
Abstract
Rice (Oryza sativa L.) pollen grains are highly sensitive to extreme temperatures. Studies on the proteomic analysis of pollen at different developmental stages are limited. In this study, to identify proteins expressed during pollen development, we performed shotgun proteomic analysis of rice anthers harvested at 8, 3, and 0 days before heading (DBH). A total of 4509 non-redundant proteins were identified in rice anthers at each of the three time points with three replications per time point. Among these, 2968 proteins were reproducible, of which 1919 were specifically expressed at a given stage of anther development. Clustering and gene ontology (GO) enrichment analyses categorized the specifically expressed proteins into four groups. Group I comprised proteins involved in cell differentiation, reproduction, and stress response; Group II contained proteins related to cell growth and carbohydrate metabolic process; Group III included proteins related to starch accumulation and lipid metabolic process; Group IV comprised proteins related to structure morphogenesis and cellular components. Proteins in Group I steadily increased during anther development, whereas those in Group I steadily decreased. Group III proteins showed the highest expression at 3 DBH, whereas Group IV proteins exhibited the lowest expression at 3 DBH.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The anther located in the stamen of the flower represents the male reproductive organ of a plant and produces the pollen required for pollination and seed set in flowering plants. Pollen development comprises several distinct stages including the pollen mother cell formation stage, tetrad stage, early young microspore stage, middle young microspore stage, early binucleate stage, late binucleate stage, and mature pollen stage (heading stage). Studies on rice (Oryza sativa L.) pollen in recent years have led to significant advances in our understanding of pollen development. A number of genes involved in pollen development have been identified in rice, and investigation of rice mutants has led to the identification of the conserved gene regulatory network involved in pollen development (Wilson and Zhang 2009).
In rice, the development of the reproductive system is highly sensitive to many environmental stresses. Microsporogenesis is the most temperature sensitive (Jagadish et al. 2007). Microarray analysis of rice plants treated with heat during pollen development revealed that several tapetum-specific genes are down-regulated by high temperature at the microspore stage, which inhibits pollen adhesion and stigma germination (Endo et al. 2009). Low temperature also affects the tapetal function during microsporogenesis. The transition time from the tetrad to the early binucleate stage is the most sensitive to cold stress because of vigorous tapetal activity (Oliver et al. 2005). Moreover, low temperature at the young microspore stage can lead to pollen sterility, thus reducing grain yield (Imin et al. 2006).
Many studies have been conducted on gene regulation and RNA expression during pollen development. Investigation of the expression patterns of recognizable classes of mRNA during pollen development showed that the pattern of mRNA expression in pollen grains is almost consistent with the pattern of starch biosynthesis (Kerim et al. 2003). Additionally, anther-specific genes induced by meiosis and plant hormones (e.g., gibberellic acid and jasmonic acid) have been isolated and characterized (Wang et al. 2005).
Proteomics analyses have been conducted in rice over the past several decades to study changes in protein expression in various organs, such as leaves, roots, and seeds, in response to biotic and abiotic stresses (Kim et al. 2014). Two-dimensional gel electrophoresis (2-DE) and MudPIT analyses revealed 1022 unique proteins in rice leaves. Proteins in rice leaf sheaths have also been isolated by 2-DE and analyzed by Edman sequencing (Shen et al. 2002). Additionally, 1350 and 877 proteins have been identified in rice roots and seeds, respectively, using 2-DE and MudPIT analyses (Koller et al. 2002).
Because pollen are critical for seed set, understanding the proteomic changes in rice pollen during various developmental stages will enhance our basic knowledge of pollen development at the molecular level and provide clues for developing varieties resistant to extreme temperatures. Therefore, in this study, we conducted shotgun proteomic analysis of rice anthers during development.
Methods
Sample preparation
Plants of the model japonica rice (Oryza sativa L. cv. Nipponbare) were grown in 15 × 30 cm rows in an experimental paddy field in Suwon, Republic of Korea. Anthers were collected at 8, 3, and 0 days before heading (DBH), representing the middle young microspore stage, the late binucleate stage, and the heading stage, respectively. Each developmental stage was determined by measuring the auricle length, which is the distance between the auricles of a leaf and the base of a panicle (Oliver et al. 2005). To ensure synchronous harvesting, anthers were harvested from spikelets located at the top of the 3 cm-wide panicles.
Protein extraction
Anthers were extracted from spikelets by vacuum suction, transferred to 1.5 ml Eppendorf tubes containing liquid nitrogen, and ground to a fine powder using two to three small ball bearings. To extract proteins from anthers, extraction buffer [8 M urea, 5 mM dithiothreitol (DTT), 1% lithium dodecyl sulfate (LDS), and 100 mM Tris (pH 8.5)] was added to the tubes. Samples were incubated at room temperature for 30 min and then centrifuged at 14,000×g for 15 min. The supernatant was filtered through a 0.45 μm membrane filter (Millipore, Billerica, MA, USA). Proteins were precipitated overnight using 20% (v/v) trichloroacetic acid (TCA) and were washed several times with cold acetone to remove unwanted by-products. The purified proteins were resuspended in a solution of 8 M urea and Tris–HCl (pH 8.5) and quantified using the 2D Quant Kit (GE Healthcare, Piscataway, NJ, USA), based on protein color reaction.
One-dimensional (1D) LDS-polyacrylamide gel electrophoresis (PAGE) and in-gel trypsin digestion
The extracted protein sample (50 μg) was mixed with NuPAGE® LDS Sample Buffer (4 ×), NuPAGE® Reducing Agent (10 ×), and deionized water (Invitrogen, Carlsbad, CA, USA), and then loaded onto the LDS-polyacrylamide gel and separated by PAGE. Subsequently, the gel was fixed in 100 ml fixing solution (40 ml deionized water, 50 ml methanol, and 10 ml acetic acid) for 10 min. Then, the gel was stained in staining solution [55 ml deionized water, 20 ml methanol, and 20 ml Stainer A and 5 ml Stainer B (Novex® Colloidal Blue Stain Kit)]; Stainer A was added first, and then Stainer B was added after 10 min. The gel was stained for approximately 12 h.
Next, in-gel digestion with trypsin was performed to identify proteins isolated by 1D LDS-PAGE. Briefly, each lane was divided into seven equal pieces, and each piece was sliced into 1 mm3 cubes and placed in a 1.5 ml Eppendorf tube. To each tube, destaining buffer (50% acetonitrile (can) in 50 mM ammonium bicarbonate (NH4HCO3) (pH 7.8)] was added to remove the Coomassie Brilliant Blue stain. Gel pieces in tubes were reduced with a reduction buffer (10 mM DTT in 25 mM NH4HCO3) at 56 °C for 45 min and then alkylated with alkylation buffer (55 mM iodoacetamide in 25 mM NH4HCO3) at room temperature in a dark environment. Then, the gel pieces were dehydrated in a SpeedVac and digested with 12.5 ng/μl trypsin in 50 mM NH4HCO3). After digestion, samples were incubated at 36 ℃ overnight. The tryptic peptides were precipitated using harvest buffer (5% formic acid in 50% ACN). Samples were then spun down for a few seconds by vortexing and incubated at room temperature for 20 min. The supernatant from each sample was dried in a SpeedVac and desalted using a Pierce® C18 spin column (Thermo Fisher Scientific, Rockford, IL, USA). Each digested peptide was analyzed by liquid chromatography–tandem mass spectrometry (LC–MS/MS).
LC–MS/MS analysis with Q Exactive
A nanoflow high-performance liquid chromatography instrument (EASY-nLC; Thermo Fisher Scientific, San Jose, CA, USA) was used online with the Q Exactive mass spectrometer and PepMap® RSLC C18 column (2 μm, 100 Å). Buffer A (0.1% formic acid) and Buffer B (ACN in 0.1% formic acid) were used for reverse phase chromatography. Samples were separated using a binary buffer system in a linear gradient (3–50% buffer B) at a flow rate of 270 nL/min. The run time for LC–MS/MS was 120 min.
MS/MS spectra data were collected using a data-dependent top 8 method, dynamically choosing the most abundant precursor ions among the survey scans (300–2000 Da) for higher-energy collisional dissociation (HCD) fragmentation. The survey scans were acquired at a resolution of 70,000 at a mass-to-charge ratio (m/z) of 200. Resolution for HCD spectra was set to 17,500 at m/z 200.
Proteomic data analysis
The MS/MS spectra data obtained by Q Exactive analysis were analyzed using Proteome Discoverer version 1.3 (Thermo Fisher Scientific, San Jose). Each peptide sequence was identified against the TIGR Rice Pseudomolecule protein database (Release v7.0; https://rice.plantbiology.msu.edu/annotation_pseudo_current.shtml). Carbamidomethylation of cysteine and oxidation of methionine represented field and variable modifications, respectively. To correct these modifications, both identifications were filtered at a false discovery rate of 1%.
Relative protein quantification
The Proteome Discoverer analysis data were calculated as normalized spectral counts (NSpC) in Microsoft Excel (Griffin et al. 2010) using the following equation:
where k represents a protein; SpC represents the total number of peptides corresponding to a single protein; L represents the protein length (i.e., number of amino acids); and n represents the total number of proteins.
Bioinformatics analysis
The molecular weight (MW) and isoelectric point (pI) of proteins were calculated using EMBOSS Pepstats < Sequence Statistics < EMBL-EBI (https://www.ebi.ac.uk/Tools/seqstats/emboss_pepstats/), which uses the EMBOSS Pepstats algorithm (https://emboss.bioinformatics.nl/cgi-bin/emboss/pepstats).
Clustering analysis and gene ontology (GO) annotation were performed using the constructed database. Clustering analysis was performed using the Genesis software version 1.7.6 (https://genome.tugraz.at). GO terms were categorized using Pantherdb (https://pantherdb.org). The GO annotations of the identified proteins were retrieved from the TIGR Rice Pseudomolecule protein database (Release V7.0). GO singular enrichment analyses were performed in agriGO (https://bioinfo.cau.edu.cn/agriGO/analysis.php) (Du et al. 2010). Composite expression analysis was performed by summing NSpC for each protein belonging to a specific functional category. (Bevan et al. 1998).
Results
Morphological changes in panicles and anthers
The morphological changes in rice panicles and anthers during development are presented in Fig. 1. With the progression of pollen development from 8 to 0 DBH, anthers gradually turned yellow (Fig. 1a), and spikelets turned dark green (Fig. 1b). Additionally, based on the appearance of spikelets, the water content of spikelets decreased gradually during pollen development, which is consistent with a previous study (Funaba et al. 2006).

Morphological changes in rice anthers and panicles at different pollen development stages. Changes in the morphology of rice anthers (a) and panicles (b) during the progression of pollen development from 8 to 0 days before heading (DBH)
Rice anther proteome
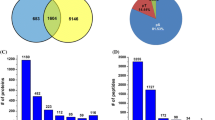
Given the small size of pollen grains, it is practically impossible to collect and extract proteins from developing pollen. Therefore, instead of collecting pure pollen, we harvested anthers to investigate pollen development. Samples used for proteome analysis in this study comprised pollen, anther wall, and tapetum, which is important for the nutrition and development of pollen grains and also acts as a source of precursors for pollen production (Piffanelli et al. 1998). Anthers were harvested at three time points (8, 3, and 0 DBH), with three biological replicates per time point. Approximately, 2500 non-redundant proteins were identified in each sample and 4509 non-redundant proteins were identified by merging all nine samples (Supplementary Table S1).
Next, we investigated the basic physiochemical properties, including pI and MW values, of 4,509 non-redundant proteins to verify that protein identification in rice anthers was unbiased (Fig. 2). The pI values of anther proteins ranged from 3.93 (LOC_Os07g41694; acidic leucine-rich nuclear phosphoprotein 32-related protein 1, putative, expressed) to 12.48 (LOC_Os01g69020; retrotransposon protein, putative, unclassified, expressed). Approximately, 45% of the identified proteins exhibited pI values ranging from 5 to 6 and 6 to 7, and the overall distribution of anther proteins identified during pollen development showed higher peaks in these ranges compared with that of the whole rice genome. Although slight skewness was detected, the distribution of anther proteins with pI values ranging from 7 to 8, 8 to 9, and 9 to 10 was similar to that of the whole rice genome. Furthermore, the pI values of more than 40% of the identified proteins were > 8. Thus, unlike the 2-DE method, no bias was detected during the resolution and identification of proteins exhibiting a wide range of pI values. The MW of proteins ranged from 2 kDa (LOC_Os02g27769, 40S ribosomal protein S27, putative, expressed) to 486 kDa (LOC_Os09g07300, BIG, putative, expressed). Distribution of the MW of the identified proteins was similar to that of the whole rice genome, despite that the MW of the identified proteins was seldom less than 4 kDa and frequently higher than 120 kDa. This was possibly a result of the shotgun proteomic approach, where proteins are identified by assembling the corresponding peptides using a mass spectrometer. Low MW proteins produce a small number of peptides, and the likelihood of these peptides being ionized and detected is less than those of high MW proteins. The detection and identification of proteins with extremely high or low MW were possibly exaggerated.

Distribution of the physiochemical properties of rice anther proteins. Distribution of the isoelectric point (pI) (a) and molecular weight (MW) (b) of 4509 non-redundant rice anther proteins compared with the entire rice genome
Comparison of rice anther proteome at different developmental stages
To compare protein expression patterns in anthers during development, the relative quantity of proteins was estimated based on spectral counts, which is a label-free method (Bridges et al. 2007; Zhu et al. 2010). Because of the analytical incompleteness phenomenon, we did not use all of the 4509 proteins in this analysis. To remove proteins detected in certain samples by random chance, we selected only proteins detected in all three replications, with more than two spectral counts at certain time points. This resulted in the selection of 2968 proteins.
The reproducibility of these 2968 proteins among the three replications was tested by correlation analysis. The R2 value of the NSpC ranged from 0.8357 to 0.8942 among the replications. Analysis of variance of the expression pattern of 2968 proteins among the three time points revealed 1919 specifically expressed proteins during rice pollen development (Supplementary Tale S2). The high number of specifically expressed proteins suggests that dynamic molecular changes occur during pollen development. The time points of harvest represent post-meiotic microgametes in fully matured pollen. The stages of pollen at 8, 3, and 0 represent the middle young microspore stage, the late binucleate stage, and the heading stage, respectively (Kerim et al. 2003).
Clustering and GO enrichment analyses
To elucidate the differences among proteins expressed at various pollen developmental stages, we performed clustering and GO enrichment analyses of the specifically expressed 1919 proteins. Clustering analysis categorized these proteins into four individual groups (Group I–IV); proteins in each group exhibited similar expression patterns (Supplementary Table S3). Most of the identified pollen proteins were related to pollen development and maturation.
GO enrichment analysis was conducted for proteins in each group to determine the enrichment of GO terms under the biological process, cellular component, and molecular function categories. In Group I, protein expression increased during pollen development, and 12 biological process GO terms were enriched (Table 1). These GO terms suggest that many proteins specific to pollen developmental processes, including reproduction, cellular process, and embryonic development process, contribute to anther development and gametogenesis. Until reaching maturity, the expression of proteins associated with mitotic division and cell differentiation, leading to the formation of generative cells and vegetative cells, was significantly increased. To characterize the global expression patterns of proteins in Group I, composite expression profiles were constructed by adding the NSpC values of each protein in each functional category (Fig. 3) (Bevan et al. 1998). Among the enriched GO terms, the composite expression profiles of proteins involved in developmental process, cell differentiation, and reproduction increased from 8 to 0 DBH, which was in accordance with morphological development (Fig. 3a–c).

Composite expression profiles of anther proteins enriched in different GO terms. a Developmental process; b cell differentiation; c reproduction; d response to stress; e response to abiotic stimulus; f regulation of anatomical structure size; g cell growth; h carbohydrate metabolic process; i starch accumulation in pollen; j lipid metabolic process; k response to stimulus; l anatomical structure morphogenesis; m cellular component organization
In Group II, protein expression decreased during pollen development. Eight GO terms were enriched under the biological process category and one under the molecular function category. Carbohydrates are a fundamental source of energy and play a crucial role in the reproductive development of plants (Zhu et al. 2015). However, as the pollen matured, carbohydrate metabolic process decreased gradually, and growth and cellular biosynthetic process decreased rapidly. The composite expression profiles of proteins involved in the regulation of anatomical structure size, cell growth, and carbohydrate metabolic process decreased to represent the completion of pollen development (Fig. 3f–h).
In Group III, protein expression increased from 8 to 3 DBH and then decreased until reaching 0 DBH. In this group, eight GO terms were enriched under the biological process category. The expression of Group III proteins was highest at 3 DBH, the late binucleate stage of pollen development, when starch is accumulated in pollen. The composite expression profiles of proteins involved in metabolic process indicated starch accumulation in pollen (Fig. 3i). The composite expression profile of proteins involved in lipid metabolic process showed the highest expression at 3 DBH (Fig. 3j, k).
In Group IV, protein expression decreased from 8 to 3 DBH and then slightly increased until maturity (0 DBH). In this group, four GO terms were enriched in the biological process category. The expression of Group IV proteins was lowest at 3 DBH. The composite expression profiles of proteins involved in anatomical structure morphogenesis and cellular component organization also represent the completion of pollen development, similar to the proteins in Group IV (Fig. 3l, m). The specific reasons why the expression of these proteins slightly increased at 0 DBH is unclear.
Discussion
To monitor protein expression patterns during pollen development, we performed shotgun proteomic analysis of rice anthers harvested at 8, 3, and 0 DBH. We constructed an in-depth rice anther proteome database by combining all of the non-redundant proteins (4509) identified at the three time points with three replications per time point. Even though we could identify total 4509 non-redundant proteins by merging all nine samples, approximately 2500 non-redundant proteins were identified in each sample. Comparison of these numbers suggests that some proteins did not overlap among samples. These were either unique proteins specifically expressed in certain samples or proteins detected randomly in certain samples; the latter is generally regarded as a phenomenon of analytical incompleteness, whereby proteins may not be detected in a single analytical run because protein concentrations in a cell vary in a complex manner (Wilkins et al. 2006). Despite the phenomenon of analytical incompleteness, we performed a more in-depth analysis of the rice proteome in anthers and identified more proteins than those in previous rice proteomic studies.
Comparison of the protein expression patterns of 2968 reproducible proteins revealed that 1919 proteins were specifically expressed at a certain developmental stage. This suggests that dynamic molecular changes occur in the rice pollen proteome during development. Clustering and GO enrichment analyses revealed that protein expression patterns were consistent with previously reported morphological and metabolic changes during pollen development. Among the he proteome expression patterns, the composite expression profile of proteins involved in carbohydrate and lipid metabolism pathways showed highest expression at 3 DBH. Pollen wall development requires lipid and polysaccharide metabolism, and lipid accumulation in anthers begins when microspores begin to expand (Paupière et al. 2014). Additionally, lipids are known to exhibit a protective role against adverse external stimuli (Paupière et al. 2014). Interestingly, the increase of proteins involved in the response to stress and abiotic stimulus (Fig. 3d, e) was detected in 3 DBH. This coincident expression patterns may suggest the possible roles in proper development of pollen grain against the unfavorable environment especially temperature stress which affect the pollen fertility. Rice pollen are highly sensitive to temperature during development (Endo et al. 2009). Although our study does not reveal the molecular function of these proteins under unfavorable temperature conditions, their role in temperature stress tolerance should be investigated further.
Overall, the proteomic data generated in this study provide fundamental information for the development of rice varieties with the ability to produce pollen and set seed under adverse environmental conditions.
References
Bevan M, Bancroft I, Bent E, Love K, Goodman H, Dean C, Bergkamp R, Dirkse W, Van Staveren M, Stiekema W (1998) Analysis of 1.9 Mb of contiguous sequence from chromosome 4 of Arabidopsis thaliana. Nature 391(6666):485–488
Bridges SM, Magee GB, Wang N, Williams WP, Burgess SC, Nanduri B (2007) ProtQuant: a tool for the label-free quantification of MudPIT proteomics data. BMC Bioinform 8(Suppl 7):S24–S24
Du Z, Zhou X, Ling Y, Zhang Z, Su Z (2010) agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res 38:W64–W70
Endo M, Tsuchiya T, Hamada K, Kawamura S, Yano K, Ohshima M, Higashitani A, Watanabe M, Kawagishi-Kobayashi M (2009) High temperatures cause male sterility in rice plants with transcriptional alterations during pollen development. Plant Cell Physiol 50:1911–1922
Funaba M, Ishibashi Y, Molla AH, Iwanami K, Iwaya-Inoue M (2006) Influence of low/high temperature on water status in developing and maturing rice grains. Plant Prod Sci 9:347–354
Griffin NM, Yu J, Long F, Oh P, Shore S, Li Y, Koziol JA, Schnitzer JE (2010) Label-free, normalized quantification of complex mass spectrometry data for proteomic analysis. Nat Biotechnol 28:83–89
Imin N, Kerim T, Weinman JJ, Rolfe BG (2006) Low temperature treatment at the young microspore stage induces protein changes in rice anthers. Mol Cell Proteomics 5:274–292
Jagadish S, Craufurd P, Wheeler T (2007) High temperature stress and spikelet fertility in rice (Oryza sativa L.). J Exp Bot 58:1627–1635
Kerim T, Imin N, Weinman JJ, Rolfe BG (2003) Proteome analysis of male gametophyte development in rice anthers. Proteomics 3:738–751
Kim ST, Kim SG, Agrawal GK, Kikuchi S, Rakwal R (2014) Rice proteomics: a model system for crop improvement and food security. Proteomics 14:593–610
Koller A, Washburn MP, Lange BM, Andon NL, Deciu C, Haynes PA, Hays L, Schieltz D, Ulaszek R, Wei J (2002) Proteomic survey of metabolic pathways in rice. Proc Natl Acad Sci 99:11969–11974
Oliver SN, Van Dongen JT, Alfred SC, Mamun EA, Zhao X, Saini HS, Fernandes SF, Blanchard CL, Sutton BG, Geigenberger P (2005) Cold-induced repression of the rice anther-specific cell wall invertase gene OSINV4 is correlated with sucrose accumulation and pollen sterility. Plant Cell Environ 28:1534–1551
Paupière MJ, Van Heusden AW, Bovy AG (2014) The metabolic basis of pollen thermo-tolerance: perspectives for breeding. Metabolites 4:889–920
Piffanelli P, Ross JH, Murphy D (1998) Biogenesis and function of the lipidic structures of pollen grains. Sex Plant Reprod 11:65–80
Shen S, Matsubae M, Takao T, Tanaka N, Komatsu S (2002) A proteomic analysis of leaf sheaths from rice. J Biochem 132:613–620
Wang Z, Liang Y, Li C, Xu Y, Lan L, Zhao D, Chen C, Xu Z, Xue Y, Chong K (2005) Microarray analysis of gene expression involved in anther development in rice (Oryza sativa L.). Plant Mol Biol 58:721–737
Wilkins MR, Appel RD, Van Eyk JE, Chung MC, Görg A, Hecker M, Huber LA, Langen H, Link AJ, Paik YK (2006) Guidelines for the next 10 years of proteomics. Proteomics 6:4–8
Wilson ZA, Zhang D-B (2009) From Arabidopsis to rice: pathways in pollen development. J Exp Bot 60:1479–1492
Zhu W, Smith JW, Huang C-M (2010) Mass spectrometry-based label-free quantitative proteomics. J Biomed Biotechnol 2010:840518–840518
Zhu W, Ma S, Zhang G, Liu H, Ba Q, Li Z, Song Y, Zhang P, Niu N, Wang J (2015) Carbohydrate metabolism and gene regulation during anther development disturbed by chemical hybridizing agent in wheat. Crop Sci 55:868–876
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2016R1D1A1B01011426) and the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT)(2017R1E1A1A01075282).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lee, Y., Kwon, Y., Kim, J. et al. Monitoring rice anther proteome expression patterns during pollen development. Plant Biotechnol Rep 14, 293–300 (2020). https://doi.org/10.1007/s11816-020-00599-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11816-020-00599-5