationship between sheath blight severity and yield loss in rice. Plant Dis 75(8):773–775
Matsumura H, Reich S, Ito A, Saitoh H, Kamoun S, Winter P, Kahl G, Reuter M, Kruger DH, Terauchi R (2003) Gene expression analysis of plant host-pathogen interactions by SuperSAGE. Proc Natl Acad Sci USA 100(26):15718–15723
Mei J, Guo Z, Wang J, Feng Y, Ma G, Zhang C, Qian W, Chen G (2019) Understanding the resistance mechanism in Brassica napus to clubroot caused by Plasmodiophora brassicae. Phytopathology 109(5):810–818
Mine A, Seyfferth C, Kracher B, Berens ML, Becker D, Tsuda K (2018) The defense phytohormone signaling network enables rapid, high-amplitude transcriptional reprogramming during effector-triggered immunity. Plant Cell 30(6):1199–1219
Mirabet V, Das P, Boudaoud A, Hamant O (2011) The role of mechanical forces in plant morphogenesis. Annu Rev Plant Biol 62:365–385
Moon S, Chandran AKN, Kim Y, Gho Y, Hong W, An G, Lee C, Jung K (2019) Rice RHC encoding a putative cellulase is essential for normal root hair elongation. J Plant Biol 62(1):82–91
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Med 5(7):621–628
Mou Z, Fan W, Dong X (2003) Inducers of plant systemic acquired resistance regulate npr1 function through redox change. Cell 113:935–944
Noctor G, Reichheld JP, Foyer CH (2018) ROS-related redox regulation and signaling in plants. Semin Cell Dev Biol 80:3–12
Pastor V, Gamir J, Camanes G, Cerezo M, Sanchez-Bel P, Flors V (2014) Disruption of the ammonium transporter AMT11 alters basal defenses generating resistance against Pseudomonas syringae and Plectosphaerella cucumerina. Front Plant Sci 5:231
Peng X, Hu Y, Tang X, Zhou P, Deng X, Wang H, Guo Z (2012) Constitutive expression of rice WRKY30 gene increases the endogenous jasmonic acid accumulation, PR gene expression and resistance to fungal pathogens in rice. Planta 236(5):1485–1498
Peng X, Wang H, Jang JC, Xiao T, He H, Jiang D, Tang X (2016) OsWRKY80-OsWRKY4 module as a positive regulatory circuit in rice resistance against Rhizoctonia solani. Rice (N Y) 9(1):63
Pooja S, Sweta K, Mohanapriya A, Sudandiradoss C, Siva R, Gothandam KM, Babu S (2015) Homotypic clustering of OsMYB4 binding site motifs in promoters of the rice genome and cellular-level implications on sheath blight disease resistance. Gene 561(2):209–218
Rao VS, Srinivas K, Sujini GN (2014) Kumar GN (2014) protein–protein interaction detection: methods and analysis. J Proteomics 4:147648
Richa K, Tiwari IM, Devanna BN, Botella JR, Sharma V, Sharma TR (2017) Novel chitinase gene LOC_Os11g47510 from indica rice Tetep provides enhanced resistance against sheath blight pathogen Rhizoctonia solani in rice. Front Plant Sci 8:596
Richa K, Tiwari IM, Kumari M, Devanna BN, Sonah H, Kumari A, Nagar R, Sharma V, Botella JR, Sharma TR (2016) Functional characterization of novel chitinase genes present in the sheath blight resistance QTL: qSBR11-1 in rice line Tetep. Front Plant Sci 7:244
Romer P, Recht S, Strauss T, Elsaesser J, Schornack S, Boch J, Wang S, Lahaye T (2010) Promoter elements of rice susceptibility genes are bound and activated by specific TAL effectors from the bacterial blight pathogen. Xanthomonas oryzae pv oryzae New Phytol 187(4):1048–1057
Seo Y, Sriariyanun M, Wang L, Pfeiff J, Phetsom J, Lin Y, Jung K, Chou HH, Bogdanove A, Ronald P (2008) A two-genome microarray for the rice pathogens Xanthomonas oryzae pv. oryzae and X. oryzae pv. oryzicola and its use in the discovery of a difference in their regulation of hrp genes. BMC Microbiol 8(1):99
Shi X, Long Y, He F, Zhang C, Wang R, Zhang T, Wu W, Hao Z, Wang Y, Wang GL, Ning Y (2018) The fungal pathogen Magnaporthe oryzae suppresses innate immunity by modulating a host potassium channel. PLoS Pathog 14(1):e1006878
Singh P, Mazumdar P, Harikrishna JA, Babu S (2019) Sheath blight of rice: a review and identification of priorities for future research. Planta 250(5):1387–1407
Streubel J, Pesce C, Hutin M, Koebnik R, Boch J, Szurek B (2013) Five phylogenetically close rice SWEET genes confer TAL effector-mediated susceptibility to Xanthomonas oryzae pv. oryzae. New Phytol 200(3):808–819
Sun Q, Li TY, Li DD, Wang ZY, Li S, Li DP, Han X, Liu JM, Xuan YH (2019) Overexpression of loose plant architecture 1 increases planting density and resistance to sheath blight disease via activation of PIN-FORMED 1a in rice. Plant Biotechnol J 17(5):855–857
Thimm O, Blasing O, Gibon Y, Nagel A, Meyer S, Kruger P, Selbig J, Muller LA, Rhee SY, Stitt M (2010) MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J 37(6):914–939
Tian X, Li X, Zhou W, Ren Y, Wang Z, Liu Z, Tang J, Tong H, Fang J, Bu Q (2017) Transcription factor OsWRKY53 positively regulates brassinosteroid signaling and plant architecture. Plant Physiol 175(3):1337–1349
Tonnessen BW, Manosalva P, Lang JM, Baraoidan M, Bordeos A, Mauleon R, Oard J, Hulbert S, Leung H, Leach JE (2015) Rice phenylalanine ammonia-lyase gene OsPAL4 is associated with broad spectrum disease resistance. Plant Mol Biol 87(3):273–286
Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25(9):1105–1111
Ulferts S, Delventhal R, Splivallo R, Karlovsky P, Schaffrath U (2015) Abscisic acid negatively interferes with basal defence of barley against Magnaporthe oryzae. BMC Plant Biol 15:7
Venu RC, Jia Y, Gowda M, Jia MH, Jantasuriyarat C, Stahlberg E, Li H, Rhineheart A, Boddhireddy P, Singh P, Rutger N, Kudrna D, Wing R, Nelson JC, Wang GL (2007) RL-SAGE and microarray analysis of the rice transcriptome after Rhizoctonia solani infection. Mol Genet Genomics 278(4):421–431
Wang H, Meng J, Peng X, Tang X, Zhou P, Xiang J, Deng X (2015) Rice WRKY4 acts as a transcriptional activator mediating defense responses toward Rhizoctonia solani, the causing agent of rice sheath blight. Plant Mol Biol 89(1–2):157–171
Wilson RA, Talbot NJ (2009) Under pressure: investigating the biology of plant infection by Magnaporthe oryzae. Nat Rev Microbiol 7(3):185–195
Xue X, Cao ZX, Zhang XT, Wang Y, Zhang YF, Chen ZX, Pan XB, Zuo SM (2016) Overexpression of OsOSM1 enhances resistance to rice sheath blight. Plant Dis 100(8):1634–1642
Yang B, Sugio A, White FF (2006) Os8N3 is a host disease-susceptibility gene for bacterial blight of rice. Proc Natl Acad Sci USA 103(27):10503–10508
Yang F, Ding X, Chen J, Shen Y, Kong L, Li N, Chu Z (2017) Functional analysis of the GRMZM2G174449 promoter to identify Rhizoctonia solani-inducible cis-elements in maize. BMC Plant Biol 17(1):233
Yang J, Luo D, Yang B, Frommer WB, Eom JS (2018) SWEET11 and 15 as key players in seed filling in rice. New Phytol 218(2):604–615
Yuan P, Zhang C, Wang ZY, Zhu XF, Xuan YH (2018) RAVL1 activates brassinosteroids and ethylene signaling to modulate response to sheath blight disease in rice. Phytopathology 108(9):1104–1113
Yu G, Wang L, Han Y, He Q (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. Omics 16(5):284–287
Zhang J, Chen L, Fu C, Wang L, Liu H, Cheng Y, Li S, Deng Q, Wang S, Zhu J, Liang Y, Li P, Zheng A (2017a) Comparative transcriptome analyses of gene expression changes triggered by Rhizoctonia solani AG1 IA infection in resistant and susceptible rice varieties. Front Plant Sci 8:1422
Zhang Y, Wang X, Rong W, Yang J, Li Z, Wu L, Zhang G, Ma Z (2017b) Histochemical analyses reveal that stronger intrinsic defenses in gossypium barbadense than in G hirsutum are associated with resistance to Verticillium dahliae. Mol Plant Microbe Int 30(12):984–996
Zhang Y, Zhao J, Li Y, Yuan Z, He H, Yang H, Qu H, Ma C, Qu S (2016) Transcriptome analysis highlights defense and signaling pathways mediated by rice pi21 gene with partial resistance to Magnaporthe oryzae. Front Plant Sci 7:1834
Zheng A, Lin R, Zhang D, Qin P, Xu L, Ai P, Ding L, Wang Y, Chen Y, Liu Y, Sun Z, Feng H, Liang X, Fu R, Tang C, Li Q, Zhang J, Xie Z, Deng Q, Li S, Wang S, Zhu J, Wang L, Liu H, Li P (2013) The evolution and pathogenic mechanisms of the rice sheath blight pathogen. Nat Commun 4:1424
Zhu T, Song F, Zheng Z (2006) Molecular characterization of the rice pathogenesis-related protein, OsPR-4b, and its antifungal activity against Rhizoctonia solani. J Phytopathol 154(6):378–384
Acknowledgments
We are immensely grateful to Prof. Bu Qingyun for providing Longjing11, Oswrky53, and OsWRKY53 OE seeds. We also greatly appreciate Prof. Wang Guo-Liang for providing Osakt1 seeds. This research was funded by THE EARMARKED FUND FOR THE CHINA AGRICULTURE RESEARCH SYSTEM, CARS-01 to S.H.W., SUPPORT PLAN FOR YONG AND MIDDLE-AGED SCIENTIFIC AND TECHNOLOGICAL INNOVATION TALENTS IN SHENYANG, RC190489 to Y.H.X. and a grant from THE NEXT-GENERATION BIOGREEN 21 PROGRAM, PJ01325901 to K.H.J.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Peng Yuan, D., Xu, X.F., Hong, WJ. et al. Transcriptome analysis of rice leaves in response to Rhizoctonia solani infection and reveals a novel regulatory mechanism. Plant Biotechnol Rep 14, 559–573 (2020). https://doi.org/10.1007/s11816-020-00630-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11816-020-00630-9