
Abstract
With the increasing prevalence of mobile devices and electric vehicles, the demand for energy storage systems has risen significantly. The current widely used lithium-ion battery architecture demonstrates the limitations of available materials and electrochemistry. Therefore, advanced battery systems, such as ultrafast charging/discharging, are necessary. Herein, we aim to elaborate on one of the most promising candidates among them, the dual-ion batteries (DIBs). Differing from conventional rocking-chair batteries, DIBs utilize both cations and anions as charge carriers, addressing rate-limiting steps and eliminating the need for ions to travel between electrodes during charge and discharge, thereby enabling ultrafast charging. In this review, we discuss the principles of ultrafast charging in DIBs, explore various types and their working mechanisms, and examine optimization strategies to enhance their performance. In addition, we highlight ongoing efforts and future perspectives in DIB development, aiming to stimulate further innovative research in this emerging field of energy storage.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
With the increasing prevalence of mobile devices and electric vehicles, the demand for energy storage systems has risen significantly. Therefore, developing rechargeable (or secondary) batteries with excellent electrochemical properties (i.e., high energy/power density, low cost, and fast-charging performance) becomes the key driving force for the current electrified life [1, 2]. In recent years, lithium-ion batteries (LIBs) have become increasingly prevalent across a wide range of energy applications from mobile devices to electric vehicles. This widespread usage is attributed to their high energy/power density, good cycling stability, and low self-discharge [3,4,5,6]. Currently, many of the cutting-edge commercial lithium-ion batteries (LIBs) utilize cathodes containing lithium (Li), nickel (Ni), and cobalt (Co). However, the availability of these materials is limited and their distribution across the globe is uneven. Furthermore, the process of recycling these materials poses challenges, being both difficult and environmentally harmful [7, 8]. In addition, a notable limitation of current lithium-ion batteries is their insufficient charging speed, which often fails to meet the commercial demands for rapid energy replenishment [9]. The main factors include the rate at which Li ions move through the electrolyte, the (de-)solvation energy at the electrode/electrolyte interface, and how well these ions move within the solid electrode material [10]. From this point of view, battery scientists are investigating advanced battery systems and new electrode materials that aim to go beyond the limitations of conventional LIBs, seeking more efficient energy storage solutions [11].
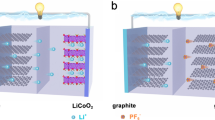
Dual-ion batteries (DIBs) stand out as one of the most promising alternatives, offering a distinct energy storage mechanism. In standard LIBs, only Li ions move back and forth between the cathode and anode during the charging and discharging process, which is referred to as the “rocking-chair” model. Generally, these Li ions are supplied by the cathode, while the electrolyte’s role is to facilitate their transport without directly contributing to the actual electrochemical energy storage process [12, 13]. In contrast, DIBs utilize both the cations and anions in the electrolyte for energy storage, offering a ‘dual’ charge storage mechanism. Taking the intercalation-type DIB as an example, it utilizes the concurrent intercalation of cations (e.g., Li+) into the anode and anions (e.g., PF6−) into the cathode, while in the subsequent discharge process, cations and anions return from the electrode to the electrolyte at the same time. This eliminates the necessity for ions to travel between electrodes, resulting in a shortened diffusion distance, enabling ultrafast charging. Generally, the intercalation potential of anions at the cathode (e.g., graphite) occurs at a high potential of > 4.2 V (vs. Li/Li+), with the calculated diffusion energy barrier within the graphite interlayer being approximately 0.2 eV [14]. Consequently, DIBs are normally able to achieve high operating voltage and rapid intercalation kinetics, exhibiting a high energy/power density and excellent rate capability (Fig. 1) [15,16,17,18,19]. In addition, due to the lower production costs and environmental benefits of using materials such as graphite and aluminum (Al), DIBs hold promise for significantly impacting the EV and large-scale energy storage markets.

Ragone plot of different energy storage systems
While DIBs offer significant advantages, the related research is still at an early stage. There is a need for further research into innovative electrode and electrolyte materials tailored for high-performance DIBs. In addition, developing effective strategies and advanced characterization techniques is crucial to deeply understanding the mechanisms underlying DIBs. Although existing reviews often focus on electrode materials and electrolytes [15, 20, 21], there is a scarcity of discussion on optimization strategies. For high-performance and ultrafast charging DIBs, the following critical problems need to be addressed: (1) cathode exfoliation due to repeated anion storage (2) decomposition of the electrolyte and other battery components under high operating voltage, and (3) mismatch reaction kinetics between the anode and cathode side. This review first discusses how dual-ion batteries (DIBs) can achieve ultrafast charging and then delve into the various types of DIBs and their mechanisms. Following this, we explore different optimization strategies and ongoing efforts to enhance the ultrafast-charging capabilities of DIBs. Finally, the discussion ends with a summary and future perspectives on DIB development to encourage further innovative research in this emerging field of energy storage.
Principles of Fast Charging in DIBs
DIBs are structured similarly to conventional LIBs, incorporating four major battery components. The primary distinction between DIBs and LIBs lies in their operational mechanisms: DIBs utilize both cations and anions within the electrolyte, whereas LIBs primarily rely on Li ions, which typically migrate from the cathode to the anode during charging. When a LIB is charged, ions move from the cathode, through the electrolyte to the anode. Crucial mechanisms governing this transit include Li ion transport (1) through the electrolyte, including Li ion solvation and desolvation, (2) across the electrode/electrolyte interphase, and (3) through the solid electrodes. Ideally, these fundamental phenomena should operate optimally to enhance battery efficiency [9, 22]. Nevertheless, actual battery operating conditions accompany a variety of side reactions that impair rate performance and reduce battery lifespan [23]. In contrast, the DIBs enable ultrafast charging compared to conventional LIBs, attributable to their innovative utilization of both types of ions within the battery system.
Conventional non-aqueous electrolytes in batteries typically consist of cyclic and linear carbonates, along with a lithium salt such as lithium hexafluorophosphate (LiPF6) [24]. In these electrolytes, the desolvation of Li+ can be impeded by the strong binding energy between the ions and solvent molecules such as ethylene carbonate [25]. Moreover, the strong coordination ability of these solvents with Li+ results in a low Li+ transference number, generally ranging from 0.3 to 0.45, which impedes rapid ion diffusion in the bulk solution [26, 27]. Alexandra J. et al., have determined the self-diffusion coefficients of solvent species, illustrating that self-diffusivity is inversely related to the particle radius through the Stokes–Einstein equation (Eq. 1) [28]:
where D is the self-diffusion coefficient, kB is the Boltzmann constant, T is the absolute temperature, η is the solvent viscosity, and r is the particle radius. Anions have much larger radii than the same-period cations. However, the solvation shell surrounding all ions is substantially larger than their bare ion radii, which significantly influences the magnitude of the diffusion coefficient [29, 30]. This impact varies depending on how tightly the ion is solvated, affecting both anions and cations. The interaction between solvent molecules and Li+ is notably stronger than with PF6− (Fig. 2a), leading to the cation traveling as a solvated complex, even in diluted electrolyte solutions [22]. In contrast, the anion drags much less solvent under similar conditions. This differential solvation structure significantly impacts diffusion rates, as evidenced by the faster diffusion of PF6− compared to Li+ in almost all electrolyte solvents, despite PF6− being the heavier species. Consistently, previous studies have shown that anion transport in solution is quicker than that of cations (Fig. 2b), likely due to the bulkiness of the Li+ solvation shell and the minimal association between anions and solvent [31, 32].

Copyright 2021, IOP Publishing. c Schematic illustration of ions diffusion pathways in DIB and LIB. d Schematic illustration of energy barriers between solvated ions, isolated ions, and intercalants, corresponding to the desolvation of the solvation structure and the insertion of ions into the electrode. Reproduced with permission [33]. Copyright 2021, Nature Publishing Group. e Energy barriers of PF6− diffusion in graphite. Reproduced with permission [14]. Copyright 2016, American Chemical Society
a Comparison of the binding energy of ethylene carbonate (EC) with Li+ and EC with PF6−. b Self-diffusion coefficients of EMC, EC, PF6−, and Li+ in 1 M LiPF6 EC/EMC (1:1 v/v). Reproduced with permission [28].
During the charging of LIBs, Li ions are released from the cathode, become solvated in the electrolyte, and then migrate to the anode/electrolyte interface, where they undergo desolvation before being incorporated into the anode. This identifies the diffusion length and desolvation energy of Li ions as the primary rate-limiting steps in LIBs [34, 35]. In contrast, DIBs supply anions and cations from the electrolyte close to their respective electrodes, thus significantly reducing the diffusion distance and facilitating ultrafast-charging capabilities, as depicted in Fig. 2c. The rate-limiting step of Li+ desolvation contributes to performance degradation in LIBs under fast-charging conditions [36]. A high desolvation energy barrier can induce significant polarization and severe lithium plating at the anode, leading to the formation of lithium dendrites which compromise battery life and safety [37, 38]. Conversely, in DIBs, both cations and anions undergo desolvation simultaneously during charging at the anode and cathode, respectively. Upon discharge, these charge carriers are de-intercalated from the anode and cathode and subsequently resolvated in the electrolyte. This desolvation process occurs only during the charging phase, in contrast to LIBs where the desolvation process occurs during both charging and discharging. Generally, anions, being larger and heavier than cations, are less solvated, which results in a lower desolvation energy barrier [39,40,41,42]. Recent studies have shown that the desolvation barriers of anions have minimal impact on cathode kinetics due to the weaker interaction between anions and solvent molecules compared to the cationic counterpart [43,44,45,46,47]. Although the solvation of Li ions in LIBs is well documented, thereof anions are very limited. The structure of the anion solvation sheath and anion–cation interactions in DIBs directly influence anion storage performance, making it crucial to understand these solvation effects. The electrochemical potential (ε) at which an anion experiences desolvation and subsequent intercalation into the cathode can be simply quantified through the calculation of the Gibbs free energy difference (∆G) between the intercalated and solvated states of the anion (Fig. 2d). This relationship is formally expressed by the following equation (Eq. 2) [33]:
where n is the number of electrons transferred in the reaction, and F is the Faraday constant. The Gibbs free energy difference comprises the sum of desolvation and intercalation energies. Due to the relatively low desolvation energy of anions, DIBs can achieve high-voltage and fast-charging conditions. To ensure the stable performance of DIBs under high-voltage and fast-charging conditions, efforts should be made to thoroughly study not only the electrode materials but also the electrolyte environment. This topic will be addressed in a later section.
Solid-state diffusion is an important parameter for characterizing the kinetic performance of batteries [48]. In batteries, the diffusion of ions is essential to the kinetic processes within electrode materials, as it substantially determines the reaction velocity and thus influences the rate performance of the electrode [49]. Contrasting with cations, which generally exhibit slower diffusion rates due to their higher charge densities, anions such as PF6− demonstrate more rapid movement through the electrode matrix. This enhanced mobility is primarily attributed to their comparatively lower energy barriers. Miyoshi et al. employed the galvanostatic intermittent titration technique (GITT) and electrochemical impedance spectroscopy (EIS) methods to calculate the diffusion constant of PF6− [14]. The chemical diffusion constant of PF6− in graphitic carbon is around 10–12 cm2·s−1, which is comparable to or slightly higher than that of Li+ in conventional cathode materials such as LiFePO4 or LiCoO2 used in LIBs. The activation energy for PF6− diffusion, obtained density functional theory (DFT) calculations, was estimated at about 0.23 eV (Fig. 2e). This value is the same or slightly lower than that for Li+ diffusion in graphite carbon and lower than that for Li+ in oxide cathode materials [50, 51].
In conclusion, DIBs offer a significant advantage by utilizing both anions and cations, effectively addressing the rate-limiting steps often encountered in LIBs. The weaker coordination between solvents and anions significantly enhances anion mobility, contrasting with the slower diffusion rates of cations. One reason for the fast-charging capabilities of DIBs is the substantially reduced diffusion distances and lower energy barriers for desolvation, which facilitate quicker ion movement within the electrolyte and across electrode/electrolyte interfaces. In addition, the efficient diffusion of anions within the cathode, despite their large molecular size, is a key factor that could revolutionize DIB performance. These characteristics underscore the potential of DIB technology to transform battery efficiency and stability, particularly in applications that demand rapid energy replenishment and durable operation.
Working Structure of Dual-Ion Batteries
DIB operates based on the principle that both anions and cations serve as carriers of charge. The different working principles of DIBs are illustrated in Fig. 3. On the basis of the operational working mechanisms, DIBs can be further divided into 3 types: insertion-type, conversion-type, and reverse DIBs. Insertion-type DIBs represent the most extensively researched category within the realm of dual-ion batteries (Fig. 3a). Graphite, commonly used as the cathode material in insertion-type DIBs, undergoes electrochemical reactions with anions at a high potential of > 4.2 V (vs. Li/Li+), which facilitates to enhanced energy density. However, the high operation voltage and repeated insertion of large-radius anions can lead to electrolyte decomposition. Therefore, high-voltage stability is indispensable for insertion-type DIBs [52,53,54]. The limited electrochemical stability window of aqueous electrolytes (i.e., 1.23 V) restricts the use of aqueous insertion-type DIBs, leading to a preference for non-aqueous systems.

The operational working mechanisms of different DIBs: a insertion-type DIBs, b conversion-insertion-type DIBs, c reverse DIBs
Conversion-type DIBs operate through anion redox reactions at the cathode and cation redox reactions at the anode. During the electrochemical process, the electrode materials in these batteries form new compounds, contributing to their exceptionally high specific capacities. Moreover, the nature of these conversion reactions enables the batteries to operate across a broader voltage window. However, significant challenges remain, as the substantial volume expansion and structural changes during cycling can negatively impact the reaction kinetics, making it difficult to achieve high energy and power densities [55]. During the charging process, anions and cations migrate from the electrolyte to the electrodes, and during discharge, these ions return to the electrolyte, similar to other DIBs. Conversion-insertion-type is one among various types of DIBs. As depicted in Fig. 3b, these batteries employ anion transport through conversion-insertion reactions. During charging, they undergo a two-step reversible process: initially, anions at the cathode undergo single-electron transfer oxidation from halide (A−) to halogen (A0). Following this, A0 inserted into the cathode, while cations are concurrently inserted into the anode host materials. The discharge process reverses these reactions [56]. This mechanism combines the benefits of conversion (i.e., fast charging) and intercalation (i.e., good reversibility), presenting a promising approach for high-performance battery design.
Reverse DIBs, like DIBs, use both anions and cations as charge carriers. In RDIBs, during charging, anions are inserted into the anode and cations into the cathode; during charging, these ions return to the electrolyte (Fig. 3c). In both types, the electrolyte is the sole charge carrier source. The intercalation of high-voltage anions, such as PF6− and TFSI−, enables high working voltages and significant energy density. However, high-voltage reactions can cause electrolyte and electrode decomposition. Thus, designing suitable electrodes and electrolytes is crucial for balancing energy density and stability. Furthermore, for rapid charging/discharging, matching reaction kinetics across battery components is essential, necessitating appropriate charge storage mechanisms, and battery configurations.
Insertion-Type DIBs
Insertion-type DIBs are the most extensively studied among DIBs. Graphite, the commonly used cathode material, reacts electrochemically with anions at high potentials, providing high energy density. Graphite’s distinctive layered structure, formed by a stacked aromatic ring with π–π interactions, enables it to accommodate both cations and anions, forming ionic graphite intercalation compounds (GICs). π-bonds in graphite can either accept or donate electrons. Cations intercalate at low potentials (e.g., Li ions intercalate at 0.01–0.2 V vs. Li/Li+), forming donor-type GICs. In contrast, anions intercalate at high potentials, resulting in the formation of acceptor-type GICs [15]. The electrochemical reactions in graphite-insertion-type DIBs with Li+ and PF6− as active ions are summarized by the following equation (Eqs. 3 and 4):
The incorporation of guest ions into the graphite host is facilitated through a “staging” mechanism. This process is governed by a delicate balance between the van der Waals forces that act among the graphene layers and the ionic repulsion that occurs within the layers of ions. This mechanism is described in the models proposed by Rüdorff and Daumas-Hérold [57] (Fig. 4a). This mechanism involves multiple stages during the formation of GICs. The stage number is determined by the number of graphene layers present between the intercalant layers [58].

Copyright 2014, Royal Society of Chemistry. b The XRD spectra of graphite cathode under 4.0 m NaFSI/EMC. Reproduced with permission [60]. Copyright 2024, Wiley–VCH. c Galvanostatic charge–discharge curve of coronene cathode. Reprinted with permission [61]. Copyright 2016, American Chemical Society. d Schematic illustration showing the working mechanism of the DIB with a Fe2(dobpdc)Ax MOF cathode. Reprinted with permission [62]. Copyright 2015, American Chemical Society
a Schematic illustration of ion insertion into graphite layer according to different models. Reproduce with permission [59].
As a practical illustration of this mechanism, Fig. 4b shows in-situ XRD data measured during the charge/discharge process of a graphite cathode using the electrolyte of 4.0 m NaFSI in ethyl methyl carbonate (EMC) electrolyte [60]. The splitting of the graphite (002) peak at 27° during the charging process indicates anion intercalation. Specifically, the incorporation of FSI− into the graphite leads to two distinct peaks corresponding to (00n + 1) and (00n + 2) reflections. Upon full charging, the positions of these peaks (marked by stage 1 at 22.76° and 34.20°) confirm the attainment of stage 1 intercalation.
Considering the other types of insertion-type DIBs, organic materials with aromatic components offer sustainability, derived from environmentally benign precursors [63,64,65,66,67]. The tunable structure of these materials allows precise control over anion electrochemical reactions, providing reasonable capacity. Nitrogen-containing organic materials are typical materials capable of anion storage, which function as an electron donor to host anions. For example, polytriphenylamine (PTPAn) was synthesized as a cathode for k-based DIBs [66]. During the charging process, the nitrogen atom in the center of the triphenylamine unit loses an electron, becoming positively charged. This positively charged nitrogen atom can then bind to a PF6− molecule. As a result, the PTPAn||Graphite full cell DIB exhibits good cycle performance within the 1.0–4.0 V range.
Polycyclic aromatic hydrocarbons (PAHs) with ordered structures and weak van der Waals forces are capable of storing anions within their intermolecular space [68, 69]. Studies have shown that the charge/discharge plateaus of these materials can be tuned by manipulating the conjugated structure of the aromatic hydrocarbon. For example, coronene polycyclic aromatic hydrocarbon exhibits highly reversible anion storage properties [61]. The coronene electrode shows flat plateaus at 4.2 V in charge and 4.0 V in discharge (Fig. 4c). Metal–organic frameworks (MOFs) present another avenue, offering diverse structures composed of metals and organic ligands for anion storage [70]. For example, Fe2(dobpdc) (dobpdc4– = 4,4′-dioxidobiphenyl-3,3′-dicarboxylate) stores anions via topotactic oxidative insertion reactions (Fig. 4d) [62]. In addition, transition metal dichalcogenides (TMDs) such as MoS2, with their graphite-like layered structures, have been reported as promising anion storage materials [71]. These advancements highlight ongoing research aimed at optimizing anion storage and improving insertion-type DIB performance.
Despite the potential of various materials for insertion-type DIBs has been studied, challenges such as volume expansion and rapid electrode degradation due to anion intercalation remain to be addressed. Consequently, developing materials with high capacity, good redox kinetics, and high stability for insertion-type DIBs is crucial. Furthermore, elucidating the mechanisms for anion hosting in these cathode materials through in-situ characterization techniques is essential for advancing novel and effective cathode materials.
Conversion-Type and Insertion–Conversion-Type DIBs
While the intercalation of anions in DIBs can achieve high voltage, it also presents challenges such as electrolyte decomposition at high voltages and difficulty using aqueous electrolytes. Therefore, it is crucial to develop cathode materials that operate at lower voltages but maintain high energy density by increasing the specific capacity of the electrode materials [72,73,74]. Conversion-type DIBs utilize reversible anion redox couples, with zinc–iodine [75] and zinc–bromine batteries [76] attracting attention due to their high energy density, abundant resources, and low cost. Due to the state of matter of iodine, iodine ions are typically utilized as carriers in flow batteries, which are promising for large-scale energy storage due to their high efficiency, safety, long-cycle life, and flexible design [77]. Li et al. reported a zinc–iodine DIB based on flow battery configuration (Fig. 5a) [75]. The electrochemical reactions in this system are as follows (Eqs. 5 and 6). The cyclic voltammetry (CV) curves clearly reveal these reactions, which correspond to the redox processes of Zn/Zn2+ and I3−/I− (Fig. 5b). Using a 3.5 M ZnI2 aqueous electrolyte, the zinc–iodine batteries exhibit a discharge energy density of 125 Wh·L−1 at 10 mA·cm−2, with capacity retention over 40 cycles:

Copyright 2015, Nature Publishing Group. c Schematic of the conversion-insertion mechanism occurring in the LBC-G composite during its oxidation in aqueous-gel electrolyte, and d CV of the LBC-G cathode at a scan rate of 0.05 mV·s−1. Reprinted with permission [78]. Copyright 2019, Nature Publishing Group
a Schematic of aqueous Zn–I flow batteries, and b CV of 0.0085 M ZnI2 on a carbon electrode at a scan rate of 50 mV·S−1. Reprinted with permission [75].
In the investigation of other conversion cathodes, copper cathode in an 8.5 mol·L−1 KOH electrolyte exhibits a high initial capacity of 300 mAh·g−1 by reversibly storing OH− [79]. However, the efficiency of the copper electrode decayed rapidly within the alkaline electrolyte. Gallagher et al. reported that the Cu electrode can reversibly store CO32− and OH− via two-phase conversion [80]. The electrochemical reaction of a two-phase conversion that involves water is supported by the single plateau charging profile as follows (Eq. 7):
Ex situ XRD data confirmed the formation of the malachite phase Cu2CO3(OH)2 showing that both CO32− and OH− anions act as charge-compensating carriers. Consequently, a copper cathode with an anode of activated carbon (AC) anode (N/P capacity ratio of 2) in a Cu||AC full cell exhibits an initial capacity over 40 mAh·g−1 at 500 mA·g−1, with a capacity retention of 49% after 50 cycles.
Despite their high capacity, conversion-type DIBs suffer from poor reversible capacity. Insertion-conversion-type DIBs combine the high capacity of conversion reactions with the excellent reversibility of intercalation mechanisms, improving performance and safety. As noted earlier, the graphite cathode demonstrates a high operating voltage in DIBs. However, its specific capacity typically remains below 130 mAh·g−1. To address this, Yang et al. developed conversion-insertion-type DIBs that merge these benefits for high-performance applications in aqueous batteries (Fig. 5c) [78]. The developed DIB features a (LiBr)0.5(LiCl)0.5-graphite cathode (LBC-G), synthesized by mixing anhydrous LiBr and LiCl with graphite. The electrochemical performance of LBC-G was evaluated in a three-electrode cell utilizing an aqueous-gel polymer electrolyte based on water-in-bisalt (WIBS). Cyclic voltammetry (CV) and charge–discharge profiles revealed two distinct reaction voltage ranges, 4.0–4.2 V for Br− insertion and 4.2–4.5 V for Cl− insertion, delivering a highly reversible discharge capacity of 243 mAh·g−1 with 82% capacity retention over 230 cycles (Fig. 5d). The two-step redox reactions are as follows (Eqs. 8 and 9):
During charging, Br− oxidize to Br0 and to intercalation into graphite, forming Cn[Br]. Further charging leads to the oxidation and intercalation of Cl−, creating the mixed intercalation compound Cn[BrCl]. Each halogen oxidation undergoes a one-electron transfer reaction, with theoretical capacities of 309 mAh·g−1 for LiBr and 632 mAh·g−1 for LiCl, along with the release of a Li ion into the bulk electrolyte. During discharging, the Cl0 and Br0 de-intercalate from the graphite structure, reduce back to halides and recombine with Li+ to form LiCl/LiBr crystals and liquefied halides outside the graphite interlayers. The corresponding LBC-G||Graphite full cell demonstrated a high energy density of 460 Wh·kg−1 with good cycling stability.
Similarly, a Zn-based conversion-insertion DIB employs a graphite cathode paired with a Zn metal anode and uses a molten hydrate electrolyte [81]. This battery system leverages dual-halogen redox couples, specifically Br0/Br− and Cl0/Cl−. The Br0/Br− redox reaction involves the adsorption/desorption of Br0 at the graphite–electrolyte interface, along with the de(intercalation) of Br0 within the graphite interlayer. Further reaction leads to a Cl0/Cl− redox couple involving similar (de)intercalation processes within the graphite. The average discharge voltage for this two-step redox system is 1.71 V, higher than the 1.60 V achieved when using only Br0/Br− redox couple. The as-described zinc-dual-halogen cell exhibits good cycling stability over 100 cycles at 250 mA·g−1. Future development directions for conversion-type DIBs include enhancing the conductivity of ionic binding products and suppressing their dissolution shuttle action through structural design. In addition, designing selective permeable membranes to inhibit the shuttle effect of dissolved anionic binding products can greatly improve the electrochemical performance of conversion-type cathodes [82].
Reverse-Type DIBs
Most DIBs used graphite as a cathode to store anions. However, graphite electrodes are typically operated at high oxidation voltage, which can cause electrolyte decomposition. To address this issue, researchers have developed novel anion storage materials that operate at lower voltages. When these low-voltage anion storage materials are paired with certain high-voltage cation-hosting materials, the ion transfer direction during the charging/discharging process in full cells is reversed compared to conventional dual-ion batteries (DIBs). The working principles of reverse dual-ion batteries (RDIBs) are analogous to those of DIBs. In DIBs, ions move from the electrolyte to the electrodes during the charging process. Conversely, in RDIBs, ions move from the electrodes to the electrolyte during charging.
For instance, a Zn-based RDIB utilizing a highly concentrated 30 m ZnCl2 aqueous electrolyte has been reported [83]. During discharge, Zn2+ are inserted into a Zn3[Fe(CN)6]2 cathode, while ZnCl42− are inserted into a ferrocene anode. During charging, Zn2+ and ZnCl42− are de-inserted from the electrodes into the electrolyte (Fig. 6a). The electrochemical reaction is as follow (Eqs. 10 and 11):

Copyright 2019, American Chemical Society. c Schematic illustration of a VTO||Li RDIB, and d CV curves of the VTO||Li RDIB at 0.5 mV·s−1. Reprinted with permission [84]. Copyright 2022, Wiley–VCH
a Schematic illustration of a Zn-based RDIB, and b voltage changes of full cells in ZnCl2 electrolytes with different salt concentration. Reprinted with permission [83].
According to the Nernst equation of anode reaction, the increase of the electrolyte potential leads to the increase of concentration and activity coefficient of anion. Accordingly, the potential of insertion/extraction potential is lower. Similarly, the potential of cation redox is increasing. Therefore, the cell voltage was enhanced in a 30 m ZnCl2 electrolyte (0.95 V) compared with a 5 m ZnCl2 electrolyte (0.60 V). The voltage of RDIBs is increased with increasing the electrolyte concentration, while the reverse phenomenon occurs in DIBs, which demonstrates the application merits of highly concentrated aqueous electrolytes in RDIBs (Fig. 6b). This effect significantly improves the energy density of RDIBs. The described full cell achieved a specific energy density of 21.0 Wh·kg−1 with a reversible capacity of 30 mAh·g−1.
To date, the configuration of RDIBs has only been realized with aqueous electrolytes. The low working potential and limited energy density have been major obstacles to the further development and broad application of these systems. In this context, Li et al. constructed high energy density RDIBs by coupling adsorption and conversion reactions (Fig. 6c) [84]. Vanadium-doped anatase (VTO) was synthesized via the hydrothermal method and used as cathode, with lithium as the anode. During the charging/discharging process, cations in the electrolyte are reversibly adsorbed and desorbed at the cathode, while a conversion reaction of Li/Cl occurs at the anode. Cyclic voltammetry defined the electrochemical behavior during the charging/discharging process, as shown in Fig. 6d. The two larger peaks at 1.65 and 2.08 V correspond to the conversion between Ti4+ and Ti3+, while the two smaller peaks at 2.1 and 2.8 V are attributed to the transformation between V4+ and V3+. The effective combination of adsorption and conversion reaction mechanisms leads to high-speed cation redox at VTO and high capacity at Li/LiCl, breaking the trade-off between power density and energy density in aqueous electrolytes. The full cell demonstrates a high-power density of 1532 W·kg−1 and a high energy density of 214 Wh·kg−1 at 1 A·g−1. The RDIB system addresses limitations of traditional electrolytes by enabling the use of cation-deficient electrodes as cathodes in metal-anode-free cells, highlighting the role of ion charge carriers in redox reactions. This approach is relevant to existing grid storage technologies like zinc–cerium and sulfur–iron redox flow batteries, where higher electrolyte concentrations also increase cell voltage. The RDIB design enhances energy storage performance and efficiency, offering significant potential for future advancements.
Optimization Strategies for Ultrafast Charging Dual-Ion Batteries
Constructing efficient DIB configurations poses significant challenges, including structural failure in cathode materials, slow reaction kinetics of anode materials, and electrolyte decomposition at high operating voltages. To achieve ultrafast-charging DIBs, it is essential to comprehensively design cathode materials, anode materials, and compatible electrolyte systems to address these obstacles. In this section, we will explore these optimization strategies in detail to develop effective approaches for achieving high-performance DIBs.
Cathode Design
In DIBs, the ability of cathodes to store anions plays a pivotal role in determining both the voltage and capacity of the batteries. Previous studies of cathode materials focused on different storage of specific anions (e.g., hexafluorophosphate (PF6−), tetrafluoroborate (BF4-), bis(trifluoromethanesulfonyl)imide (TFSI−), and bis(fluorosulfonyl)imide (FSI−)) [85,86,87,88]. However, the relatively large radius of these anions often results in unsatisfactory capacity and structure collapse. Furthermore, during the anion intercalation at high voltage, solvent molecules are also likely to co-intercalate into the cathode materials [89, 90]. To enhance DIB efficiency, especially for fast charging, the structural and compositional makeup of cathode materials must be refined. Efforts to improve electrochemical kinetics for ultrafast charging include interlayer engineering, reinforcing materials with functional binders, and applying anion-inactive coatings.
Adjusting the spacing between layers in cathode materials facilitates easier movement and storage of anions, thus improving the battery’s overall rate capability and stability. It has been found that cathodes with a large planar size contribute to improved structural stability during the repetitive (de)intercalation process of anions. Consequently, expanded graphite (EG) cathode exhibits enhanced performances with a high specific capacity and good rate capability [94]. In addition, Li et al. reported molybdenum disulfide (MoS2) with large layer spacing (~ 6.16 Å) serves as an effective cathode for anion storage in Li-based DIBs (Fig. 7a) [71]. Such a structure with enlarged interlayer distance significantly enhances the diffusion kinetics and maintains excellent stability across a wide voltage range (~ 5 V). The diffusion coefficient (Ds) of anions, calculated based on Fick’s second law, rages from 10–14 cm2·s−1 to 10–9 cm2·s−1, which is substantially higher than that of PF6− in graphite (10–14 cm2·s−1 to 10–18 cm2·s−1) (Fig. 7b). The cycling performance of intercalating difluoro(oxalate)borate (DFOB−) demonstrates a specific capacity 98 mAh·g−1, maintaining 88% capacity after 500 cycles at a fast-charging rate of 2 mA·cm2. MoS2||graphite DIBs exhibit a high specific capacity of 135 mAh·g−1 over 50 cycles. Similar to this idea, various studies have modified the morphology of graphite to increase the number of active sites for anion storage and enhance rate performance. For example, porous nanoflake graphite, synthesized by cathodically polarizing amorphous carbons in molten CaCl2 at 1100 K, delivers a high capacity of 116 mAh·g−1 at a rate of 1.8 A·g−1 and retains 92% capacity when cycled at a high rate of 10 A·g−1 [95]. In addition, the graphite exfoliation issue can also be solved by the chemical link of graphite surface groups. For example, selective incorporation of carboxylic anhydride functional groups between graphite layers can achieve a stabilizing effect on the crystal structure [96].

Copyright 2020, Elsevier. c Schematic of graphite electrodes using the AZACA binder, and d rate performance evaluation of DIB cells using different binders at 3.0–5.3 V. Reproduced with permission [91]. Copyright 2023, Wiley–VCH. e Schematic illustration of Li||SMG DIB. Reproduced with permission of [92]. Copyright 2019, Wiley–VCH. f Schematic diagram of the LTO-modified MCMB g Cycle performance of LTO-modified MCMB at 5 C. Reprinted with permission of [93]. Copyright 2019, Wiley–VCH
a Schematic illustration of MoS2 cathode with expanded interlayer, and b the diffusion coefficient (Ds) of MoS2 cathode calculated by the GITT method. Reproduced with permission [71].
In DIBs, the binder must operate effectively under high-voltage conditions and suppress the volume expansion. Polyvinylidene fluoride (PVdF) is a commonly used high-voltage cathode binder due to its high oxidative resistance, attributed to electron-withdrawing groups (EWGs) (-F) in its backbone [97]. However, PVdF not fully covering highly active carbon black accelerates electrolyte degradation at high voltages [98]. To overcome this, poly (vinylidene fluoride-co-hexafluoropropylene) (PVdF-HFP) has been utilized in graphite cathodes for DIBs [99]. This binder not only enhances interfacial stability compared to PVdF binders but also improves the stability of the cathode–electrolyte interface through its abundant fluorine groups. Additional research aimed at enhancing the chemical binding and adsorption energy between cathode materials has explored the use of binders such as polyacrylic acid (PAA), sodium carboxymethyl cellulose (CMC), and alginate (Alg) [100]. For instance, Alg, a bioderived aqueous binder, shows superior performance compared to other binders. Alg-based graphite exhibits no significant structural deformation, while PVdF-based cathodes suffer from severe material loss from the current collector [101]. This is explained by Alg’s high mechanical strength and good interaction with active materials [102]. Alg-based graphite cathodes deliver superior rate performance and cycle stability, with a capacity of 97.8 mAh·g−1 and 87 mAh·g−1 at high rates of 10 C and 50 C, respectively. These cathodes also maintain 80% of their initial capacity after 1000 cycles at 10 C. Despite their advantages, commercial hydrophilic binders such as PAA, CMC, and Alg have linear chain structures that struggle to withstand significant stress, highlighting the need for stress delocalization strategies [103]. To address this, Kang et al. developed a novel polymeric binder and cohesive graphite combination that exhibits stable behavior at anion redox potentials greater than 5 V [91]. The binder was synthesized through the copolymerization of methyl acrylate and vinyl benzyl chloride with potassium polyacrylate (PAAK) moiety, providing excellent electrochemical stability and self-healing via ion–dipole interactions. After subsequent azide modification and exchange reactions, the binder linked to graphite through azacyclic compounds (AZCA binder) enables deeper and reversible anion intercalation, resulting in longer-lasting and fast-charging capabilities (Fig. 7c). The AZCA binder effectively disperses mechanical stress during repeated cycling, establishing a robust conductive network for the effective transport of anions and electrons. As a result, AZCA-graphite cathode achieved a reversible capacity of 97.5 mAh·g−1 at an ultrafast rate of 50 C (Fig. 7d). In addition, the long-term cycling performance of the AZCA binder showed 83% retention of its initial capacity and a saturated CE of 99.1% at 3500 cycles at 10 C.
To improve the structural integrity of the cathode materials in DIBs, constructing advanced protective layers on the cathode surface is essential. A solid electrolyte interphase (SEI) layer is typically formed to maintain long-term cycling stability and fast-charging capability [104]. Li et al. introduced a surface-modification strategy to protect the graphite cathode by generating an artificial SEI layer through a constant current/discharging process in the potential window of 0.3–2.0 V (Fig. 7e) [92]. The SEI layer significantly improves structural stability, protecting graphite from anion solvation effects and deposition from electrolyte decomposition. Consequently, the SEI-modified graphite (SMG) cathode demonstrated superior cycling stability with 96% capacity retention after 500 cycles at 200 mA·g−1. To further enhance cathode integrity, recent studies have focused on coating anion-inactive materials to facilitate the formation of successive SEI layers and prevent electrolyte decomposition. For example, Wu et al. deposited a thin amorphous Al2O3 layer on graphite, which acted as a stable artificial SEI, preventing electrolyte decomposition product deposition [105]. As a result, Li-Al2O3 deposited graphite DIBs achieved 80% capacity retention over 2700 cycles at 200 mA·g−1 and an acceptable capacity of around 80 mAh·g−1 at a fast-charging rate of 2000 mA·g−1. Another study demonstrated that a robust Li4Ti5O12 (LTO) coating on a graphite surface is a feasible method to further improve cyclic stability (Fig. 7f) [93]. The graphite cathode was chemically coated with an LTO layer, which acts as a skeleton and provides electrocatalytic active sites for in-situ generation of a favorable cathode–electrolyte interphase (CEI) layer (Fig. 7g). This LTO-CEI-reinforced structure cathode exhibited excellent cyclability, with 85.1% capacity retention after 2000 cycles, even at a high cutoff potential of 5.4 V at 5C.
Anode Design
Anode materials in DIBs face significant challenges, including limited specific capacity and sluggish reaction kinetics, which contribute to a mismatch with the cathode. For ultrafast-charging DIBs, it is crucial to reinforce anode reaction kinetics to co-determine overall cell performance, including rate capabilities, specific capacity and safety, in conjunction with cathode materials and other components. Strategies such as developing porous architectures, integrating highly conductive carbon materials, and crafting nanostructured crystals have proven effective in enhancing the electrochemical performance of anodes. Specifically, creating anode materials with porous structure is a common strategy to enhance reaction kinetics by providing ample cation storage sites and promoting fast diffusion kinetics. Commonly used materials include three-dimensional porous dipotassium terephthalate (pK2TP) and redox-active conjugated porous polymers [19, 106, 107]. In addition, Song et al. developed an omnidirectional porous Al wire anode that can significantly buffer the volume change during the cycle, and maintain good electrical contact and electrode integrity [108]. The porous structure was fabricated using an electrochemical etching method and can be observed in a scanning electron microscope (SEM) image (Fig. 8a). Upon charging, Li ions react with porous Al anode to form Al–Li alloys. The initial specific discharge capacity of the porous Al anode is 120 mAh·g−1. It demonstrates good cycling stability, retaining a specific capacity of 138 mAh·g−1 after 200 cycles at 1 C (Fig. 8b). Furthermore, in rate capability tests, the porous Al anode exhibited high structural stability at a high current density of 20 C. Similarly, Al foil is transformed into a porous structure and coated with amorphous carbon [109]. This porous structure alleviates mechanical stress caused by Al volume changes during electrochemical cycling and shortens ion diffusion lengths. The carbon-coated layer buffers Al volume changes and mitigates undesirable surface reactions. Consequently, the synergistic effect of the porous conductive structure and carbon layer in the pAl/C anode allows the pAlC-Graphite full cell to exhibit a high reversible capacity of 104 mAh·g−1 at 2 C, with 88% capacity retention after 200 cycles (Fig. 8c).

Copyright 2019, Elsevier c Cycle performance of the 3D porous Al foil coated with a carbon layer (pAl/C) at 2 C. Reprinted with permission [109]. Copyright 2016, Wiley–VCH d Schematic illustration of LNTC interwoven with CNTs through π–π superposition effect, and e rate performance of the LNTC@CNTs||EG at different current densities. Reproduced with permission [110]. Copyright 2024, Wiley–VCH. f Schematic illustration of the MoS1.5Te0.5@C nanocables, and g rate performance profiles of the MoS1.5Te@C nanocables||EG dual-ion batteries at various rate (from 0.1 to 5 A·g−1). Reproduced with permission [111]. Copyright 2022, Nature Publishing Group. h TEM and EDX-elemental mapping images of α-Fe2O3, and i cycling capability of Li-DIB and Na-DIB at 500 mA·g−1. Reproduced with permission [112]. Copyright 2022, Elsevier
a SEM images of the porous Al wire with a cross-sectional view shown in the inset, and b cycling stability of the pristine Al wire and porous Al wire at 1 C. Reprinted with permission [108].
Integrating anode materials with conductive carbon elements is an effective strategy to boost electronic conductivity and accelerate cation transfer rates within the anode [113, 114]. For example, black phosphorous nanoparticles dispersed in carbon have been used as anode materials for DIB [115]. Using a LiTFSI in DMC electrolyte, the BP-C||graphite DIB demonstrates good rate performance and cycling stability. However, these dispersion strategies can sometimes lead to unexpected structure compromises, hindering the achievement of optimal electrochemical performance. To address this, Su et al. developed a new composite anode, tetralithium 1,4,5,8-naphthalenetetracarboxylate (LNTC) interwoven by carbon nanotubes (CNTs), forming the LNTC@CNTs composite for Li ion storage [110]. The naphthalene interwoven with CNTs enhances electron delocalization through the π-π superposition effect (Fig. 8d). This conjugated structure not only boosts reaction kinetics but also reinforces Li ion storage stability. As a result, LNTC@CNT@||EG full cell exhibits a discharge capacity of 122 mAh·g−1 and excellent cycling stability with a capacity retention of 84.2% after 900 cycles at 200 mA·g−1. Moreover, the discharge capacity returns to 122 mAh·g−1, when the current density recovers from 1000 mA·g−1 back to 50 mA·g−1, indicating its ultrafast-charging capability (Fig. 8e). Similarly, Liu et al. reported a source-template synthetic strategy to fabricate nanowire-in-nanotube MoS1.5Te0.5@C structures with an in-situ grown carbon film coating (Fig. 8f) [111]. The carbon surrounding the MoS1.5Te0.5 nanocables acts as a protective layer, facilitating stable SEI, enhancing electrical conductivity, and decreasing charge transfer resistance. Due to these compositional advantages, the MoS1.5Te0.5@C demonstrates an improved Na-ion storage. Consequently, MoS1.5Te0.5@C||EG full cell with a 3 M NaPF6-based electrolyte delivers reversible capacities of 214.2, 207.8, 195.9, 175.8 and 150.2 mAh·g−1 at current densities 0.1, 0.2, 0.5, 1 and 2 A·g−1, respectively (Fig. 8g). Even at a high current rate of 5 A·g−1, the full cell achieves an acceptable capacity of 100.9 mAh·g−1, indicating its ultrafast-charging capability.
Designing a nanostructured anode is an effective strategy for achieving ultrafast charging. Nanostructures are permeable and possess high resistance to volume changes, which can reduce the energy barrier for ion diffusion and ensure good cycling stability [116]. In addition, the large surface area provides numerous active sites and enhances the wettability of the electrolyte, further improving the overall performance of the battery [117]. For instance, Co3O4 nanosheets grown on carbon fiber paper (CFP) enhance electrolyte immersion and shorten the ionic transport pathway [118]. This Co3O4/CFP electrode exhibits good rate capability across current densities ranging from 50 and 500 mA·g−1. In another study, Tong et al. introduced core/shell aluminum carbon nanospheres (nAl@C) as anode material for DIBs [119]. These nanospheres accommodate mechanical strain and stress, preventing pulverization. In addition, the conductive carbon layer enhances the conduction of both Li ions and electrons, which supports ultrafast charging. The DIB with nAl@C nanospheres anode demonstrated superior rate performance 88 mAh·g−1 at 15 C and maintained 95.1% capacity retention after 1000 cycles. Another strategy involves combining porous and nanostructures, such as synthesizing nanocubic structured α-Fe2O3 through a nanostructure engineering strategy [112]. This design suppresses volume expansion and structural degradation. The ordered nanocubic structure with a large specific area facilitates rapid ion diffusion and the quick formation of a stable SEI layer. The synthesized α-Fe2O3 features microporous and mesoporous structures with pore sizes between 1 and 30 nm. Transmission electron microscope (TEM) images reveal a regular cubic structure ~ 70 nm wide, with iron and oxygen elements evenly distributed (Fig. 8h). As an anode for Li-DIB and Na-DIB, it demonstrates outstanding long-cycle performance, maintaining stability for 520 cycles at a current density of density 500 mA·g−1 (Fig. 8i).
Electrolyte Design
The electrolyte is crucial in DIBs, as it is the only source of charge carriers, acting as both ion transport medium and active material. A variety of solvents, salts, and additives are chosen to withstand high voltages and ensure stable electrochemical performance. Typically, anions with smaller ionic radii, such as PF6−, BF4−, AlF4−, and FSI−, are preferred to enhance ion diffusion kinetics and reduce cathode material decomposition. For example, FTSI−, being smaller than TFSI-, shows easier intercalation into graphite and higher discharge capacity at the same cutoff voltage [120]. However, this principle does not apply to all anions. For instance, large-sized bis(pentafluoroethylsulfonyl)imide (BETI−) and small-sized FSI− are unsuitable for realizing high capacities, and the intercalation capacities follow the order: FTFSI− > TFSI− > FSI− > BETI−. These results suggest that besides size (BETI−: 0.94 nm > TFSI−: 0.8 nm > FTFSI−: 0.65 nm > FSI−: 0.54 nm), factors like the “solvation effect” should be considered in the DIB’s performance [121]. Different anions are predicted to enhance the diffusivity, reversibility, and chemical stability of electrolytes. Each anion possesses unique characteristics, making the selection of the appropriate anion crucial for optimizing the performance of electrolytes in DIBs. To achieve superior performance, hybridization anion strategies are commonly used. For example, Shin et al. [122] utilized an anion-hybridization strategy by combining PF6− with BF4− in the electrolyte (Fig. 9a). PF6− suppresses electrolyte decomposition by forming a fluorine-rich SEI layer. Adding BF4− with a small ionic radius to LiPF6 electrolytes lowers the anion diffusion barrier in graphite. With 5% LiBF4, these DIBs showed a reversible capacity of 90.5 mAh·g−1 at 15 C and good cycling stability for 500 cycles at 5 C (Fig. 9b).

Copyright 2019, Royal Society of Chemistry. c Comparison of the specific discharge capacities of Li||Graphite DIBs in the 20th Cycle. Reproduced with permission [123]. Copyright 2017, Elsevier. d Photographs for 4.0 m LiFSI in TMS and, e LSV test LiFSI in different electrolyte concentrations. Reprinted with permission [18]. Copyright 2021, Wiley–VCH f LUMO and HOMO of electrolyte components, and g cycle performance of graphite||LTO DIBs at 20 C. Reprinted with permission [124]. Copyright 2023, Elsevier
a Schematic illustration of the aluminum–graphite tri-ion (Li+/PF6−/BF4−) battery configuration and charging/discharging mechanism, and b Cycling performance of the battery for 500 cycles at 5C with 0% LiBF4 for comparison. Reproduced with permission [122].
As mentioned earlier, the electrolyte serves as the sole source of charge carriers in DIBs. Therefore, adjusting the electrolyte concentration is crucial for optimizing DIB performance. The concentration of the electrolyte significantly influences the solvation structure of ions, electrochemical performance, and the electrolyte/electrode interface in DIBs. Generally, higher electrolyte concentrations improve electrochemical performance, reversibility, and stability due to an increased number of charge carriers [125]. This improvement is largely because reducing free solvent molecules helps suppress the solvent co-intercalation phenomenon, which contributes to enhanced Coulombic efficiency (CE) and cycle stability in DIBs. In addition, the high-concentration electrolyte enhances intercalation capacity according to the Nernst equation. For example, Heckmann et al. found that the reversible capacity of dual-graphite DIBs with highly concentrated electrolyte (HCE) significantly increased from 50 mAh·g−1 (1 M LiPF6/EMC and 1 M LiPF6/DMC) to 90 mAh·g−1 (3.5 M LiPF6/EMC and 4 M LiPF6/DMC) with a higher cutoff voltage of 5 V [123]. Moreover, higher electrolyte concentrations improved CE during cycling and showed good cycle retention across various current densities (50–1000 mA·g−1). However, despite the superior performance of HCEs, DIBs with PF6-based concentrated electrolytes remain unsatisfactory. PF6− salts have limited solubility in most solvents, especially for NaPF6 and KPF6, where concentrations are typically under 1 M [126, 127]. Increased PF6− concentrations lead to dramatically high viscosity and lower ionic conductivity. To address this, imide-based salts such as TFSI− and FSI− are used to achieve higher concentrations, due to weaker cation–anion interactions. For example, a 7.5 m LiFSI dissolved in EC/DMC (1:1 v/v) electrolyte demonstrated higher reversible capacity and rate performance compared to batteries with 0.8 m to 4.0 m electrolytes [54]. In another study, Tong et al. [18] developed a 4.0 m LiFSI in tetramethylene sulfone (TMS) for DIBs (Fig. 9c). Compared to the dilute electrolyte, the high concentrated electrolyte shows high oxidation potential ≈ 6.0 V (Fig. 9d). The Li||graphite DIB retained 94.7% capacity after 1000 cycles at 200 mA·g−1 and exhibited discharge capacities of 110.2, 108.1, 106 and 103.6 mAh·g−1 at current densities of 200, 300, 400 and 500 mA·g−1, respectively, indicating good rate capabilities. High-concentration electrolytes ensure good electrochemical performance in DIBs but often suffer from degraded ionic conductivity due to increased viscosity. Recently, Han et al. proposed a binary electrolytes system of TMS:DMC (1:1 v/v) [128]. In this system, DMC and FSI− engaged in the K+-solvation structure, while TMS associates with FSI−, forming a stable interphase at the electrolyte/electrode surface and enhancing oxidation resistance for reversible FSI− intercalation. The combination of TMS’s high dielectric constant (ε = 43.4) and DMC’s low dielectric constant (ε = 3.1) results in a high solubility, low viscosity electrolyte. Consequently, a K-based dual-graphite DIB assembled with a 6.9 m electrolyte system achieved superior rate performance at 5 C and maintained around 100 mA·g−1 discharge capacity with approximately 90% retention over 400 cycles at 1 C. Additional research has focused on solvents containing electron-withdrawing groups, such as fluorine and chloride, to enhance the oxidation stability of electrolytes. For instance, the use of partially fluorinated carbonate solvents like methyl 2,2,2-trifluoroethyl carbonate (FEMC) has proven effective [129]. In zinc–graphite DIBs, FEMC has demonstrated a high Coulombic efficiency of 98.5% over 800 cycles at a 2C rate, with a robust discharge capacity of 156 mAh·g−1 [130]. This high performance is attributed to the formation of a stable, solvent-derived fluorinated CEI layer that protects the graphite surface and interlayer from exfoliation and over-expansion. Similarly, chlorine-substituted EMC (Cl-EMC) has been used in sodium-based dual-ion batteries [60]. Replacing a hydrogen atom in EMC with a chlorine atom significantly reduces the electron density on the oxygen atom, resulting in a lower HOMO level than EMC and helping to suppress electrolyte decomposition. Moreover, the electrolyte compatibility with both the cathode and anode is improved through the formation of Cl-containing interface layers. Consequently, Na-graphite dual-ion batteries utilizing Cl-EMC achieve a discharge capacity of 104.6 mAh·g−1 within a voltage range of 3.0–5.0 V, with virtually no capacity decay observed over 900 cycles at a 1C.
Electrolytes composed of pure salts and solvents often struggle to meet the high oxidative and reductive stability requirements for DIBs. Moreover, there are conflicting requirements for the electrolyte systems at the cathode and anode sides. Therefore, the development of suitable additives is crucial to improve the DIB’s electrochemical performance by stabilizing the electrolyte/electrode interface and limiting electrolyte decomposition during fast charging. For instance, the vinylene carbonate (VC) additive is commonly used in DIBs to provide a stable SEI on the electrodes, enhancing the cycling stability of DIBs [131]. Fluoroethylene carbonate (FEC) constructs a uniform cathode electrolyte, suppressing electrolyte decomposition and increasing CE at high current density [132, 133]. Another additive, lithium difluoro(oxalate)borate (LiDFOB), forms a thin LiF-rich SEI layer on the electrode [124]. LiDFOB has the lowest LUMO level and highest HOMO level among solvent components, leading to easier decomposition at both the cathode and anode (Fig. 9e). Adding LiDFOB to 2 M LiPF6 in FEMC maintains stable cycle performance for over 8000 cycles at 20 C (Fig. 9f). In addition, tetraethylammonium tetrafluoroborate (TEABF4) reduces undesirable irreversible capacity and gas formation in DIBs [134]. The TEA+ added electrolyte weakens Lewis acidity, decreasing irreversible capacity and gas formation at the LTO anode side.
Summary and Perspectives
Dual-ion batteries (DIBs) represent a significant advancement in energy storage technology, distinguished by their capacity to employ both anions and cations as charge carriers. This capability facilitates potentially ultrafast charging and higher energy densities, substantially enhancing battery efficiency and performance in applications such as electric vehicles and mobile devices. Furthermore, DIBs present a cost-effective and environmentally friendly alternative, leveraging materials that are more abundant and less detrimental than those used in LIBs [82]. However, the study of electrochemical reactions and the behavior of anions in DIBs is still nascent, highlighting the urgent need for the development of robust methods for fundamental anion analysis to address these challenges effectively. In this review, we have delineated the foundational principles, working mechanisms, and recent advancements in dual-ion batteries, with a particular emphasis on their rapid charging capabilities (Fig. 10).

Copyright 2020, Elsevier. Reproduced with permission of [91]. Copyright 2023, Wiley–VCH. Reproduced with permission of [93]. Copyright 2019, Wiley–VCH. Graphs for anode design. Reprinted with permission of [108]. Copyright 2019, Elsevier. Reproduced with permission of [110]. Copyright 2024, Wiley–VCH. Reproduced with permission of [112]. Copyright 2022, Elsevier. Graphs for electrolyte design. Reproduced with permission of [122]. Copyright 2019, Royal Society of Chemistry. Reprinted with permission [18]. Copyright 2021, Wiley–VCH. Reproduced with permission [124]. Copyright 2023, Elsevier
Overview of strategies for ultrafast-charging DIBs. Graphs for cathode design. Reproduced with permission [71].
Although considerable progress has been achieved in recent years, the current electrochemical performance of DIBs remains in its developmental stages. Due to the simultaneous use of both anions and cations from the electrolyte in DIBs, several challenges arise. At the cathode, the structural failure resulting from repeated anion storage and the phenomena of solvent co-intercalation must be addressed. At the anode, sluggish reaction kinetics require enhancement to align with the cathode materials and to rectify ion imbalances. Moreover, the decomposition of the electrolyte under high operating voltages presents a significant barrier that must be overcome. To realize the full potential of high-performance and ultrafast-charging DIBs, these issues necessitate focused research and innovative solutions.
-
(1)
Cathode Optimization Strategy
For cathodes, the capacity to store anions significantly influences both the voltage and capacity of batteries. However, commonly used cathode materials undergo considerable volume changes due to anion insertion. Therefore, it is essential to develop materials that feature interlayer engineering to adjust the spacing between layers, enhancing the movement and storage of anions. Moreover, the incorporation of chemical links, such as carboxylic anhydride functional groups between layers, can stabilize the crystal structure. Functional binders also play a crucial role in preserving the integrity of the cathode structure, effectively addressing issues like structural failure and the detrimental effects of solvent co-intercalation. In addition, the application of anion-inactive coatings can further enhance the structural stability and electrochemical performance of the cathode materials.
-
(2)
Anode Optimization Strategy
In the development of DIBs, anode design is essential for addressing key challenges such as limited capacity and slow reaction kinetics. To improve overall cell performance, strategies such as integrating conductive carbon materials, developing porous architectures, and crafting nanostructured anodes have been pivotal. Porous structures like omnidirectional porous aluminum wire help buffer volume changes and maintain electrical integrity, enhancing mechanical stability and ion diffusion. The incorporation of conductive carbons such as carbon nanotubes boost electronic conductivity and accelerates ion transfer, crucial for ultrafast charging. Nanostructured anodes, including Co3O4 nanosheets and aluminum carbon nanospheres, increase surface area for better ion interaction, support rapid ion diffusion, and stabilize the anode during cycling. These innovations are instrumental in optimizing anode performance, ensuring compatibility with cathode materials, and enhancing the safety and efficacy of DIBs.
-
(3)
Electrolyte Optimization Strategy
In dual-ion batteries, the electrolyte serves as the exclusive source of charge carriers, rendering the optimization of the electrolyte composition crucial for enhancing overall battery performance. This strategy involves a meticulous selection of solvents, salts, and additives, tailored to ensure that the electrolyte can withstand high operational voltages and maintain stable electrochemical performance. While anions with smaller ionic radii are generally preferred to enhance ion diffusion kinetics and minimize cathode material decomposition, this principle is not universally applicable. Consequently, employing an anion-hybridization strategy often proves effective in meeting the comprehensive performance requirements of the battery. Furthermore, high-concentration electrolytes are advantageous as they reduce the presence of free solvent molecules, thereby enhancing Coulombic efficiency and cycle stability, and augmenting the electrolyte’s capacity for ion intercalation. The strategic incorporation of specific additives, such as vinylene carbonate and fluoroethylene carbonate, plays a pivotal role in stabilizing the electrolyte/electrode interface and preventing decomposition under rapid charging conditions. Overall, this holistic approach not only optimizes the ionic conductivity and stability of the electrolyte but also adeptly addresses the dynamic operational demands of DIBs, ensuring an optimal balance between high performance and long-term durability.
-
(4)
Integrative Approaches for Full Cell Strategy
DIBs utilize both anions and cations as charge carriers, making the balance between kinetics and ion movement crucial in their development. These cells are prone to degradation mechanisms such as anion trapping or lithium cation trapping, which can lead to significant capacity loss, lithium metal plating, and associated safety concerns. The anode, in particular, tends to experience more side reactions, including electrolyte decomposition, SEI formation, and irreversible trapping by structural defects. In this context, an N/P ratio greater than 1 is often chosen to minimize the risk of lithium metal plating and irreversible trapping [115]. Furthermore, strategies often involve using active metals as anodes to replenish cations, addressing the depletion caused by repeated SEI formation and structural defects [135]. In addition, DIBs are characterized by fast anion intercalation kinetics at the cathode, which is beneficial for achieving high rate capabilities. To fully exploit this advantage, the rational design and optimization of anode materials with favorable kinetics are essential for developing DIBs with high rate performance.
Data availability
The data that support the findings of this study are available from the corresponding author on reasonable request.
References
J. Shin, Y. Kim, J.M. Lee, Korean J. Chem. Eng. 40, 1850 (2023)
S. Jayasubramaniyan, H.W. Lee, Korean J. Chem. Eng. 40, 488 (2023)
L. Kugler, Commun. ACM 62, 1 (2019)
J.B. Goodenough, K.S. Park, J. Am. Chem. Soc. 135, 1167 (2013)
G.E. Blomgren, J. Electrochem. Soc. 164, A5019 (2017)
S. Choi, J. Kang, J. Ryu, S. Park, Macromol. Res. 28, 1175 (2020)
S. Lee, A. Manthiram, ACS Energy Lett. 7, 3058 (2022)
B.E. Murdock, K.E. Toghill, N. Tapia-Ruiz, Adv. Energy Mater. 11, 2102028 (2021)
A. Tomaszewska, Z. Y. Chu, X. N. Feng, S. O'Kane, X. H. Liu, J. Y. Chen, C. Z. Ji, E. Endler, R. H. Li, L. S. Liu, Y. L. Li, S. Q. Zheng, S. Vetterlein, M. Gao, J. Y. Du, M. Parkes, M. G. Ouyang, M. Marinescu, G. Offer, B. Wu, Etransportation 1 (2019)
B. Wang, J. Ryu, S. Choi, X.H. Zhang, D. Pribat, X.L. Li, L.J. Zhi, S. Park, R.S. Ruoff, ACS Nano 13, 2307 (2019)
B. Park, S. Lee, D.Y. Han, H. Jang, D.G. Seong, J.K. Yoo, S. Park, Y. Oh, J. Ryu, Appl. Surf. Sci. 614, 156188 (2023)
F.W. Ming, H.F. Liang, G. Huang, Z. Bayhan, H.N. Alshareef, Adv. Mater. (2021). https://doi.org/10.1002/adma.202004039
Y.C. Lyu, X. Wu, K. Wang, Z.J. Feng, T. Cheng, Y. Liu, M. Wang, R.M. Chen, L.M. Xu, J.J. Zhou, Y.H. Lu, B.K. Guo, Adv. Energy Mater. (2021). https://doi.org/10.1002/aenm.202000982
S. Miyoshi, T. Akbay, T. Kurihara, T. Fukuda, A.T. Staykov, S. Ida, T. Ishihara, J. Phys. Chem. C 120, 22887 (2016)
X.L. Zhou, Q.R. Liu, C.L. Jiang, B.F. Ji, X.L. Ji, Y.B. Tang, H.M. Cheng, Angew. Chem. 59, 3802 (2020)
C.L. Jiang, L. Xiang, S.J. Miao, L. Shi, D.H. Xie, J.X. Yan, Z.J. Zheng, X.M. Zhang, Y.B. Tang, Adv. Mater. (2020). https://doi.org/10.1002/adma.202070135
C.Y. Wei, D.C. Gong, D.H. Xie, Y.B. Tang, ACS Energy Lett. 6, 4336 (2021)
X.Y. Tong, X.W. Ou, N.Z. Wu, H.Y. Wang, J. Li, Y.B. Tang, Adv. Energy Mater. (2021). https://doi.org/10.1002/aenm.202100151
A. Yu, Q.G. Pan, M. Zhang, D.H. Xie, Y.B. Tang, Adv. Funct. Mater. (2020). https://doi.org/10.1002/adfm.202001440
Y.M. Sui, C.F. Liu, R.C. Masse, Z.G. Neale, M. Atif, M. AlSalhi, G.Z. Cao, Energy Storage Mater. 25, 1 (2020)
M. Zhang, X.H. Song, X.W. Ou, Y.B. Tang, Energy Storage Mater. 16, 65 (2019)
M. Weiss, R. Ruess, J. Kasnatscheew, Y. Levartovsky, N.R. Levy, P. Minnmann, L. Stolz, T. Waldmann, M. Wohlfahrt-Mehrens, D. Aurbach, M. Winter, Y. Ein-Eli, J. Janek, Adv. Energy Mater. (2021). https://doi.org/10.1002/aenm.202101126
J. Ryu, D.Y. Han, D. Hong, S. Park, Energy Storage Mater. 45, 941 (2022)
H. Li, Joule 3, 911 (2019)
Y.X. Yao, N. Yao, X.R. Zhou, Z.H. Li, X.Y. Yue, C. Yan, Q. Zhang, Adv. Mater. (2022). https://doi.org/10.1002/adma.202206448
S. Zugmann, M. Fleischmann, M. Amereller, R.M. Gschwind, H.D. Wiemhöfer, H.J. Gores, Electrochim. Acta 56, 3926 (2011)
A. Mallarapu, V.S. Bharadwaj, S. Santhanagopalan, J. Mater. Chem. A 9, 4858 (2021)
A.J. Ringsby, K.D. Fong, J. Self, H.K. Bergstrom, B.D. McCloskey, K.A. Persson, J. Electrochem. Soc. 168, 080501 (2021)
D.L. Chao, H.J. Fan, Chem 5, 1359 (2019)
B. Tansel, Sep. Purif. Technol. 86, 119 (2012)
C. Capiglia, Y. Saito, H. Kageyama, P. Mustarelli, T. Iwamoto, T. Tabuchi, H. Tukamoto, J. Power. Sources 81, 859 (1999)
A.V. Cresce, M. Gobet, O. Borodin, J. Peng, S.M. Russell, E. Wikner, A. Fu, L.B. Hu, H.S. Lee, Z.C. Zhang, X.Q. Yang, S. Greenbaum, K. Amine, K. Xu, J. Phys. Chem. C 119, 27255 (2015)
Z.D. Huang, Y. Hou, T.R. Wang, Y.W. Zhao, G.J. Liang, X.L. Li, Y. Guo, Q. Yang, Z. Chen, Q. Li, L.T. Ma, J. Fan, C.Y. Zhi, Nat. Commun. (2021). https://doi.org/10.1038/s41467-021-23369-5
J. Holoubek, H.D. Liu, Z.H. Wu, Y.J. Yin, X. Xing, G.R. Cai, S.C. Yu, H.Y. Zhou, T.A. Pascal, Z. Chen, P. Liu, Nat. Energy 6, 303 (2021)
N. Zhang, T. Deng, S.Q. Zhang, C.H. Wang, L.X. Chen, C.S. Wang, X.L. Fan, Adv. Mater. (2022). https://doi.org/10.1002/adma.202107899
S.G. Jung, M.S. Rani, J. Bae, S.K. Kang, G.H. Park, J. Ji, W.B. Kim, Korean J. Chem. Eng. 41, 2029 (2024)
S. Ahmed, I. Bloom, A.N. Jansen, T. Tanim, E.J. Dufek, A. Pesaran, A. Burnham, R.B. Carlson, F. Dias, K. Hardy, M. Keyser, C. Kreuzer, A. Markel, A. Meintz, C. Michelbacher, M. Mohanpurkar, P.A. Nelson, D.C. Robertson, D. Scoffield, M. Shirk, T. Stephens, R. Vijayagopal, J.C. Zhang, J. Power. Sources 367, 250 (2017)
Z. Li, J. Huang, B.Y. Liaw, V. Metzler, J.B. Zhang, J. Power. Sources 254, 168 (2014)
J. Holoubek, Y.J. Yin, M.Q. Li, M.Y. Yu, Y.S. Meng, P. Liu, Z. Chen, Angew. Chem. 58, 18892 (2019)
J.W. Chen, Y. Peng, Y. Yin, Z. Fang, Y.J. Cao, Y.G. Wang, X.L. Dong, Y.Y. Xia, Angew. Chem. 60, 23858 (2021)
Q.Y. Dou, N.Z. Wu, H. Yuan, K.H. Shin, Y.B. Tang, D. Mitlin, H.S. Park, Chem. Soc. Rev. 50, 6734 (2021)
D.D. Yu, Q.N. Zhu, L.W. Cheng, S. Dong, X.H. Zhang, H. Wang, N.J. Yang, ACS Energy Lett. 6, 949 (2021)
D. D. Yu, K. X. Li, G. Y. Ma, F. Ru, X. K. Zhang, W. Luo, P. F. Hu, D. Chen, H. Wang, Chemsuschem (2023)
H. Jeong, L. Cheng, J. Power. Sources 602, 234227 (2024)
L.J. Jia, H.F. Hu, X.M. Cheng, H. Dong, H.H. Li, Y.Z. Zhang, H. Zhang, X.Y. Zhao, C.H. Li, J. Zhang, H.Z. Lin, J. Wang, Adv. Energy Mater. (2024). https://doi.org/10.1002/aenm.202470036
Z.D. Huang, X.L. Li, Z. Chen, P. Li, X.L. Ji, C.Y. Zhi, Nat. Rev. Chem. 7, 616 (2023)
X. Shi, J.H. Xie, J. Wang, S.L. Xie, Z.J. Yang, X.H. Lu, Nat. Commun. (2024). https://doi.org/10.1038/s41467-023-44615-y
C.H. Doh, B.S. Jin, J.H. Lim, S.I. Moon, Korean J. Chem. Eng. 19, 749 (2002)
Y.X. Chen, J. Key, K. O’Regan, T.F. Song, Y.S. Han, E. Kendrick, Chem. Eng. J. 450, 138275 (2022)
G.C. Guo, D. Wang, X.L. Wei, Q. Zhang, H. Liu, W.M. Lau, L.M. Liu, J. Phys. Chem. Lett. 6, 5002 (2015)
M. Zhu, Y. F. Huang, G. R. Chen, M. Lu, A. A. Nevar, N. Dudko, L. Y. Shi, L. Huang, N. V. Tarasenko, D. S. Zhang, Chem. Eng. J. 468 (2023).
X.F. Xu, K. Lin, D. Zhou, Q. Liu, X.Y. Qin, S.W. Wang, S. He, F.Y. Kang, B.H. Li, G.X. Wang, Chem 6, 902 (2020)
J.A. Read, A.V. Cresce, M.H. Ervin, K. Xu, Energy Environ. Sci. 7, 617 (2014)
L. Xiang, X.W. Ou, X.Y. Wang, Z.M. Zhou, X. Li, Y.B. Tang, Angew. Chem. 59, 17924 (2020)
R. Borah, F.R. Hughson, J. Johnston, T. Nann, Mater Today Adv 6, 100046 (2020)
T. Placke, M. Winter, Joule 3, 1184 (2019)
W. Rüdorff, U. Hofmann, Z. Anorg. Allg. Chem. 238, 1 (1938)
H. Zhang, Y. Yang, D.S. Ren, L. Wang, X.M. He, Energy Storage Mater. 36, 147 (2021)
C. Sole, N.E. Drewett, L.J. Hardwick, Farad. Disc. 172, 223 (2014)
Y. Lin, J. Shang, Y. Liu, Z. Wang, Z. Bai, X. Ou, Y. Tang, Adv. Mater. 36, e2402702 (2024)
I.A. Rodríguez-Pérez, Z.L. Jian, P.K. Waldenmaier, J.W. Palmisano, R.S. Chandrabose, X.F. Wang, M.M. Lerner, R.G. Carter, X.L. Ji, ACS Energy Lett. 1, 719 (2016)
M.L. Aubrey, J.R. Long, J. Am. Chem. Soc. 137, 13594 (2015)
M. Rajesh, F. Dolhem, C. Davoisne, M. Becuwe, Chemsuschem 13, 2364 (2020)
W. Tang, H.Y. He, J.Y. Shi, B. Cao, C.L. Yang, C. Fan, Org. Electron. 75, 105386 (2019)
S. Chen, T. Jia, G.Y. Zhou, C.J. Zhang, Q. Hou, Y.H. Wang, S.L. Luo, G. Shi, Y.H. Zeng, J. Electrochem. Soc. 166, A2543 (2019)
L. Fan, Q. Liu, Z. Xu, B.G. Lu, ACS Energy Lett. 2, 1614 (2017)
M.E. Speer, M. Kolek, J.J. Jassoy, J. Heine, M. Winter, P.M. Bieker, B. Esser, Chem. Commun. 51, 15261 (2015)
S.Y. Dong, Z.F. Li, I.A. Rodríguez-Pérez, H. Jiang, J. Lu, X.G. Zhang, X.L. Ji, Nano Energy 40, 233 (2017)
I.A. Rodríguez-Pérez, C. Bommier, D.D. Fuller, D.P. Leonard, A.G. Williams, X.L. Ji, ACS Appl. Mater. Interfaces 10, 43311 (2018)
S. Dühnen, R. Nölle, J. Wrogemann, M. Winter, T. Placke, J. Electrochem. Soc. 166, A5474 (2019)
C. Li, B.G. Lao, Z.K. Li, H.B. Yin, Z.Y. Yang, H.Y. Wang, D.L. Chen, X.L. Zhang, Y.J. Xu, C.H. Sun, Energy Storage Mater. 32, 1 (2020)
M.A. Reddy, M. Fichtner, J. Mater. Chem. 21, 17059 (2011)
R.G. Zhang, C. Ling, F. Mizuno, Chem. Commun. 51, 1487 (2015)
Z.Q. Cao, H.B. Hu, D. Ho, Adv. Funct. Mater. (2022). https://doi.org/10.1002/adfm.202111805
B. Li, Z.M. Nie, M. Vijayakumar, G.S. Li, J. Liu, V. Sprenkle, W. Wang, Nat. Commun. (2015). https://doi.org/10.1038/ncomms7303
L.J. Gao, Z.X. Li, Y.P. Zou, S.F. Yin, P. Peng, Y.Y. Shao, X. Liang, Iscience 23, 101348 (2020)
W. Lee, G. Park, D. Schröder, Y. Kwon, Korean J. Chem. Eng. 39, 1624 (2022)
C.Y. Yang, J. Chen, X. Ji, T.P. Pollard, X.J. Lü, C.J. Sun, S. Hou, Q. Liu, C.M. Liu, T.T. Qing, Y.Q. Wang, O. Borodin, Y. Ren, K. Xu, C.S. Wang, Nature 569, 245 (2019)
J. Duay, T.N. Lambert, M. Kelly, I. Pineda-Dominguez, J. Electrochem. Soc. 166, A687 (2019)
T.C. Gallagher, C.Y. Wu, M. Lucero, S.K. Sandstrom, L. Hagglund, H. Jiang, W. Stickle, Z.X. Feng, X.L. Ji, Angew. Chem. (2022). https://doi.org/10.1002/anie.202203837
H.W. Liu, C.Y. Chen, H. Yang, Y. Wang, L.L. Zou, Y.S. Wei, J.L. Jiang, J.C. Guo, W. Shi, Q. Xu, P. Cheng, Adv. Mater. (2020). https://doi.org/10.1002/adma.202004553
M. Zhang, W.Y. Zhang, F. Zhang, C.S. Lee, Y.B. Tang, Sci. China Chem. (2024)
X.Y. Wu, Y.K. Xu, C. Zhang, D.P. Leonard, A. Markir, J. Lu, X.L. Ji, J. Am. Chem. Soc. 141, 6338 (2019)
Y. Li, J. Zhang, Q.Y. Yuan, Y.J. Wu, J.B. Han, Adv. Mater. Interfaces (2022). https://doi.org/10.1002/admi.202200925
I. Cameán, B. Lobato, N. Cuesta, A.B. García, Batter. Supercaps (2024). https://doi.org/10.1002/batt.202300508
Q. Li, K.X. Ma, C. Hong, Z. Yang, C.Z. Qi, G.Z. Yang, C.X. Wang, Energy Storage Mater. 42, 715 (2021)
K.I. Kim, L.T. Tang, P. Mirabedini, A. Yokoi, J.M. Muratli, Q.B. Guo, M.M. Lerner, K. Gotoh, P.A. Greaney, C. Fang, X.L. Ji, Adv. Funct. Mater. (2022). https://doi.org/10.1002/adfm.202112709
K.V. Kraychyk, P. Bhauriyal, L. Piveteau, C.P. Guntlin, B. Pathak, M.V. Kovalenko, Nat. Commun. (2018). https://doi.org/10.1038/s41467-018-06923-6
J.A. Seel, J.R. Dahn, J. Electrochem. Soc. 147, 892 (2000)
T. Placke, G. Schmuelling, R. Kloepsch, P. Meister, O. Fromm, P. Hilbig, H.W. Meyer, M. Winter, Z. Anorg. Allg. Chem. 640, 1996 (2014)
J. Kang, S. Lee, J. Hwang, S. Kim, S. Lee, S. Yoo, J.W. Han, J.H. Ryu, J. Ryu, S. Park, Adv. Mater. (2023). https://doi.org/10.1002/adma.202306157
W.H. Li, Q.L. Ning, X.T. Xi, B.H. Hou, J.Z. Guo, Y. Yang, B. Chen, X.L. Wu, Adv. Mater. (2019). https://doi.org/10.1002/adma.201804766
X.Q. Han, G.J. Xu, Z.H. Zhang, X.F. Du, P.X. Han, X.H. Zhou, G.L. Cui, L.Q. Chen, Adv. Energy Mater. (2019). https://doi.org/10.1002/aenm.201804022
A.O. Yu, D.C. Gong, M. Zhang, Y.B. Tang, Chem. Eng. J. 401, 125834 (2020)
J.J. Peng, N.Q. Chen, R. He, Z.Y. Wang, S. Dai, X.B. Jin, Angew. Chem. 56, 1751 (2017)
M. W. Zhang, Y. Pei, W. W. Liu, R. L. Liang, Y. P. Deng, Z. W. Chen, A. P. Yu, Nano Energy 81 (2021)
J. Kang, D.Y. Han, S. Kim, J. Ryu, S. Park, Adv. Mater. 35 (2023)
J.N. Liang, D.C. Chen, K. Adair, Q. Sun, N.G. Holmes, Y. Zhao, Y.P. Sun, J. Luo, R.Y. Li, L. Zhang, S.Q. Zhao, S.G. Lu, H. Huang, X.X. Zhang, C.V. Singh, X.L. Sun, Adv. Energy Mater. 11 (2021)
A. Kotronia, W.W.A. van Ekeren, H.D. Asfaw, K. Edström, Electrochem. Commun. 146 (2023)
J. Kang, J.Y. Kwon, D.Y. Han, S. Park, J. Ryu, Appl. Phys. Rev. 11 (2024)
G. Wang, M.H. Yu, J.G. Wang, D.B. Li, D.M. Tan, M. Löffler, X.D. Zhuang, K. Müllen, X.L. Feng, Adv. Mater. 30 (2018)
I. Kovalenko, B. Zdyrko, A. Magasinski, B. Hertzberg, Z. Milicev, R. Burtovyy, I. Luzinov, G. Yushin, Science 334, 75 (2011)
J.Y. Kwon, J. Lyu, E. Kim, H. Park, J.E. Jeong, J.C. Kim, J. Ryu, J. Mater. Chem. A 12, 15996 (2024)
H. Wang, J. Ryu, Y.Y. Shao, V. Murugesan, K. Persson, K. Zavadil, K.T. Mueller, J. Liu, ChemElectroChem 8, 3013 (2021)
W.H. Li, Y.M. Li, X.F. Liu, Z.Y. Gu, H.J. Liang, X.X. Zhao, J.Z. Guo, X.L. Wu, Adv. Funct. Mater. 32 (2022)
H.G. Wang, Q. Li, Q. Wu, Z.J. Si, X.L. Lv, X.T. Liang, H.L. Wang, L. Sun, W.D. Shi, S.Y. Song, Adv. Energy Mater. (2021). https://doi.org/10.1002/aenm.202100381
S. Sarkar, T.K. Dutta, B.P. Mandal, A. Patra, Chem. Commun. 60, 5010–5013 (2024)
C.H. Song, Y.P. Li, H. Li, T. He, Q. Guan, J. Yang, X.L. Li, J.L. Cheng, B. Wang, Nano Energy 60, 285 (2019)
X.F. Tong, F. Zhang, B.F. Ji, M.H. Sheng, Y.B. Tang, Adv. Mater. 28, 9979 (2016)
Y.Q. Su, J. Shang, X.C. Liu, J. Li, Q.G. Pan, Y.B. Tang, Angew. Chem. (2024)
Y.J. Liu, X. Hu, J.W. Li, G.B. Zhong, J. Yuan, H.B. Zhan, Y.B. Tang, Z.H. Wen, Nat. Commun. (2022). https://doi.org/10.1038/s41467-022-28176-0
H.Z. Wu, L. Li, W.H. Yuan, Chem. Eng. J. 442, 136259 (2022)
J. Ryu, T.W. Chen, T. Bok, G. Song, J. Ma, C. Hwang, L.L. Luo, H.K. Song, J. Cho, C.M. Wang, S.L. Zhang, S. Park, Nat. Commun. (2018). https://doi.org/10.1038/s41467-018-05398-9
Lee, H., Kang, S. O. and An, G. H., Korean J. Chem. Eng. (2024).
J.M. Wrogemann, L. Haneke, T. Ramireddy, J.E. Frerichs, I. Sultana, Y.I. Chen, F. Brink, M.R. Hansen, M. Winter, A.M. Glushenkov, T. Placke, Adv. Sci. (2022). https://doi.org/10.1002/advs.202201116
J. Ryu, D. Hong, M. Shin, S. Park, ACS Nano 10, 10589 (2016)
Y.J. Rho, Y.J. Yoo, W.H. Ryu, Korean J. Chem. Eng. 40, 461 (2023)
L. Sui, X.Y. Shi, T. Deng, H. Yang, H. Liu, H. Chen, W. Zhang, W.T. Zheng, J. Energy Chem. 37, 7 (2019)
X.F. Tong, F. Zhang, G.H. Chen, X.Y. Liu, L. Gu, Y.B. Tang, Adv. Energy Mater. (2018). https://doi.org/10.1002/aenm.201701967
P. Meister, V. Siozios, J. Reiter, S. Klamor, S. Rothermel, O. Fromm, H.W. Meyer, M. Winter, T. Placke, Electrochim. Acta 130, 625 (2014)
K. Beltrop, P. Meister, S. Klein, A. Heckmann, M. Grünebaum, H.D. Wiemhöfer, M. Winter, T. Placke, Electrochim. Acta 209, 44 (2016)
K. Shin, F. Zhang, X.W. Ou, N.Z. Wu, C.S. Lee, Y.B. Tang, J. Mater. Chem. A 7, 10930 (2019)
A. Heckmann, J. Thienenkamp, K. Beltrop, M. Winter, G. Brunklaus, T. Placke, Electrochim. Acta 260, 514 (2018)
H.K. Zhu, S.Y. Dong, Y. Zhao, L. Pui-Kit, D.Y.W. Yu, J. Power Sources 592 (2024)
J. Li, K. Hui, D.A. Dinh, S. Wu, X. Fan, F. Chen, K. Hui, Mater. Today Sustain. 19, 100188 (2022)
M.H. Sheng, F. Zhang, B.F. Ji, X.F. Tong, Y.B. Tang, Adv. Energy Mater. 7 (2017)
L.S. Kremer, T. Danner, S. Hein, A. Hoffmann, B. Prifling, V. Schmidt, A. Latz, M. Wohlfahrt-Mehrens, Batter. Supercaps 3, 1172 (2020)
C.J. Han, H.Y. Wang, Z.L. Wang, X.W. Ou, Y.B. Tang, Adv. Mater. (2023). https://doi.org/10.1002/adma.202300917
M.C. Smart, B.V. Ratnakumar, V.S. Ryan-Mowrey, S. Surampudi, G.K.S. Prakash, J. Hub, I. Cheung, J. Power. Sources 119, 359 (2003)
S.W. Tao, B. Demir, A. Baktash, Y.T. Zhu, Q.B. Xia, Y.L. Jiao, Y.Y. Zhao, T.E. Lin, M. Li, M. Lyu, I. Gentle, L.Z. Wang, R. Knibbe, Angew. Chem. (2023). https://doi.org/10.1002/anie.202307208
X.L. Zhang, Y.B. Tang, F. Zhang, C.S. Lee, Adv. Energy Mater. (2016). https://doi.org/10.1002/aenm.201670064
Y. Wang, Y.J. Zhang, Q.H. Duan, P.K. Lee, S. Wang, D.Y.W. Yu, J. Power. Sources 471, 228466 (2020)
F. Zhang, B. Ji, X.F. Tong, M.H. Sheng, X.L. Zhang, C.S. Lee, Y.B. Tang, Adv. Mater. Interfaces (2016). https://doi.org/10.1002/admi.201670112
R. Nozu, E. Suzuki, O. Kimura, N. Onagi, T. Ishihara, Electrochim. Acta 337, 135711 (2020)
Z.J. Cheng, Q. Yao, C. Zheng, F.L. Zhang, K.P. Song, Q.Y. Dong, J. Pan, J. Yang, Adv. Funct. Mater. (2024). https://doi.org/10.1002/adfm.202310449
Acknowledgements
This work was supported by the Research Fund of Korea Institute of Materials Science (PNK9720) and Korea Toray Science Foundation.
Funding
Korea Institute of Materials Science, PNK9720, Jaegeon Ryu, Korea Toray Science Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Heo, J.J., Ryu, J. A Critical Review of Ultrafast Charging Dual-Ion Batteries. Korean J. Chem. Eng. (2024). https://doi.org/10.1007/s11814-024-00255-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11814-024-00255-6