
Abstract
As bis(4-hydroxyphenyl)methanone (BHP) is one of the most common UV light stabilizers (UVLS), but exhibits endocrine disrupting toxicity, this study aims to develop useful sulfate radical-based techniques to eliminate BHP from water by activating peroxymonosulfate (PMS). Hence, while cobalt (Co) exhibits efficacy as a transition metal for the activation of PMS, the utilization of manganese/cobalt (Mn/Co) bimetallic oxides presents an even more encouraging prospect as heterogeneous catalysts for PMS activation. In this study, we have successfully produced N-doped carbon-supported Mn/Co nanoparticles (NCMC) with a distinctive hollow-engineered nanostructure. The synthesis involved the utilization of Co-MOF as a precursor, followed by easy etching and Mn doping to achieve the desired composition of Mn/Co bimetallic oxide nanoparticles. The inclusion of Mn dopant facilitates the integration of Mn/Co nanoparticles into the hollow-structured N-doped carbon matrix. NCMC demonstrates much higher activity compared to NCC and the benchmark catalyst, Co3O4 NP, in terms of PMS activation for the degradation of BHP. The findings of the eco-toxicity study indicate that the degradation of BHP by NCMC + PMS does not yield hazardous or extremely toxic byproducts, so establishing NCMC as a potentially effective heterogeneous catalyst for activating PMS in the degradation of BHP.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Bis(4-hydroxyphenyl)methanone (BHP) is a benzophenone molecule which has attracted considerable interest due to its wide range of applications and frequent occurrence in various aquatic environments [1]. Nevertheless, there exists very few investigations pertaining to the elimination of BHP from aqueous solutions [2, 3]. The primary aim of this study is to develop efficient techniques for the decomposition of BHP in aqueous solutions and elucidate the degradation process of this UV light stabilizer.
In conventional practice, Advanced Oxidation Processes (AOPs) are utilized to generate reactive oxygen species (ROS) possessing high oxidation potentials, hence facilitating the oxidation process of pollutants. There is an increasing focus on AOPs that utilize sulfate radicals (SO4·–) because of the high oxidation potential of the sulfate radical anion (SO4·–) (i.e., 2.5–3.1 V). Currently, there exists a broad range of commercial precursors of SO4·–. This involves the utilization of peroxymonosulfate (PMS), which requires activation in order to expeditiously release SO4·–.
The activation of PMS can be accomplished by a range of techniques, including heat, light radiation, ultrasound, and catalysts [4]. The employment of heterogeneous catalysts is largely acknowledged as the most promising strategy for activating PMS among the several approaches available. Transition metals, including cobalt (Co), iron (Fe), manganese (Mn), and nickel (Ni), have demonstrated notable efficacy in the activation of PMS [5]. Recent studies have shown that the combination of Co with other metals in bimetallic composites might have synergistic effects, resulting in improved catalytic activity [6, 7]. Due to its high abundance in the Earth’s crust, Mn has garnered significant attention as a potential catalyst for activating PMS. In particular, bimetallic oxides containing both Mn and Co have emerged as viable candidates for this purpose [8,9,10,11,12]. For instance, Dung et al. [13] documented the synthesis of cobalt manganese oxide (CMO) nanoparticles (NPs) as catalysts to activate PMS for the degradation of organic dyes.
Hence, the immobilization or embedding of CMO or Co/Mn NPs onto substrates is a viable approach, particularly owing to the favorable characteristics of carbon materials in terms of availability and stability. Consequently, carbonaceous substrates have been widely employed for the support and embedding of Co/Mn NPs [14]. In addition, carbon substrates have the potential to be doped with hetero-atoms, such as nitrogen, in order to enhance electron transport and catalytic activity through the provision of additional reactive sites [15]. Hence, the development of a hybrid catalyst for the incorporation of CMO NPs into N-doped carbon substrates holds great potential.
In addition, the utilization of templates including specific constituents, namely metals and organics, subsequent to direct carbonization, presents a very favorable approach for the production of such a hybrid catalyst. Metal–organic frameworks (MOFs) have garnered significant attention due to their intricate structures and incorporation of metal/organic components [16]. Among these MOFs, zeolitic Co-MOF stands out as a particularly promising template, primarily due to the ligand of imidazolate, which may be converted into N-doped carbon [17]. The introduction of Mn into cobalt metal–organic frameworks (Co-MOFs) would result in the formation of manganese/cobalt nanoparticles (NPs) following thermal treatments [18].
Hence, in this study, a straightforward and practical method is proposed for the fabrication of a hollow structure in Co-MOF by selectively removing the inner region, resulting in the formation of a box-shaped hollow morphology with thin layers. Upon the introduction of Mn onto the “nanobox”, subsequent heat treatment would result in a thin layer of N-doped carbon-supported Mn/Co (NCMC). The degradation of BHP utilizing PMS activated by NCMC would be thoroughly examined in order to determine the catalytic activities of NCMC.
Experimental
The process of fabricating NCMC was executed based on the illustration presented in Fig. 1. The supporting information contains more details of the synthesis process, degradation of BHP by PMS, analytical techniques, and computational chemistry.

Preparation scheme for NCMC
In order to quantify the degradation kinetics, the utilization of the pseudo-first-order rate law would be applied to determine the observed rate constant, denoted as k [19, 20].
Results and Discussion
Properties of NCMC
Morphologies and Compositional Analyses
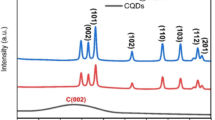
The morphology of the zeolitic Co-MOF was firstly investigated as NCMC was generated from the Co-MOF. In Fig. 2a, the typical cubic morphology of the zeolitic Co-MOF can be well-characterized by its distinct faces and well-defined edges. The transmission picture of the Co-MOF (Fig. 2b) confirms that the original Co-MOF structure had a cubic shape and a solid texture. Figure 3a provides additional evidence that the X-ray diffraction (XRD) pattern of the pristine zeolitic Co-MOF was well-matched to the reported pattern [21, 22], confirming the successful formation of Co-MOF.

Electronic microscopic images of a, b solid Co-MOF; c–e NCMC

a XRD patterns; b N2 sorption isotherms, c pore size distributions of NCMC and NCC; d saturation magnetization of NCMC
After the post treatment with tannic acid and doping with Mn2+, followed by carbonization, the resulting product (Fig. 2c) maintained its cubic shape. However, the faces of the resultant product displayed noticeable roughening and were adorned with a significant number of NPs. More importantly, the initial solid configuration was transformed into a hollow configuration characterized by exceedingly thin outer layers, as evidenced by the observed fracture. The transmission picture (Fig. 2d) provides further confirmation that the solid texture of the pristine Co-MOF had undergone a transformation into a hollow texture.
The presence of tannic acid, which includes several hydroxyl groups bonded by single bonds, facilitates the formation of a structure. This is due to the release of free protons (H+) from tannic acid, disrupting the Co–N bonds present in the Co-MOF. Simultaneously, the formation of fresh N–H bonds and the coordination of a Co-tannic acid complex would occur, resulting in the transformation of the Co-MOF coordination framework into a network of coordinated Co2+ ions and tannic acid. The coordination compounds of Co-tannic acid would thereafter undergo formulation and accumulation on the surface, resulting in the formation of shells. Consequently, the formation of the hollow structure is achieved by a continuous process of etching and re-coordination. Following the doping of Mn2+, the subsequent adsorption and deposition of Mn2+ ions onto the Co-tannic acid coordinated shells would be achieved.
Moreover, the closer-view in Fig. 2d further reveals that the outer layers of the resultant product was certainly decorated by numerous NPs. Its corresponding XRD pattern was then measured in Fig. 3a, which exhibited a different pattern, demonstrating that the Co-MOF had been transferred to another material through the etching, doping and carbonization. A few noticeable peaks can be noted on 17.9°, 31.8°, 36.5°, 38.8°, 41.6°, 47.6°, 49.2°, 53.4°, and 59.1°, ascribed to the formation of mixed metal spinel of cobalt manganese oxide (CMO), (Mn,Co)(Mn,Co)2O4, according to JCPDS PDF #18-0408 and #18-0410 [23]. While the thermal treatment was implemented in N2 atmosphere, the presence of NO3− from both Co(NO3)2 and Mn(NO3)2 might act as an oxidant to convert these metal species into metal oxides [24]. Nevertheless, due to the limited content of NO3−, partial Co from the Co-MOF was transferred to the metallic Co rather than cobalt oxides as several peaks of Co0 can be observed at 44.2°, 51.6°, and 75.8° based on JCPDS PDF #15-0806. Moreover, the organic ligand of Co-MOF was also carbonized to produce a significant amount of carbon nitride (CN) at 15.9° (JCPDS PDF #50-0664), whereas a considerable fraction of “carbon” was also observed at 26.0°.
In view of these XRD patterns, Co-MOF had been changed to a hybrid material comprised of carbon, N-doped carbon, Co0 and, more importantly, CMO NPs, formulating a composite of N-doped carbon-supported Mn/Co (NCMC). Figure 4 further displays the mapping analysis of NCMC nanocubes, which reveals the uniform distribution of C, O, Mn, Co and N all over these NCMC nanocubes. This validates that the etching, and Mn doping were homogeneously applied on Co-MOF nanocubes during the preparation process.

a–f mapping analysis of NCMC
On the other hand, an analogue to NCMC was also prepared without the doping of Mn2+. As shown in Fig. S1 (please see the supporting information), even without the Mn2+ doping, the etching and carbonization treatment had also successfully converted Co-MOF to the hollow-structured material. The corresponding transmission image further confirmed unraveled that the faces of the resulting product (without the Mn2+ doping) also contained many fine NPs on its surfaces but the sizes of these NPs seemed much smaller. Figure 3a also displays its XRD pattern which was also different than Co-MOF with a series of notable peaks at 26.3°, 44.2°, 51.6° and 76.1°. Specifically, the signal on 26.3° could be attributed to carbon owing to carbonization of organic molecules, while other peaks were ascribed to Co0 (JCPDS Card# 15-0806), formulating the N-doped carbon-supported Co (NCC).
These comparisons further reveal that NCMC seemed to contain the relatively large NPs in contrast to NCC possibly because the Mn2+ was doped and inserted into the coordination and caused disturbance to the Co-coordinated structure.
Physical and Chemical Properties
As NCMC exhibited a unique hollow structure with the composition of NPs, its textural property was then further analyzed in Fig. 3b, and the N2 sorption curve could be classified as a type IV adsorption accompanied by a noticeable hysteresis, suggesting that NCMC was porous, especially mesoporous. The large hysteresis also suggested that the pore structures in NCMC was more complex possibly because differently-sized NPs of Mn/Co NPs were embedded into the carbon matrix, creating variously-sized pores. The presence of mesopores was confirmed by its pore distribution (Fig. 3c). The relatively high N2 sorption also enabled NCMC to show a high surface area as 187 m2/g and its pore volume was determined as 0.20 cc/g.
Figure 3c also reveals the N2 sorption of NCC, which was actually similar to that of NCMC but with a much smaller N2 sorption volume. Therefore, NCC merely showed a surface area of 26 m2/g and the total pore volume of NCC volume was merely 0.045 cc/g. The relatively superior textural properties in NCMC were possibly owing to the fact that NCMC exhibited much more coarse faces and the surficial textures were obviously more porous according to its TEM image (Fig. 2d) in comparison to that of NCC (Fig. S1b). In addition, NPs formed on NCMC surfaces were multiple-sized and these clusters of hetero-sized NPs might create more and different pores as validated in the pore size distribution (Fig. 3c).
On the other hand, as NCMC consisted of metallic Co, it shall exhibit magnetic properties and hence it would be interesting to examine the magnetism of NCMC. In Fig. 3d, NCMC exhibited a significantly high magnetization, and its saturation magnetization reached 64 emu/g, which enabled NCMC to be easily controllable for uniform dispersion and quick collection as shown in the inset.
In this study, X-ray photoelectron spectroscopy (XPS) examinations were conducted on NCMC and NCC samples, as seen in Fig. 5. The C1s curve of NCMC (Fig. 5a) was seen to exhibit numerous bands at energy levels of 284.0 eV, 286.6 eV, and 288.5 eV, that may be attributed to the C–C, C–N, and C=O functional groups, respectively [25]. Moreover, the Co2p band of NCMC may also be acquired and then resolved into several bands (Fig. 5b). The band seen at 778.8 eV might be ascribed to the presence of Co0, whereas the bands at 780.0 and 795.6 eV were associated with Co3+ species [26]. Additionally, the bands at 782.0 and 798.6 eV can be assigned to Co2+ species.

a–d XPS analyses of NCMC; fractions of N species in e NCMC and f NCC
Moreover, the Mn2p curve (Fig. 5c) would be examined in order to illustrate the presence of several bands. The spectral peaks seen at 641.0 and 652.6 eV were attributed to the presence of Mn2+ ions, whereas the peaks at 642.3 and 654.0 eV were potentially indicative of Mn3+ ions. Additionally, it has been suggested that the bands seen at 645.2 and 655.4 eV might be attributed to the presence of Mn4+ [27]. Furthermore, the N1s present in NCMC would be depicted in the figure. Multiple bands at energy levels of 398.3, 399.3, and 400.5 eV were noted, corresponding to the presence of pyridinic N, pyrrolic N, and quaternary N [28].
The XPS examination of NCC was also conducted, as seen in Fig. S2. Its C1s band was subjected to analysis, which resulted in the identification of several bands associated with C–C, C–N, and C=O bonds [25]. Furthermore, the band seen at 778.5 eV was determined to originate from Co0, whereas the bands at 780.2 and 796.3 eV were shown to correspond to Co2+ species [26]. The N1s spectrum exhibited many peaks at energy levels of 398.3, 399.3, 400.5, and 402.5 eV, corresponding to the presence of pyridinic N, pyrrolic N, quaternary N, and N–O group, respectively [28].
In addition to conducting XPS examinations, Raman spectroscopy was also employed to examine the chemical distinctions between NCMC and NCC by elucidating their structural fingerprints. The Raman spectra of NCMC and NCC (Fig. 6a), exhibit comparable patterns characterized by prominent peaks at 192, 474, and 680 cm−1. These peaks can be ascribed to the F2g, Eg, and A1g vibration modes in the Co metal species [29]. The obtained data provide confirmation that both NCMC and NCC contained Co.

a Full-survey Raman spectra of NCMC and NCC; b, c, regional Raman spectra of NCMC and NCC; and d Fractions of G and D bands in NCMC and NCC
Degradation of BHP by NCMC + PMS System
The evaluation of BHP elimination using PMS activated by NCMC is depicted in Fig. 7. However, in light of the potential removal of BHP by adsorption, an investigation was conducted to examine the adsorption of BHP onto NCMC. In Fig. 7a, the concentration of BHP did not exhibit significant changes in the presence of NCMC, indicating that the adsorption process was ineffective in removing BHP. On the other hand, the usage of PMS alone resulted in a just minor reduction of BHP in 30 min, indicating that PMS without activation could not effectively destroy BHP.

a Comparison of degradation of BHP and b corresponding rate constants of BHP degradation; c comparison of PMS consumption and d rate constants of PMS consumption (Catalyst = 200 mg/L, PMS = 200 mg/L,T = 30 °C)
Nevertheless, the combination of NCMC and PMS resulted in a rapid and near-complete elimination of BHP in 30 min. This observation implies that NCMC has the potential to activate PMS and effectively eradicate BHP in aqueous solutions and possible synergy might exist between NCMC and PMS [5, 6]. In order to establish a comparison, commercial Co3O4 NP (as shown in Fig. S3) and NCC were also employed for activating PMS in order to eliminate BHP. In Fig. 7a, the Co3O4 nanoparticles combined with PMS resulted in a Ct/C0 value of 0.38 (equivalent to the removal of 62% of BHP) in 30 min. On the other hand, the combination of NCC and PMS achieved a Ct/C0 value of 0.2 (equivalent to the removal of 80% of the target compound) in 30 min. However, the degradation efficiencies of Co3O4 NP and NCC for BHP were much lower compared to that of NCMC. On the other hand, the variation of total organic carbon (TOC) concentration of BHP by using Co3O4, NCC and NCMC were then measured in Fig. S4. NCMC was validated to decompose BHP and cause the mineralization of BHP and reduction in TOC. In comparison to NCC and Co3O4, NCMC also enabled a much higher extent of TOC removal, indicating the much superior catalytic capability of NCMC.
In order to conduct more quantitative comparisons for these catalysts in degrading BHP, the pseudo first-order rate law was then employed. The resulting rate constants for BHP degradation were then compiled and presented in Fig. 7b. The rate constant (k) obtained from the NCMC + PMS reaction was determined to be 0.678 min−1, which is much higher than the rate constants observed for Co3O4 NP + PMS (0.047 min−1) and NCC + PMS (0.097 min−1). This finding provides further evidence of the superiority of NCMC over both Co3O4 NP and NCC in terms of reaction rates.
Given the significantly accelerated degradation of BHP by NCMC, it would be of interest to investigate into the consumption of PMS by NCMC in compared to other catalysts. Therefore, the variation in PMS concentration during BHP degradation was further examined in Fig. 7c. During the degradation of BHP, the concentration of PMS reduced significantly in the presence of NCMC. Approximately 75% of the PMS had undergone decomposition by NCMC in 30 min. In contrast, the consumption of PMS during the degradation of BHP by NCC was significantly lower, approximately 60%, and exhibited a slower rate. The rate constant obtained for PMS consumption by NCMC was found to be 0.196 min−1, which was significantly higher than the rate constants obtained for NCC (0.044 min−1) and Co3O4 NP (0.026 min−1). This observation validates that the consumption of PMS by NCMC was more effective and faster compared to NCC and Co3O4 NP, thereby enabling a faster degradation of BHP. The restricted catalytic efficiency of NCC and commercially available Co3O4 can be attributed to their diminished surface area resulting from significant aggregation, as seen in Fig. S3. Therefore, the quantity of active sites that are accessible for catalytic reactions was decreased.
Furthermore, the activation of PMS is attributed to the surface reactions of catalysts with PMS. Therefore, the surface chemistry of catalysts is of significant importance in the activation of PMS [9,10,11,12]. Although both NCMC and NCC included Co species, NCMC was distinguished by the presence of Mn2+, Mn3+, and Mn4+ ions, which provided extra reactive sites for the activation of PMS [9,10,11,12, 30]. Prior researches have indicated that the utilization of dual or multiple-metallic compositions might result in an increased number of reactive sites for facilitating PMS activation, thus leading to enhanced catalytic activity [12, 30]. NCMC had distinct proportions of nitrogenous species, with pyridinic N, pyrrolic N, and quaternary N contributing 29%, 50%, and 21%, respectively. In contrast, NCC consisted of pyridinic N, pyrrolic N, and quaternary N in proportions of 48%, 20%, and 22%, respectively. The activation of PMS has been confirmed to occur at both pyridinic N and pyrrolic N sites [31]. It was observed that the total proportion of pyridinic N and pyrrolic N in NCMC (79%) was substantially higher compared to that in NCC (68%). The presence of significantly higher proportions of pyridinic and pyrrolic nitrogen fractions in NCMC was anticipated to contribute to an increased number of active sites in NCMC compared to NCC. Consequently, NCMC exhibited enhanced catalytic activity in the activation of PMS during BHP degradation.
Moreover, Fig. 7b shows that the A1g band of NCC was located at 673 cm−1, however in CFMC, the center of this band had shifted from 673 to 648 cm−1. The band shift, identical to the one seen in the case of F2g, is also evident in Fig. 7c. The band center of NCC was seen at 188 cm−1, whereas the F2g band center of NCMC exhibited a downward shift to 182 cm−1. The observed shifts in A1g and F2g can be attributed to the coordination of cobalt species, indicating the presence of defect sites or oxygen vacancies in NCMC [32]. The presence of defect sites or oxygen vacancies had the potential to enhance the ability of NCMC to uncover more catalytically active surfaces [32], which might potentially lead to improvements in the catalytic activities of PMS activation.
Furthermore, it is worth noting that two further spectral peaks were seen at wavenumbers of 1331 cm−1 and 1587 cm−1, which were attributed to the D and G bands associated with the carbon structures present in NCMC and NCC. The magnitudes of the D and G bands serve as indicators of the levels of defect and graphitization in catalysts [33, 34]. The carbonaceous matrix seen in NCMC (Fig. 7d) exhibited a greater level of defectiveness compared to the carbonaceous matrix in NCC. The presence of carbonaceous defects has been identified as potential active sites for activating PMS. It is probable that the increased proportion of the D band in NCMC is also responsible for facilitating greater catalytic activity in NCMC [35].
Furthermore, the activation of PMS would also involve redox processes of both PMS and catalysts. Consequently, the electrochemical characteristics of catalysts play a significant role in controlling PMS activation [9,10,11,12]. Given the much higher activity of NCMC compared to NCC in the degradation of BHP, it was informative to investigate the electrochemical distinctions between NCMC and NCC.
Firstly, the results of cyclic voltammetry (CV) analyses for NCMC and NCC are presented in Fig. 8a. NCC exhibited a relatively small CV curve in this potential range, with less prominent redox peaks. On the other hand, NCMC demonstrated a bigger CV curve, displaying distinct redox peaks. The specific capacitance of NCMC was then calculated as 53.7 F/g, which was much greater than 14.1 F/g in NCC. This comparison confirms that NCMC possessed superior redox characteristics and was expected to facilitate quicker interfacial processes compared to NCC [36]. Additionally, Fig. 8b illustrates the determination of the linear sweep voltammogram (LSV) curves of NCMC. At a current level of 1 mA, the NCMC system required a starting potential of 0.48 V, but the NCC system necessitated a higher starting potential of 0.83 V. This discrepancy suggests that electrical transportation in NCMC was comparatively more efficient than in NCC. Furthermore, an analysis of the charge transfers in both catalysts was conducted by examining their Nyquist plots (Fig. 8c). When comparing NCC and NCMC, it was observed that NCMC exhibited a significantly narrower semi-circle in the high-frequency area. This suggests that NCMC possessed a higher degree of charge transfer and lower resistivity compared to NCC [37]. The results of these tests indicate that NCMC, with its superior porosity characteristics and more active surfaces, exhibited more favorable electrochemical behaviors. Consequently, NCMC had much greater catalytic activity for activating PMS and degrading BHP.

Electrochemical properties of NCMC and NCC: a CV curves, b LSV curves, c EIS Nyquist plots
Other Factors on Degradation of BHP by NCMC + PMS
Given the capability of NCMC + PMS to effectively remove BHP from water, it was imperative to explore additional elements that influenced the degradation of BHP, including temperature and pH. Figure 9a illustrates the results of BHP degradation by NCMC + PMS at temperatures ranging from 30 to 50 °C. At a temperature of 40 °C, the rate constant for the removal of BHP.

Effects of various factors on BHP degradation by NCMC + PMS: a temperature (Catalyst = 200 mg/L, PMS = 200 mg/L, pH = 7), b, c pH and corresponding rate constants, d, e scavengers and corresponding rate constants; f recyclability (Catalyst = 200 mg/L, PMS = 200 mg/L,T = 30 °C)
increased from 0.678 min−1 at 30 °C to 0.909 min−1, resulting in a significantly accelerated reaction rate. Upon reaching a temperature of 50 °C, the degradation of BHP accelerated at an increased rate, resulting in an elevated k value of 1.248 min−1. This observation suggests that higher temperatures had a favorable impact on the removal of BHP, due to enhanced mass transfer at elevated temperatures. The occurrence of this phenomenon may be attributed to the fact that elevated temperatures often augment the reaction kinetics by supplying additional energy to the molecules involved in the reaction. This results in a greater frequency of successful collisions and an enhanced production of reactive species.
In order to conduct additional comparisons between NCMC and other processes and methodologies for BHP degradation, the activation energy (Ea) of BHP was also determined by using the Arrhenius equation, where the k values at different temperatures were modeled according to the following law:
The calculated activation energy (Ea) for the reaction was found to be 24.8 kJ/mol, which was much lower than the Ea value (i.e., 68 kJ/mol) reported for the degradation of BHP [2, 3]. This result suggests that NCMC appeared as a promising alternative activator for PMS in the removal of BHP.
Figure 9b further illustrates the investigation of the impact of pH on the removal of BHP by NCMC + PMS. At pH 7, which represents a neutral environment, the quick removal of BHP with k = 0.678 min−1 was achieved. In a more acidic situation with pH 5, a significantly slower degradation of BHP was observed, as evidenced by a decrease in the k to 0.297 min−1. The degradation of BHP was even slower with a reduced rate constant (k = 0.257 min−1) when the starting state was more acidic, specifically at pH 3. The observed negative impact in the acidic state may be attributed to the presence of a relatively large concentration of H+ ions in the acidic environment, which can potentially interact with both SO4·‒ and ·OH radicals, leading to their neutralization as described by the following equations [38,39,40]:
Consequently, the acidic environment would result in a reduced presence of radicals during the degradation process of BHP, thereby leading to a decrease in the efficiency of BHP degradation.
Furthermore, it has been observed that the presence of an acidic environment might result in a decrease in the reactivity of PMS [41]. Consequently, the activation of PMS becomes more challenging, ultimately leading to a less effective degradation of BHP.
The BHP degradation was also significantly influenced a pH 9, which created an alkaline environment. This alkaline state resulted in a slower degradation rate of BHP, as seen by the observed rate constant of k = 0.118 min−1. The degradation of BHP was decelerated with k = 0.073 min−1, when the pH was adjusted to 11. The data also indicated that the alkaline condition appeared to have a slight inhibitory effect on the degradation of BHP by NCMC + PMS. This could be attributed to the possibility that the NCMC surface becomes more negatively charged in the presence of BHP, leading to increased repulsion between BHP, NCMC, and SO52‒. As a result, the generation of radicals was constrained [39, 40]. Additionally, since the pHzpc of NCMC was 8.7 (Fig. S5) and the pKa value of PMS is 9.4, under extremely basic conditions, the electrostatic repulsion between NCMC and PMS became more pronounced, thereby constraining the reaction between PMS and NCMC [41].
Additional investigation on the BHP degradation using NCMC + PMS in different aqueous settings might provide valuable insights. In Fig. 9d, the testing medium consisted of tap water, seawater, and BHP. Additionally, DI water was included in the experiment. Significantly, the introduction of tap water had a discernible impact on the initiation of BHP degradation, resulting in a considerably diminished rate constant of k = 0.128 min−1. Nevertheless, the entire elimination of BHP was achieved in 30 min. Given that tap water contains many components, such as ions and solids, it was noteworthy that the overall degrading effectiveness of BHP remained adequate, as BHP was still completely removed. Despite the decreased k of 0.119 min−1, the degradation of BHP in seawater was still achieved even in the presence of various ions and minerals in seawater.
In particular, chloride (Cl) has the potential to be found in seawater, which might hinder the degradation efficiency because Cl‒ might react with SO4·‒ to produce chloride radicals (i.e., Cl·, Cl2·‒) [42], that have lower oxidation power compared to SO4·‒. Consequently, the efficacy of eliminating BHP was reduced in seawater. However, NCMC + PMS remained an efficient and viable approach for the degradation of BHP in both tap water and seawater.
Recyclability of NCMC for BHP Elimination
As NCMC activated PMS to efficiently BHP, the recyclability of NCMC to eliminate PMS over multiple cycles was then investigated. Figure 9f displays that BHP was capable of degrading BHP over five continuous cycles without considerable changes by the spent NCMC. The concentrations of Mn and Co in the supernatant solution at the end of the recyclability test (after the five cycles) had then monitored. The concentrations of Mn and Co in were 0.02 and 0.07 mg/L, respectively, that were much less than the concentration of NCMC added in the beginning (i.e., 200 mg/L).
These results indicate that the spent NCMC remained stable and useful to activate PMS for degrading BHP. The XRD pattern of the spent NCMC was also analyzed in Fig. S6 and the pattern was comparable to that of the pristine NCMC. Additionally, the SEM image of the used NCMC has been also added as Fig. S6, showing that the reused NCMC still retained its morphology and hollow structure. These results suggest that NCMC was a stable and durable heterogeneous catalyst.
Degradation Mechanism by NCMC + PMS
In order to get a deeper understanding of the contributions of reactive oxygen species (ROS) to the degradation of BHP, a comprehensive analysis of probe agents was then conducted. Tert-butanol (TBA) can serve as a suitable probe agent for the detection of hydroxyl radicals (·OH), whereas methanol can be employed to probe the existence of sulfate radicals (SO4·‒) and hydroxyl radicals (·OH). Figure 10a indicates that the introduction of TBA led to a significantly slower degradation of BHP, as seen by a drop in its rate constant from 0.037 to 0.018 min−1. This decrease suggests the presence of ·OH, which was likely contributing to the degradation of BHP. The introduction of methanol appeared to significantly limit the degradation of BHP, as shown by its low k of just 0.002 min−1. This suggests that both SO4·‒ and ·OH species coexisted concurrently during the degradation of BHP. In addition, an additional investigative agent known as benzoquinone (BQ) was used in order to ascertain the presence of the superoxide (O2·‒) species. Figure 10b indicates that the inclusion of BQ resulted in a decelerated degradation of BHP, with a k of 0.021 min−1. This observation potentially implied the potential occurrence of O2·‒ generation during the degradation process of BHP. Furthermore, an additional probe agent, NaN3, was subsequently utilized to examine the existence of singlet oxygen (1O2), a non-radical species. The inclusion of NaN3 resulted in a relatively sluggish degradation of BHP, with a k of 0.011 min−1, suggesting that the occurrence of 1O2 might also be attributed to NCMC + PMS during BHP degradation, which involved a non-radical degradation pathway.

a Effects of various scavengers and b corresponding rate constants; EPR analyses: c DMPO, d DMPO in methanol, e TEMP, f NCCentrations of ROS; and FFA consumption by NCMC + PMS (Catalyst = 200 mg/L, PMS = 200 mg/L,T = 30 °C).
In order to conduct a more comprehensive examination into the presence of reactive oxygen species (ROS), the generation of sulfate radicals (SO4·‒) and hydroxyl radicals (·OH) at different time intervals would be assessed using semi-quantitative methods that include the use of specialized probe agents [2, 3, 43]. As seen in Fig. 10c, the presence of SO4·‒ and ·OH was verified by the generation of para-benzoquinone (p-BQ) and para-hydroxybenzoic acid (p-HPA). This observation suggests a progressive rise in the concentrations of SO4·‒ and ·OH throughout the course of the study. Nevertheless, it was noted that the amount of SO4·‒ exceeded that of ·OH, and this result was consistent to the observation obtained from scavenger tests.
Furthermore, the quantification of singlet oxygen (1O2) generation might be implemented by establishing a correlation with the consumption of furfuryl alcohol (FFA) during the reaction with 1O2 (k = 1.2 × 108 M−1 s−1) [44]. Figure 10c illustrates the consumption of FFA at different reaction times for PMS alone and NCMC + PMS. The absence of NCMC in PMS would lead to a minimal consumption of FFA, implying a very low generation of singlet oxygen (1O2). Nevertheless, the use of NCMC + PMS has been seen to expedite the depletion of FFA, demonstrating that NCMC promoted the decomposition of PMS, hence triggering the production of singlet oxygen (1O2) for the non-radical elimination of BHP.
Additionally, electron paramagnetic resonance (EPR) was used to analyze ROS produced by NCMC + PMS. 5,5-Dimethyl-1-pyrroline N-oxide (DMPO) and 2,2,6,6-tetramethylpiperidine (TEMP) were then employed as spin-trapping agents. Initially, no discernible feature was obtained when DMPO was added in the absence of NCMC, as shown in Fig. 10d. When DMPO, PMS, and NCMC were present simultaneously, a noticeable pattern was then observed, corresponding to DMPO-SO4 and DMPO-OH, as displayed in Fig. 10d. This signifies that SO4·‒ and ·OH might occur from NCMC + PMS, hence contributing to BHP degradation. Moreover, the same experiment was conducted in MeOH to see whether superoxide (Fig. 10e), O2·‒, would be produced from NCMC + PMS, and a notable signal was observed and ascribed to DMPO-O2, confirming the production of O2·‒.
Furthermore, an alternative spin-trapping agent, TEMP, was used; however, no discernible signal was seen for TEMP and Oxone. After TEMP, PMS, and NCMC were present together, however, a unique triplet pattern was obtained, corresponding to TEMP-1O2 (Fig. 10f) [45], demonstrating that 1O2 existed and contributed to BHP degradation via the non-radical route. These investigations further validated that BHP degradation by NCMC + PMS involved with a number of ROS (SO4·‒, ·OH, O2·‒, 1O2) as illustrated in Fig. 11.

Illustration for the degradation mechanism of BHP by NCMC + PMS
The generation of these ROS might be derived through the following equation in the presence of NCMC:
Firstly, PMS would be dissociated to afford HSO5−, which then reacts with the catalyst, NCMC, for generating SO4·−. Meanwhile, SO4·− might further reacts with water molecules to produce ·OH. On the other hand, HSO5− would also react with H2O to produce H2O2. The additional reaction between ·OH and H2O2 might result in HO2·−, which would be then dissociated into O2·−. Upon the reaction of O2·− and H2O, the singlet oxygen (1O2).
Degradation Pathway of BHP by NCMC and PMS
Since NCMC can efficiently activated PMS to degrade BHP, it was important to further investigate the BHP elimination route. For exploring the BHP degradation pathway by ROS, the computational chemistry using the Density Function Theory (DFT) was adopted to investigate the molecular susceptibility in BHP. The molecular structure of BHP was be geometrically-optimized as shown in Fig. 12a. Accordingly, Fig. 12b and c displays the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of BHP. In general, the HOMO regions would attract electrophilic attacks, and both SO4·‒ and ·OH are considered to exhibit electrophilic nature and 1O2 is a strongly electrophilic species. Therefore, the HOMO located at the benzene ring of BHP seemed to easily receive electrophilic attacks from these ROS. Figure 12d also illustrates the electrostatic potential (ESP)-mapped iso-surface of BHP, which also indicates that the electron-rich region (the blue-colored zones) of BHP would be prone to electrophilic reactions.

a Optimized structure; b, c HOMO and LUMO; d ESP; e–g electron density mapped by f−, f0, f+; and h Fukui indices of BHP
For distinguishing the probable site of BHP for gaining attacks, the Fukui indices of BHP were then computed and listed in Fig. 12h. In particular, f–, f0, and f+ represent the electrophilic attack, the radical attack, and the nucleophilic attack, respectively. Since 1O2 is a highly reactive electrophilic species, the sites of BHP with relatively high and positive values of f– would tend to receive the electrophilic attacks. Specifically, O1, O3, C6, C12, C14, and C16 have relatively high f– values, suggesting that these sites might undergo the electrophilic attacks. However, since O1 and O3 are saturated, C12 and C14 were even more possible sites to accept initial attacks.
To determine the decomposition pathway of BHP by NCMC + PMS, the mass spectrometry of decomposition products from BHP degradation (Fig. S7 and Table S1) was then analyzed and combined with the above-mentioned theoretical knowledge to propose the degradation process in Fig. 13. Initially, the benzene ring of BHP, where the HOMO resides, would be subjected to a ring-opening attack to form an intermediate, M1, which would subsequently be degraded to yield M2. In response to the continual ring-opening reaction, M2 would disintegrate further to produce M3. Next, M3 would undergo further decomposition to generate M4, which is then constantly degraded to generate M5, M6, and, eventually M7.

A proposed degradation process of BHP based on the detected intermediates
Eco-toxicity Evaluation of BHP Degradation Intermediates
Based on the decomposition process of BHP by NCMC + PMS, it was also crucial to evaluate the eco-toxicity of intermediates generated from BHP elimination particularly for aquatic ecology. Herein, the Ecological Structure Activity Relationships (ECOSAR) Predictive Model is then adopted here to examine variations in toxicity of degradation intermediates from BHP based on acute toxicity (LC50fish/daphnia and EC50green algae) and chronic toxicity (ChVfish/daphnia/green algae) as shown in Fig. 14a and b, respectively. based on the Globally Harmonized System of Classification and Labelling of Chemicals (Table S2), the data of LC50 of these intermediates from the Ecological Structure Activity Relationships (ECOSAR) software could be then categorized into the four levels of toxicity (i.e., very toxic, toxic, harmful and non-harmful). Figure 14b displays values of Log LC50 of these intermediates as well as their categories. In the case of green algae, the toxicity of pristine BHP was relatively high (i.e., very low dosage of LC50). As BHP was decomposed, the toxicities of the initial intermediates (M1 and M2) seemed to decrease substiantially, and then toxicities were continuously and considerably reduced in M3–M7.

Eco-toxicity evaluation of BHP degradation by the ECOSAR: a acute toxicities; b chronic toxicities
Similarly, the same tendency was also observed in the cases of fish and daphnia as the initial toxicity of BHP was high, and gradually decreased along the decomposition process. These results suggest that while BHP could be considered as a harmful chemical, its degradation intermediates by NCMC + PMS were mostly non-harmful, demonstrating that NCMC + PMS was useful to degrade BHP without the occurrence of toxic/very toxic intermediates, and also detoxify BHP along the decomposition.
Conclusion
In this study, we propose NCMC as a heterogeneous catalyst for the activation of PMS. NCMC is facilely fabricated to possess a distinctive hollow-engineered nanostructure, and it was composed primarily of Co/Mn oxide NPs by using the zeolitic Co-MOF as a precursor after the facile etching and Mn doping treatments. The Mn doping embedded Mn/Co NPs NPs into the hollow-structured N-doped carbon substrate, making NCMC exhibit the higher meso-porosity, active N species and more superior electrochemical properties than its analogue without Mn dopants, NCC. Thus, NCMC exhibited a considerably larger activity than NCC and the benchmark catalyst, Co3O4 NP, for PMS activation to degrade BHP. Additionally, NCMC + PMS also demonstrated a considerably lower activation energy (Ea) for the degradation of BHP compared to the reported value. Furthermore, NCMC + PMS maintained its high efficacy in removing BHP across various water media. NCMC also demonstrated consistent composition and sustained its activity during several degradation cycles. The mechanism of BHP decomposition has been thoroughly investigated and radical and non-radical routes have been revealed. In accordance to the eco-toxicity assessment, BHP degradation by NCMC + PMS did not produce the formation of toxic and extremely toxic byproducts during its decomposition process. This ensures that NCMC would be an advantageous catalyst for PMS activation to degrade BHP and other emerging contaminants in water.
References
M.M.P. Tsui, J.C.W. Lam, T.Y. Ng, P.O. Ang, M.B. Murphy, P.K.S. Lam, Environ. Sci. Technol. 51, 4182 (2017)
J.-Y. Yin, W.D. Oh, E. Kwon, B.X. Thanh, S. You, H. Wang, K.-Y.A. Lin, Colloids Surf. A 625, 126891 (2021)
J.-Y. Yin, H. Wang, K.-P. Yu, J. Lee, K.-Y.A. Lin, J. Water Process Eng. 44, 102282 (2021)
Q. Chen, Y. Liu, Y. Lu, Y. Hou, X. Zhang, W. Shi, Y. Huang, J. Hazard. Mater. 422, 126929 (2022)
S. Giannakis, K.-Y.A. Lin, F. Ghanbari, Chem. Eng. J. 406, 127083 (2021)
L. Chang, X. Xie, X. Zhang, H. Chai, Y. Huang, Sep. Purif. Technol. 322, 124360 (2023)
F. Li, H. Xia, Q.-Q. Ni, Diamond Relat. Mater. 120, 108669 (2021)
L. Hu, R. Zhang, L. Wei, F. Zhang, Q. Chen, Nanoscale 7, 450 (2015)
W.-J. Liu, E. Kwon, N.N. Huy, T.C. Khiem, G. Lisak, T. Wi-Afedzi, C.-C. Wu, F. Ghanbari, K.-Y.A. Lin, J. Taiwan Inst. Chem. Eng. 133, 104253 (2022)
W.-J. Liu, E. Kwon, B. Xuan Thanh, T. Cong Khiem, D. Dinh Tuan, J.-Y. Lin, T. Wi-Afedzi, C. Hu, S. Sirivithayapakorn, K.-Y.A. Lin, Sep. Purif. Technol. 295, 120945 (2022)
W.-J. Liu, Y.-K. Park, H.M. Bui, N.N. Huy, C.-H. Lin, S. Ghotekar, T. Wi-Afedzi, K.-Y.A. Lin, J. Alloys Compd. 937, 165189 (2022)
Z. Liu, X. Sun, Z. Sun, Chemosphere 308, 136291 (2022)
N.T. Dung, T.V. Thu, T. Van Nguyen, B.M. Thuy, M. Hatsukano, K. Higashimine, S. Maenosono, Z. Zhong, RSC Adv. 10, 3775 (2020)
C. He, C. Tang, W.-D. Oh, J. Environ. Chem. Eng. 10, 107874 (2022)
X.G. Duan, H.Q. Sun, Y.X. Wang, J. Kang, S.B. Wang, ACS Catal. 5, 553 (2015)
S. Wu, J. Liu, H. Wang, H. Yan, Int. J. Energy Res. 43, 697 (2019)
X.-W. Zhang, M.-Y. Lan, F. Wang, C.-C. Wang, P. Wang, C. Ge, W. Liu, Chem. Eng. J. 450, 138082 (2022)
J.Y. Lin, J. Lee, W.D. Oh, E. Kwon, Y.C. Tsai, G. Lisak, S. Phattarapattamawong, C. Hu, K.Y.A. Lin, J. Colloid Interface Sci. 602, 95 (2021)
X.-Y. Jiang, E. Kwon, H.-C. Chang, N.N. Huy, X. Duan, S. Ghotekar, Y.-C. Tsai, A. Ebrahimi, F. Ghanbari, K.-Y. Andrew Lin, Sep. Purif. Technol. 308, 122789 (2023)
W.-J. Liu, Y.-K. Park, H.M. Bui, N.N. Huy, C.-H. Lin, S. Ghotekar, T. Wi-Afedzi, K.-Y.A. Lin, J. Alloys Compd. 937, 165189 (2023)
K.-Y.A. Lin, H.-A. Chang, J. Taiwan Inst. Chem. Eng. 53, 40 (2015)
D.D. Tuan, K.-Y.A. Lin, Chem. Eng. J. 351, 48 (2018)
H. Muneam, S. Hadi, M.M. Kareem, S.D. Jackson, Int. J. Ind. Chem. 7, 93 (2016)
C. Fang, B. Min, I. Angelidaki, Appl. Biochem. Biotechnol. 164, 464 (2011)
G. Li, J.H. Sun, W.P. Hou, S.D. Jiang, Y. Huang, J.X. Geng, Nat. Commun. (2016). https://doi.org/10.1038/ncomms10601
Z. Hasan, D.W. Cho, C.M. Chon, K. Yoon, H. Song, Chem. Eng. J. 298, 183 (2016)
Y. Luo, Y. Zheng, J. Zuo, X. Feng, X. Wang, T. Zhang, K. Zhang, L. Jiang, J. Hazard. Mater. 349, 119 (2018)
D.Y. Osadchii, A.I. Olivos-Suarez, A.V. Bavykina, J. Gascon, Langmuir 33, 14278 (2017)
Y. Wang, X. Wei, X. Hu, W. Zhou, Y. Zhao, Catal. Lett. 149, 1026 (2019)
X. Sun, H. Qi, S. Mao, Z. Sun, Chem. Eng. J. 423, 130169 (2021)
J. Miao, W. Geng, P.J.J. Alvarez, M. Long, Environ. Sci. Technol. 54, 8473 (2020)
Z. Wang, W. Wang, L. Zhang, D. Jiang, Catal. Sci. Technol. 6, 3845 (2016)
D.I.T. Oyekunle, B.B. Wu, F. Luo, J. Ali, Z.Q. Chen, Chem. Eng. J. 421, 129818 (2021)
T. Zeng, H.Y. Zhang, Z.Q. He, J.M. Chen, S. Song, Sci. Rep. (2016). https://doi.org/10.1038/srep33348
S. Qu, Y. Yuan, X. Yang, H. Xu, A.K. Mohamed, J. Zhang, C. Zhao, L. Liu, B. Wang, X. Wang, J. Rinklebe, Y.C. Li, S. Wang, Chem. Eng. J. 441, 135864 (2022)
T. Cong Khiem, X. Duan, W.-J. Liu, Y.-K. Park, H. ManhBui, W.-D. Oh, S. Ghotekar, Y. FaiTsang, K.-Y. Andrew Lin, Chem. Eng. J. 453, 139699 (2022)
Y. Li, F.-M. Li, X.-Y. Meng, S.-N. Li, J.-H. Zeng, Y. Chen, ACS Catal. 8, 1913 (2018)
H.T. Nguyen, J. Lee, E. Kwon, G. Lisak, B.X. Thanh, W.D. Oh, K.-Y.A. Lin, J. Colloid Interface Sci. 591, 161 (2021)
D.D. Tuan, C. Khiem, E. Kwon, Y.F. Tsang, S. Sirivithayapakorn, B.X. Thanh, G. Lisak, H. Yang, K.-Y.A. Lin, Chemosphere 294, 133441 (2022)
D.D. Tuan, E. Kwon, S. Phattarapattamawong, B.X. Thanh, T.C. Khiem, G. Lisak, H. Wang, K.-Y.A. Lin, J. Environ. Chem. Eng. 10, 106989 (2022)
W. Guo, S. Su, C. Yi, Z. Ma, Environ. Prog. Sustain. Energy 32, 193 (2013)
T. Zhang, Y. Chen, Y. Wang, J. Le Roux, Y. Yang, J.-P. Croué, Environ. Sci. Technol. 48, 5868 (2014)
T.C. Khiem, X. Duan, W.-J. Liu, Y.-K. Park, H.M. Bui, W.-D. Oh, S. Ghotekar, Y.F. Tsang, K.-Y.A. Lin, Chem. Eng. J. 453, 139699 (2023)
S. Mostafa, F.L. Rosario-Ortiz, Environ. Sci. Technol. 47, 8179 (2013)
Z.-Y. Choong, M.F. Gasim, K.-Y.A. Lin, T.S. Hamidon, H. Hussin, W.-D. Oh, Chem. Eng. J. 451, 138958 (2023)
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chen, YC., Jiang, XY., Lin, JY. et al. MOF-Derived Bimetal-Embedded Carbon with Etched Morphologies as an Efficient Activator of Peroxymonosulfate for Eliminate Emerging Contaminants. Korean J. Chem. Eng. 41, 1815–1831 (2024). https://doi.org/10.1007/s11814-024-00108-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11814-024-00108-2