
Abstract
Blends of cocoa butter with soybean oil (CB/SO) or canola oil (CB/CO) were crystallized at either of two agitation rates (100 or 1,000 rpm) and at two process temperatures (14 or 17 °C) in a scraped surface heat exchanger (SSHE). The physical properties were characterized at the SSHE output and during storage (14 and 28 days) at 15 °C. At the SSHE output, the CB/CO and CB/SO systems that had been processed at 100 rpm presented a more solid-like character than systems processed at 1,000 rpm despite the fact that the former systems contained a higher solid fat content than the latter. The degree of secondary crystallization increased with increasing shear rate. Nevertheless, the polymorphic behavior of cocoa butter crystals resembled the behavior observed under static isothermal crystallization conditions. At the SSHE output, systems of either blend contained a mixture of β′ and β crystals. During storage, β′ converted to β in both blends, although it did so to a higher extent in the CB/CO systems. Crystal ripening, observed in the CB/CO blend, provided stability to the systems during storage. In contrast, the CB/SO system increased its hardness by a slow sintering process. The polymorphism and hardness evolution in the blends under study were found to be associated with the molecular compatibility of the triacylglycerols in the cocoa butter and the vegetable oils tested.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
During the past few years, the food industry has been working toward replacing partially hydrogenated fats, which contain high amounts of trans fatty acids, with trans-free fats in their products. This transition is driven by the growing awareness of the detrimental effects of trans fatty acids on human health [1] as well as the labeling requirements and regulations imposed by several countries on the trade of products containing trans fats [2]. Blending of high-melting fats (HMF) and low-melting fats (LMF) is one of the most feasible strategies for producing trans-free shortenings with functional properties (melting profile, texture, mouthfeel) that are similar to those of the partially hydrogenated fats in current use. The effects of LMF composition on LMF–HMF blend crystallization have been studied with a focus on the crystallization kinetics and phase behavior of the HMF. Within this framework, Wright et al. [3] described important changes in the polymorphism and crystallization kinetics of anhydrous milk fat as a function of the molecular complementarity of the HMF and LMF (i.e., canola oil and the low-melting fraction of milk fat) used in a blend. Nevertheless, little attention has been devoted to the effects of LMF on the microstructural and textural properties of dispersions of HMF crystals. Shi et al. [4] and Pérez-Martínez et al. [5] evidenced that even in the absence of mixed crystal formation, the triacylglycerol (TAG) composition of the liquid fat phase influenced the polymorphism, microstructure, and, therefore, the rheological properties of the crystallized blends in LMF–HMF blends with a high LMF concentration (60–70% LMF/HMF). Pérez-Martínez et al. [5] reported important differences in the crystallization kinetics and microstructural parameters, i.e., crystal morphology, fractal dimension, and solid fat content (SFC), in binary blends of cocoa butter and soybean oil or cocoa butter and canola oil crystallized under isothermal conditions. Most importantly, Pérez-Martínez et al. [5] identified different relationships among the microstructural and rheological parameters (yield stress and storage modulus) for blends with similar LMF content. Such differences were related to the molecular complementarity among the LMF and HMF TAGs, more specifically to the amount of mono- and diunsaturated fatty acids present in the blended fats. Thus, it is important to investigate the extent to which molecular interactions between the TAGs of HMF and of LMF affect the crystallization process and the functional properties of crystallized blends under industrial production conditions.
The industrial production of lipid-based products, such as chocolates, coatings, margarines, and butters, occurs under dynamic conditions (i.e., agitation), usually in scraped surface heat exchangers (SSHEs), where the product is precrystallized. Emerging from the SSHE, the product is molded and stored in chambers in which crystallization is completed. SSHEs are preferred as precrystallization units because they have the capacity to heat and/or cool highly viscous fluids. In such units, the product is pumped through an annular gap between the shaft and the tubular heat exchanger. During the process, the shaft rotates, the blades continuously agitate the product, and the blades scrape the tubular surface to remove product from the surface, to avoid crystal crust formation, and to enhance the heat transfer rate. In the SSHE, most crystallization occurs at the inner surface of the heat exchanger and in the solution near the surface (i.e., the inner wall tube), where high shearing and supercooling conditions coexist. Under such conditions, the crystals are usually very small because a large number of nuclei form under the high supercooling and crystal attrition conditions [6].
Several studies on the crystallization of fats or lipid-based foods in SSHEs have been conducted [7–9]. Bolliger et al. [7] evaluated the influence of agitation and outlet temperature on milk chocolate tempering (tempermeter and calorimetric measurements). During storage, milk chocolate that had precrystallized in the SSHE achieved a bloom stability similar to that achieved using a conventional temper machine. An SSHE was used by Miskandar et al. [8] to process palm oil margarine. They found that the β′ crystal stability in margarine improved during storage upon lowering of the process temperature below 20 °C. They also reported an increase in the consistency during storage at 28 °C, independent of the prevailing crystal polymorphic structure (β′ or β) in the margarine. More recently, Kloek et al. [9] studied the physical properties of hydrogenated palm oil/sunflower oil blends crystallized in an SSHE. In this study, the residence time and outlet temperature used to achieve blend crystallization were varied, and the polymorphic structure and the mechanical properties were measured during storage. The crystallized blends required different amounts of time to reach the final texture after crystallization in the SSHE, depending on whether the blend was partly or completely crystallized in the SSHE. In the partly crystallized blend system, crystallization in storage caused rapid sinterization, whereas completely crystallized systems slowly recrystallized and showed an increased consistency during storage. These studies demonstrated that the SSHE conditions controlled the crystallization process and, thus, the physical properties of fat-based foods. However, additional investigations are required to determine the effects of the processing conditions, in particular the shear rate, on the melting behavior, polymorphism, microstructure, and textural properties of the crystallized systems. Moreover, it is of fundamental importance to characterize the extent to which crystallization on the bench scale and under static conditions translates to crystallization conducted under pilot plant conditions (i.e., under shearing in continuous systems). Within this framework, the objective of this research was to systematically study the effects of shear rate, process temperature, and vegetable oil composition on the parameters associated with the microstructure and physical properties of cocoa butter/vegetable oil blends crystallized in an SSHE.
Materials and Methods
A single batch of unrefined cocoa butter (CB) was obtained from PEALPAN (San Luis Potosí, Mexico). Refined, bleached, and deodorized soybean oil (SO) and canola oil (CO) were kindly donated by Coral Internacional S.A. de C.V. (San Luis Potosí, Mexico). CB/vegetable oil blends were prepared in a 30% (w/w) blend solution. Fats were heated to 80 °C for 20 min, weighed on a 20-kg-capacity mechanical scale (Ohaus Florham park, NJ, USA), and blended prior to crystallization in the SSHE. TAG profiles for the CB and vegetable oils were determined by HPLC, as reported by Pérez-Martínez et al. [10].
Crystallization in an SSHE
The configuration of the SSHE (FT25BBPA; Armfield Ltd., Ringwood, UK) used in this investigation is shown in Fig. 1. The melted blend (input temperature 80 °C) was fed at 120 mL/min by a progressive cavity pump into the first of two crystallization barrels (0.35 m length and 0.045 m2 contact area) connected in series. The residence time of the blend in the SSHE was estimated to be ~8 min. Both barrels were operated at the same agitation rate by four-blade scrapers. As shown in Fig. 1, the SSHE was cooled by brine recirculation in the barrel jackets. The process temperature (T p) used to crystallize the blends was controlled at the SSHE output using a proportional–integral–derivative (PID) controller. Relative supercooling, determined by the difference between the input temperature (60 °C) and the T p, was tested at two levels (T p = 14 or 17 °C). Additionally, the agitation rate was set to either 100 rpm or 1,000 rpm in each experiment.

Diagram of the SSHE used in this research. Continuous lines and dashed lines represent blend and cooling fluid flow pathways, respectively. The position of blend temperature measurement at the SSHE output (T p) is marked. The arrow indicates one of the four scraper blades
Samples of the crystallized blends were collected at the SSHE output (Fig. 1) and quickly analyzed to obtain the melting profile by differential scanning calorimetry (DSC), the SFC by pulsed NMR spectroscopy (pNMR), a rheological analysis by oscillatory tests, and the microstructure by polarized light microscopy (PLM). An identical set of samples was stored at 15 °C, and the melting profiles, SFC, microstructure, released oil, and hardness were determined after 14 and 28 days. The experiment was set up to test the full factorial combination of T p and agitation rate, and the resulting treatments were distributed randomly in duplicate. The effects of T p, agitation rate, and storage time on the measured parameters were established for each blend by analysis of the variance and contrasts among the treatment means using STATISTICA ver. 7.1 (StatSoft, Inc., Tulsa, OK). Unless otherwise stated, mean value ± standard deviation of the studied variables will be reported throughout the “Results and Discussion” section.
SFC
SFC was determined using a pNMR instrument (Minispec mq20, 20 MHz; Bruker Analytik, Rheinstetten, Germany) equipped with a jacketed measuring probe connected to a recirculation bath maintained at 15 ± 0.1 °C (Brookfield 505; Brookfield, MA, USA). The pNMR was calibrated daily prior to measurements using SFC standards of 0, 30.4, and 70.3% (Bruker Analytik, Rheinstetten, Germany). The SFC of the crystallized blends was determined by filling 4-mL pNMR tubes from the sample stream at the SSHE output (while the sample was still pourable) using a handcrafted hose connection. The filled tubes were analyzed and stored at 15 °C for further analysis.
Melting Profile by DSC
The melting profiles of the crystallized blends were determined using a differential scanning calorimeter (Q1000; TA Instruments, New Castle, DE, USA) equipped with an autosampler and refrigerated cooling unit (TA Instruments, New Castle, DE, USA). Prior to each measurement, the DSC was calibrated to obtain a baseline using an empty cell with the heat flow in T4P mode. The temperature and melting enthalpy of the phase changes were calibrated using indium (melting temperature 156.6 °C and ΔH = 28.45 J/g). Empty aluminum pans were used as a reference. Ten-milligram samples were weighed, crimped in aluminum pans, and immediately placed in the DSC cell equilibrated at T = 13 °C. Immediately thereafter, the melting exotherms were obtained by heating from 13 to 80 °C at a rate of 5 °C/min. Temperatures at the maximum (T Pk) and at the end (T e) of the endotherm were calculated from the first derivative of the heat flow determined by the DSC software (Universal Analysis 2000 ver. 4.2E; TA Instruments–Waters LLC), according to Toro-Vazquez et al. [11].
Oscillatory Rheometry
The phase angle (δ) of the crystallized blends at the SSHE output was determined using a temperature-controlled rheometer (Physica USD 200; Physica, Stuttgart, Germany) fitted with parallel plates 50 mm in diameter (PM31, Paar Physica). The upper plate was attached to the measuring unit, and temperature control was achieved using a Peltier system located in the lower plate. Crystallized samples were gently poured onto the lower plate (T = 15 °C), and the upper plate was positioned to leave a 1-mm gap with respect to the lower plate using the auto-gap function of the software (UDS 200 ver. 2.21; Physica Messtechnik GmbH, Stuttgart, Germany). All sample excess was trimmed from the plate’s edge, and a deformation sweep from 0.05 to 10% was applied at an angular frequency of 10 rad/s. All rheological results were determined within the linear viscoelastic region.
PLM
Microphotographs of crystallized systems were collected using a polarized light microscope (Olympus BX5; Olympus Optical Co., Ltd., Tokyo, Japan) equipped with a temperature-controlled stage (LTS 330; Linkam Scientific Instruments, Waterfield, UK) connected to a temperature control station (LTS350; Linkam Scientific Instruments, Waterfield UK) that was cooled with liquid nitrogen. Images were acquired using a color video camera (BX60F/PCB35; Olympus Optical Co., Ltd., Tokyo, Japan) coupled to the Linksys software ver. 1.3.1 (Linkam Scientific Instruments, Waterfield, UK). To guarantee a uniform sample thickness, two coverslips were placed on a glass microscope slide leaving a 2.2-cm gap between them. The sample was placed in the gap, and a coverslip was placed on the sample, leaving a final sample thickness of ~0.16 mm. Next, the slide was placed on the stage (T = 15 °C), and microphotographs were collected under polarized light at a constant light intensity for all systems.
Hardness Measurements
The hardness values of the crystallized blends were measured using a texture analyzer (TA-XT plus; Stable Microsystems Ltd., Surrey, UK) equipped with a 45°-angle Perspex conical probe (Stable Microsystems Ltd., Surrey, UK). Plastic cups (3 cm height, 6 cm top diameter, 5 cm bottom diameter) were filled with 10 mL of the crystallized blend at the SSHE output and immediately stored at 15 °C until analysis. The parameters of the texture analyzer were set as follows: measuring force, compression; pretest speed, 2.0 mm/s; test speed, 1.0 mm/s; post-test speed, 1.0 mm/s; distance, 10 mm; trigger type, auto (3 g). The maximum force of penetration was considered to define the sample hardness. Five determinations were performed per sample.
Oil Release Measurements
The amount of oil released by the crystallized blend after storage (15 °C) was measured at 20 °C. Aliquots of crystallized blends were transferred into Eppendorf tubes (~1 g per tube) and stored at 20 °C for 30 min, followed by centrifugation at 2,700×g for 15 min in a temperature-controlled centrifuge (Z382 K; Hermle Labortechnik GmbH, Wehingen, Germany). The released oil was drained by inverting the tubes for 5 min, and the Eppendorf tube walls were wiped with tissue paper to remove any residual oil. The percentage of oil released was determined by the weight difference according to:
Results and Discussion
The TAG profiles of the CB, CO, and SO used in this research are shown in Table 1. In CB, symmetrical TAGs, such as saturated-oleic-saturated (i.e., StOSt, POP, POSt), were 87.1%, and diunsaturated asymmetrical TAGs (i.e., POO, StOO) were 6.8%. In contrast, vegetable oils had large amounts of triunsaturated TAGs. In CO, 87.3% of the total content was composed of OOO, LOO, LLnEo, LLPo, and LLO, in order of decreasing concentration. In SO, 58.0% of TAGs were composed of LLL, LLO, LOO, LLnO, OOO, and LLnL, in order of decreasing concentration, and 28.8% were diunsaturated asymmetric TAGs containing one palmitic acid (i.e., LLP and PLO). It is worth noting that the concentrations of TAGs in CB and CO with an oleic acid esterified at the sn-2 position were 93.6 and 68.52%, respectively. In contrast, SO presented only 17.38% of these kinds of TAGs, but it had a concentration of diunsaturated TAGs containing one palmitic acid of more than sixfold that observed in CO. In previous studies of crystallization under static conditions, Pérez-Martínez et al. [5, 10] reported that vegetable oil TAGs composition affected the organization of the crystal network, the types of interactions among CB crystals, and, therefore, the rheology of the crystallized system. Recently, Dibildox-Alvarado et al. [12] revealed important effects of liquid-phase TAGs composition on the lamellar structure of crystallizing tripalmitin or tristearin in blends with triolein, soybean oil, or high oleic safflower oil. In this study, low-melting fats with TAGs containing at least one palmitic acid induced an increase in microviscosity (i.e., liquid state structuring) previous to the nucleation step of the saturated TAGs. According to the molecular modeling analysis, this phenomenon was more pronounced in blends with TAGs having more energetically favorable interactions (i.e., tripalmitin and PLL, PLO and POO). Within this context, TAGs interactions between CB with SO must be stronger than those between CB and CO.
Melting Profile Analysis
The melting profiles of the blends collected at the SSHE output (t = 0 days) displayed at least two overlapping endotherms with a maximum at 24–25 °C (Fig. 2). Such melting behavior was observed for blends and pure CB samples that were crystallized under static and isothermal conditions [5, 13]. On this basis, it was established that the TAGs from CB were the only TAGs that crystallized in the studied blends. Additionally, the endotherms correlated with the melting of β′ and β CB crystals (Fig. 2). The lowest melting endotherm corresponded to the β′ polymorph, whereas the highest melting endotherm corresponded to the β polymorph of the CB crystals (Fig. 2). The β′/β ratio in the crystallized blends was qualitatively established by visual comparison of the melting endotherms. Thus, at the SSHE output, the CB/SO blend yielded higher amounts of the stable β polymorph than did the CB/CO blend. In the CB/CO blend, the maximum peak of the melting thermogram (T Pk) was lower (P < 0.05) for the blends crystallized at 100 rpm (24.18 ± 0.43 °C) than for those crystallized at 1,000 rpm (24.64 ± 0.44 °C). Similarly, in the CB/SO blends, the system that was crystallized at 100 rpm and 17 °C yielded a T Pk value (23.10 ± 0.16 °C) that was lower than those of the other CB/SO systems (24.42 ± 0.16 °C). The reduced T Pk in systems crystallized at 100 rpm was attributed to a higher concentration of the β′ form in these systems.

Melting thermograms of CB/CO (a) and CB/SO (b) blends at the SSHE output
During storage, all blends displayed an increase in the endotherm corresponding to the β crystalline form, indicating that a β′ to β polymorphic transition took place. The rate of this transition was followed through the changes in the melting thermograms. After 14 days of storage, all CB/CO systems increased their T Pk values by approximately 2 °C to reach T Pk = 26.17 ± 0.18 °C. Subsequently, during the last 14 days of storage (t = 28 days), all systems showed an increase in T Pk of approximately 0.2 °C to reach a final T Pk = 26.40 ± 0.1 °C. In contrast, after 14 days of storage, all CB/SO blends reached a value of T Pk (25.95 ± 0.28 °C) that was slightly lower than that of the CB/CO systems. The endotherm corresponding to the β polymorph increased to a greater extent in the CB/CO systems. This trend was also observed after 28 days of storage (Fig. 3), when the T pk of the CB/SO systems that crystallized at 14 °C increased to 26.37 ± 0.29 °C, and 26.73 ± 0.34 °C for the systems crystallized at 17 °C. The higher rate of CB polymorphic transitions observed in CB/CO blends was also observed by Pérez-Martínez et al. [5] for blends crystallized in the absence of shear and under either dynamic or isothermal conditions [5]. The rate of polymorphic transitions is influenced by interfacial molecular mobility [14] and the rate of exchange of TAGs between the crystalline and liquid phases during crystal ripening [15, 16]. Thus, CB/CO systems were expected to present higher molecular mobility and/or exchange rate than CB/SO systems. According to Smith et al. [16], during crystal ripening TAGs exchange between crystal and liquid phase occurs at certain hotspots (i.e., kinks and defects on the crystal surface). Some molecular interactions among TAGs from crystals and liquid-phase compounds (i.e., diacylglycerols) can limit molecular mobility and even block these hotspots. Within this context, interactions at the hotspots among TAGs from CB crystals and TAGs from vegetable oil used in the blend could modulate polymorphic transitions in the studied systems. On the basis of the findings of Dibildox-Alvarado et al. [17] regarding the liquid structure of TAGs, the slower polymorphic transitions in CB/SO systems could be ascribed to limited mobility at the hotspots caused by an increase in microviscosity due to interactions of the palmitic acid moiety of TAGs containing palmitic acid from SO (i.e., LLP, PLO) with main TAGs from CB (i.e., StOSt, POP, POSt). Such a limiting effect was not possible in CB/CO systems given the limited content of TAGs containing palmitic acid in CO. Thus, it is clear that the degree of molecular complementarity between the TAGs of CB and the vegetable oil in the blend was a determining factor in the systems’ polymorphism.

Melting thermograms of CB/CO (a) and CB/SO (b) crystallized after 28 days of storage at 15 °C
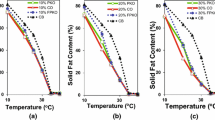
Solid Fat Content in Crystallized Blends
Both agitation and T p affected the SFC of the CB/SO and CB/CO systems at the SSHE output (SFC0). CB/CO systems were affected only by the agitation rate (P < 0.05): systems processed at 1,000 rpm reached 15.79 ± 1.04% and systems crystallized at the lower shearing 6.27 ± 0.61%. On the other hand, the SFC in CB/SO systems increased as agitation increased and T p was reduced (P < 0.05). The highest SFC was observed in the CB/SO system processed at 1,000 rpm and 14 °C (18.52 ± 0.95%), whereas the system processed at the same agitation rate and 17 °C yielded an SFC of 10.02 ± 0.82%. In contrast, the system crystallized at 100 rpm and 14 °C had an SFC of 13.29 ± 0.23%, whereas the system processed at the same agitation rate and 17 °C presented the lowest SFC (4.55 ± 1.37%). Higher values of SFC0 at 1,000 rpm were expected because an increase in the scraping frequency (agitation rate) enhanced the heat transfer efficiency and the rate of secondary nucleation at the inner barrel surface. As mentioned, no significant differences in SFC0 were observed in the CB/CO blends as a function of T p, which, at first glance, appeared to be incorrect. Nevertheless, it is worth remembering that T p only represented the supercooling conditions in the bulk flow of an SSHE, and most crystallization occurred within a few millimeters of the inner barrel surface, where the highest supercooling conditions existed. The supercooling conditions in this zone were not determined because the surface temperature was not measured and no information on the factors affecting heat transfer (i.e., shear rate, blend viscosity, heat capacity of the blend, crystal crust formation on the barrel’s inner surface, T p oscillatory behavior) was collected.
During storage, all CB/SO systems yielded a constant value of SFC (17.20 ± 0.72%; P < 0.05). In contrast, the CB/CO systems reached maximum values of SFC = 16.56 ± 0.78 °C after 14 days of storage (P < 0.05). Thereafter, all CB/CO systems yielded a decrease in SFC to reach SFC = 15.30 ± 0.76% (P < 0.05) after 28 days in storage. Such a decline in SFC was associated with the dissolution of unstable crystals (i.e., sub-micrometric crystals); similar behavior was observed in the long-term storage of tempered chocolate and chocolate equivalent [18]. The SFC decline in CB/CO systems could be facilitated by a high interfacial molecular mobility of CB TAGs at the crystal hotspots.
Systems’ Rheology at the SSHE Output
The effects of the agitation rate and T p on the rheological behavior of each system at the SSHE output are presented in Fig. 4. Both blend systems that had been crystallized at 1,000 rpm reached higher values of SFC0 than systems crystallized at 100 rpm. Thus, one would expect that systems crystallized at 1,000 rpm would show a more solid consistency (i.e., lower δ) than those crystallized at 100 rpm. Contrary to expectations, the systems crystallized at 100 rpm presented a more solid-like behavior than those crystallized at 1,000 rpm. The solid-like behavior increased as a function of T p, although an increase in T p implied a decrease in SFC0. For both blends, the systems crystallized at 100 rpm and T p = 17 °C displayed greater elasticity (δ ≈ 1.3°). Previous reports of the crystallization of CB and chocolate [7] described an increase in system viscosity as a function of SFC. However, viscosity cannot be compared with oscillatory rheometry parameters (i.e., δ) because oscillatory rheometry is carried out under static conditions that preserve the system’s crystal network microstructure (i.e., crystal agglomerates, fractal geometry), whereas viscosity measurements disrupt the crystal network as it is carried out under shearing conditions. Thus, the values of δ showed that the fat crystal network was highly perturbed by shear flow in the SSHE at the agitation rate of 1,000 rpm (i.e., avoiding the crystal aggregation process), but was preserved in those systems crystallized at 100 rpm. The highly solid-like behavior of blends crystallized at 100 rpm was consistent with the presence of crystal aggregates (highly birefringent spots indicated by the arrows in Fig. 5), as observed by visual inspection of PLM images. On the other hand, the more fluid-like consistency observed in samples processed at 1,000 rpm was characterized by the absence of aggregates. The effect of the agitation rate on crystal aggregate development resulted from two opposing phenomena caused by shearing of the crystallized samples: crystal aggregation versus crystal aggregate disruption. Mild agitation increased the frequency of crystal collisions, thereby increasing crystal aggregation. However, high agitation rates (i.e., 1,000 rpm) increased the number of particle collisions. If a crystal aggregate collides with another particle, the aggregate may break down, reducing the crystal size of aggregates as well as the size of the fragments detached from the aggregates [19].

Phase angles (δ) of CB/CO (a) and CB/SO (b) crystallized blends at the SSHE output (mean value and standard errors)

Polarized light microphotographs for the 30% CB/CO (a, b, c, d) and CB/SO (d, f, g, h) blends crystallized at 100 rpm and T p = 14 °C (a, d) or T p = 17 °C (c, f), and crystallized at 1,000 rpm and T p = 14 °C (b, g) or T p = 17 °C (d, h). The arrows indicate the position of the crystal aggregates
Blend Hardness During Storage
The hardness values of the crystallized blends during storage are shown in Table 2. In general, systems processed at T p = 17 °C had a higher hardness than systems processed at T p = 14 °C. Only the CB/CO blend processed at 1,000 rpm and T p = 17 °C yielded a hardness comparable to that of the systems processed at T p = 14 °C. The agitation rate significantly affected sample hardness (P < 0.05) only for those systems that were processed at T p = 17 °C. Systems that were crystallized at this T p and processed at 100 rpm developed the highest hardness within each blend. As previously discussed, within each blend, all crystallized systems achieved similar SFC values and comparable amounts of β′ and β crystals during storage (Fig. 4). Hence, the influence of T p and agitation rate on system hardness was not related to differences in SFC or to the sample polymorphism. The hardness of systems processed at T p = 17 °C correlated with the extent of crystallization during the earlier period of storage (SFC0–SFC14). Under low supercooling conditions (T p = 17 °C), the increase in SFC observed after the partly crystallized systems left the SSHE was ascribed to the growth of existing crystals rather than to additional nucleation. Thus, crystal growth promoted the formation of solid bridges among the crystals and crystal aggregates (i.e., agglomeration) from which the fat crystal network was strengthened. According to Brunsteiner et al. [20] the agglomerative strength (degree of solid bridge formation and strength among crystals) was proportional to the liquid-phase supersaturation in the vicinity of the aggregates. In our case, the lower SFC0 in the systems crystallized at T p = 17 °C resulted in a higher supersaturation of the liquid phase that increased the network solid bridge formation during the system setting. The properties of samples crystallized at 14 °C were not correlated with the SFC levels after processing. At this degree of supercooling, crystallization during storage led to new crystal formation, which reduced solid bridge formation in the fat crystal network.
The time-dependent system hardness was affected by the oil used in the blend. The hardness of the CB/CO systems did not increase after 14 days of storage, implying that recrystallization and crystal ripening events (i.e., β′ → β crystal transformation) occurred to a greater extent during the early period of storage. On the other hand, all CB/SO systems had a significant increase in hardness (i.e., a stronger fat crystal network), promoted by crystal sintering. Such processes occur over relatively long periods through solubilization and recrystallization of compound crystals [9, 21]. Thus, the time required to achieve a constant hardness value in the CB/SO systems depended on the molecular mobility at the crystal’s hotspots. According to Dibildox-Alvarado et al. [17], an increase of microviscosity due to the formation of mixed lamellar structures formed among SO and CB TAGs could limit the recrystallization process. Specifically, segregation of CB TAGs from the mixed lamellar structure delayed the crystallization events (i.e., sintering, β′ → β transition), causing an increment in the time required to achieve a constant hardness value in the CB/SO systems.
Oil Loss
Within each blend, the amount of released oil decreased with the extent of crystallization during the earlier period of storage (SFC0–SFC14). Thus, the CB/SO systems crystallized at T p = 14 °C (i.e., lower values of hardness and lower sintering) released greater amounts of oil than systems crystallized at 17 °C. The effects of agitation rate were significant only in systems crystallized at 14 °C, and a greater quantity of oil was released for the CB/SO systems processed at 100 rpm. On the other hand, oil loss in the CB/CO system was affected by both the agitation rate and T p. The CB/CO system crystallized at 100 rpm and 14 °C had the highest quantity of oil released, whereas the system crystallized at 100 rpm and 17 °C produced a lower quantity of released oil.
The amount of oil released by the crystallized systems was correlated with the blend hardness (Fig. 6). All crystallized systems with a hardness above ~400 g f released as much 2.5% oil, independent of the blend, crystallization conditions, or length of storage. In contrast, systems with hardness below ~400 g f released oil in quantities that were inversely proportional to the system hardness. According to Jahaniaval et al. [22], the oil released by the crystallized systems is related to the phase interactions and to the wetting properties of disrupted crystals. However, the parameter that defined the amount of oil released in this test appears to have been the capacity of the fat crystal network to withstand collapse under an applied force (i.e., centrifugal force). Although this relationship did not relate the microstructural properties of the fat crystal network to the oil released, it did reveal that the capacity of the crystal network to resist pressures exerted by a column of its own mass decreased oil loss in the system.

Oil loss in the crystallized systems as a function of system hardness after 14 and 28 days of storage. All CB/CO and CB/SO systems were included in the fits
Conclusions
The physical properties of the CB/vegetable oil blends crystallized in an SSHE was determined not only by processing conditions but also by the composition of the liquid phase of crystallized blends. Molecular interactions among TAGs in the crystalline phase and TAGs in the liquid phase of the crystallized systems affected the rate of polymorphic changes and recrystallization events of CB crystals during storage. In particular, it was hypothesized that TAGs in a liquid phase containing palmitic acid limited the molecular mobility of CB TAGs slowing down the polymorphic changes as well as the recrystallization of CB during storage. The quicker β′ to β transition drove the CB/CO systems to reach a constant hardness value during storage. Additionally, the texture in crystallized systems during storage was mainly affected by the effect of T p on the crystallization events after processing in the SSHE. Thus, at the lower supercooling (T p = 17 °C), the fat crystal network was strengthened by the formation of crystalline bridges under quiescent conditions. In contrast, systems processed at the higher supercooling (T p = 14 °C) presented a reduced solid bridge formation in the crystal network, thus resulting in softer textures than systems processed at the lower supercooling.
It was demonstrated that the rheology of the systems at the SSHE output processed at the lower agitation rate presented a solid-like behavior owing to the formation of crystal aggregates. In contrast, the higher agitation rate crystal aggregates were disrupted and these systems presented a fluid-like consistency. Nevertheless, the consistency of the systems at the SSHE output was not a determining factor in the system’s texture during storage.
References
Wolfram G (2003) Dietary fatty acids and coronary heart disease. Eur J Med Res 8:321–324
Kodali DR, List GR (2005) Trans fats alternatives, 8th edn. AOCS Press, Champaign
Wright AJ, McGauley SE, Narine SS, Willis MW, Lencki RW, Marangoni AG (2000) Solvent effects on the crystallization behavior of milk fat fractions. J Agric Food Chem 48:1033–1040
Shi Y, Liang B, Hartel RW (2005) Crystal morphology, microstructure, and textural properties of model lipid systems. J Am Oil Chem Soc 82:399–408
Pérez-Martínez D, Alvarez-Salas C, Charó-Alonso C, Dibildox-Alvarado E, Toro-Vazquez JF (2007) The cooling rate effect on the microstructure and rheological properties of blends of cocoa butter with vegetable oils. Food Res Int 40:47–62
Rao CS, Hartel RW (2006) Scraped surface heat exchangers. Crit Rev Food Sci Nutr 46:207–219
Bolliger S, Breitschuh B, Stranzinger M, Wagner T, Windhab EJ (1998) Comparison of precrystallization of chocolate. J Food Eng 35:281–297
Miskandar MS, Che-Man YB, Yusuff MSA, Rhaman RA (2002) Effect of scraped-surface tube cooler temperatures on the physical properties of palm oil margarine. J Am Oil Chem Soc 79:931–936
Kloek W, van Vliet T, Walstra P (2005) Mechanical properties of fat dispersions prepared in a mechanical crystallizer. J Texture Stud 36:544–568
Pérez-Martínez D, Alvarez-Salas C, Morales-Rueda JA, Toro-Vazquez JF, Charó-Alonso M, Dibildox-Alvarado E (2005) The effect of supercooling on crystallization of cocoa butter-vegetable oil blends. J Am Oil Chem Soc 82:471–479
Toro-Vazquez JF, Dibildox-Alvarado E, Charó-Alonso E, Herrera-Coronado V, Gomez-Aldapa CA (2002) The Avrami index and the fractal dimension in vegetable oil crystallization. J Am Oil Chem Soc 79:855–866
Dibildox-Alvarado E, Laredo T, Toro-Vazquez JF, Marangoni AG (2010) Pre-nucleation structuring of TAG melts revealed by fluorescence polarization and molecular mechanics simulations. J Am Oil Chem Soc 87:1115–1125
Toro-Vazquez JF, Pérez-Martínez D, Dibildox-Alvarado E, Charo-Alonso M, Reyes-Hernandez J (2004) Rheometry and polymorphism of cocoa butter during crystallization under static and stirring conditions. J Am Oil Chem Soc 81:195–202
Campos R, Ollivon M, Marangoni AG (2010) Molecular composition dynamics and structure of cocoa butter. Cryst Growth Des 10:205–217
Löfborg N, Smith P, Furó I, Bergenstål B (2001) Molecular exchange in thermal equilibrium between dissolved and crystalline tripalmitin by NMR. J Am Oil Chem Soc 80:1187–1197
Smith KW, Smith PR, Furó I, Pettersson ET, Cain FW, Favre L, Talbot G (2007) Slow recrystallization of tripalmitoylglycerol from MCT oil observed by 2H MNR. J Agric Food Chem 55:8585–8588
Dibildox-Alvarado E, Marangoni A, Toro-Vazquez JF (2010) Pre-nucleation structuring of triacylglycerols and its effect on the activation energy of nucleation. Food Biophys 5:218–226
Sonwai S, Rouseau D (2006) Structure evolution and bloom formation in tempered cocoa butter during long term storage. Eur J Lipid Sci Technol 108:735–745
Sonntag RC, Russell WB (1887) Structure and breakup of flocs subjected to fluid stresses. J Colloid Interface Sci 115:378–389
Brunsteiner M, Jones AG, Pratola F, Price SL, Simmons SJR (2005) Toward a molecular understanding of crystal agglomeration. Cryst Growth Des 5:3–16
Johansson D, Bergenstål B (1995) Sintering of fat crystal networks in oil during postcrystallization processes. J Am Oil Chem Soc 72:911–920
Jahaniaval F, Kakuda Y, Abraham V (2002) Oil binding capacity of plastic fats: effects of intermediate melting point TAGS. J Am Oil Chem Soc 79:389–394
Acknowledgments
The present research was supported by the National Council for Science and Technology of Mexico (CONACYT) through the grant number 48273-Z.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Pérez-Martínez, J.D., Reyes-Hernández, J., Dibildox-Alvarado, E. et al. Physical Properties of Cocoa Butter/Vegetable Oil Blends Crystallized in a Scraped Surface Heat Exchanger. J Am Oil Chem Soc 89, 199–209 (2012). https://doi.org/10.1007/s11746-011-1904-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11746-011-1904-y