
Abstract
The crystallization kinetics of milk fat were studied under non-isothermal and simulated adiabatic conditions using pulsed NMR spectroscopy. Isothermal experiments confirmed that when milk fat is shock cooled to below the α melting point it crystallizes in two steps due to the different crystallization kinetics of α and β′ modifications. In non-isothermal experiments, the fat samples were heated early during the plateau between steps to a temperature above the α melting point and β′ crystals formed more rapidly than in isothermal conditions. Fresh α crystals are believed to melt and form lamellar units containing triglyceride molecules with high degrees of isomorphism and these units can accelerate the nucleation and growth rates of β′ polymorph crystals. The crystallization behavior changed when the heating occurred late in the plateau and the α crystals are believed to have demixed, which allowed them to transform to β′ crystals directly in the solid state. Under simulated adiabatic conditions the rate of β′ crystallization was increased by a factor of 2–3 over the isothermal case. These findings were used to infer approaches to process difficult fat blends in scraped-surface heat exchanger plants.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Milk fat consists of a wide range of triglycerides including short-chain fatty acids that result in a broad melting range (−40 to 38 °C). Hence, the fat is often only partially crystallized during processing and under typical usage conditions. This is important as many of the properties perceived by the customer, such as hardness, plasticity and slump, are related to the state of the 3-dimensional crystal network in the product [1].
Most researchers have found at least α and β′ polymorphs in milk fat [2, 3]. In addition, γ and β modifications have been observed, but these polymorphs appear to be related to the time/temperature history of the sample. Milk fat is known for its strong β′ tendency due to its broad range of fatty acid content [4]. Although thermodynamically unfavored, less stable polymorphs can be formed by rapid cooling fat due to their faster crystallization kinetics [5]. Less stable polymorphs also have lower melting points than more stable polymorphs of the same composition. Van Beresteyn [2] found that all α polymorph crystals for milk fat disappeared above 22 °C. The more recent availability of time-resolved X-ray diffraction has enabled a systematic study of milk fat crystallization to be undertaken [6–8]. These researchers examined the effects of cooling rate and subsequent heating on polymorphic form and transitions.
The solid fat content (SFC) of partially crystalline fats is routinely measured using the direct method on a pulsed NMR instrument (AOCS Cd 16b-93). This method requires a multiplication or F-factor to compensate for detector dead-time. Alderliesten, van den Enden and Human (unpublished) found that the required F-factor can vary from 1.4 to 1.5 for the β′ polymorph and 1.1 to 1.3 for α crystals. This property can be used as an indicator of polymorphic transformations [9]; however, the F-factor can also be affected by fat composition and temperatures, which can complicate interpretation.
During manufacturing plastic fat products, a molten fat mix is cooled in scraped-surface heat exchangers (SSHE) and pin-workers [10]. The SSHE units have multiple functions including heat transfer, crystallization and high-shear mixing [11]. The residence time in the SSHE units is quite short relative to the crystallization kinetics of most fats including milk fat. The pin-workers allow crystallization to proceed under conditions of gentle working, which prevent the formation of a strong crystal network and hence a hard brittle product.
Various fat blends, including milk fat fractions, have different crystallization kinetics that can affect their behavior inside a crystallization plant. In order to characterize this behavior, it is common to shock cool a sample of the fat blend and then measure the progression of crystallization under isothermal conditions using pulsed NMR instrument; however, crystallization occurs approximately 20 times slower in an NMR tube than in a SSHE. This can be attributed to such factors as the high nucleation rates existing at the walls of SSHE barrels and the effect of mixing on the mass transfer of appropriate species to the crystal growth sites. The difference in speeds brings into question the validity of using isothermal NMR to indicate behavior inside the crystallization plant.
The majority of the residence time in a crystallization plant occurs in the pin-workers. There is no heat transfer surface in a pin-worker so the crystallization is occurring almost adiabatically with a small amount of mechanical energy input. The present work investigated whether the difference between isothermal and adiabatic crystallization can explain some of the variation in crystallization kinetics between laboratory measurements and in-plant performance.
Experimental Procedures
A typical summer anhydrous milk fat (AMF) sample was obtained from Fonterra (Palmerston North, New Zealand). Fifty NMR tubes were filled to 4 cm height with molten AMF and preheated to 65 °C for 15 min. The tubes were then transferred to a rack and the rack was placed in a Haake water bath. Placing the tubes directly in water resulted in a much faster thermal time constant than using a temperature controlled NMR cell. At regular intervals, a tube was removed from the bath, dried and inserted into a Bruker MQ20 Minispec NMR instrument. To effect a step change in temperature the rack of tubes was transferred to a second bath held at a new temperature. For simulated adiabatic runs, the NMR computer was connected to the water bath using an RS232 port to allow manipulating the temperature set point.
An application program was written for the NMR instrument using the ExpSpel language. The SFC was measured by the direct method using a 90° pulse and the standard algorithm supplied by Bruker. In addition, the free induction decay (FID) curve was measured every 0.4 μs during the period 7–100 μs and saved to disk. A second program was written in the Delphi language that processed the FID data off-line and fitted a combination of Gaussian and linear functions to the FID curve in order to determine the time constant of the solid-state decay (τ):
where
- v :
-
is the FID measurement
- t :
-
is the time
- g, h & i:
-
are fitted constants
For the simulated adiabatic experiments, the water bath temperature set point T SP was increased in proportion to the SFC as the crystallization progressed. The multiplication factor (0.5 °C/%) was an estimate of the ratio of latent heat of crystallization to specific heat capacity determined from historical differential scanning calorimetric data. It is difficult to obtain a precise value because the ratio is affected by fat composition and polymorphic form.
where T i is the initial fat temperature
Results and Discussion
Isothermal Crystallization
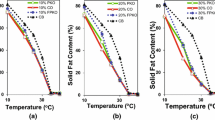
Initially, AMF was crystallized under isothermal conditions over a range of temperatures (10, 15, 22 °C) (Fig. 1). When AMF was rapidly cooled to temperatures >22 °C, the fat crystallized in a single step following a sigmoidal curve. X-ray diffraction has been used to show that only β′ crystals are formed under such conditions [3]. Hence, the β′ polymorph resulted in a solid-state time constant τ of 13 μs at these temperatures.

Isothermal crystallization curves at 10 °C( a), 15 °C (b) and 22 °C (c)
The slow initial start of the single step (i.e. at 22 °C), sometimes called induction time, was due to the limited nucleation rate and the slow initial growth rate of the crystals due to two factors. Firstly, the nuclei were so small that there was little facet surface area for growth to occur on. If one assumes that the growth rate of a facet is constant at a fixed sub-cooling, the McCabe ΔL law [12], the mass of the crystal would increase proportionally with time to the third power. Secondly, due to the free energy associated with the high degree of curvature of a small crystal, the driving force for crystal growth of a small crystal was reduced. The asymptotic end of the sigmoidal curve was due to the reduction in sub-cooling, which was the driving force for crystallization.
When the AMF was rapidly cooled to a temperature <22 °C and crystallized isothermally, it did so in two steps as previously observed [13, 14]. The slow β′ sigmoid was still present but a rapid step appeared due to the formation of α crystals. During the α crystallization phase, the time constant τ had a value of 18 μs, which reduced to 13 μs during the second step confirming the occurrence of α to β′ transformation.
The α crystallization phase occurred rapidly relative to the heat transfer rate from the fat sample to the water bath. If it were possible to instantaneously cool the NMR tube to the measurement temperature, α crystallization would be even more rapid. Fitting an equation to this portion of the curve results in parameters that include a contribution from the heat transfer kinetics as well as the crystallization dynamics of the fat [14].
From the set of isothermal crystallization curves, we found that once the crystallization temperature was reduced sufficiently to generate α crystals, the time required for β′ crystallization was similar for all temperatures. This can be explained by noting that the melting point for an α polymorph crystal was on average 10–12 °C below that for β′ crystals with the same composition [15]. Once the crystallization temperature was reduced into the α polymorphic region, the driving force for β′ nucleation and growth was relatively constant at sub-cooling temperatures of 10–12 °C. The β′ crystallization takes longer at very low temperatures, which is what was expected due to the lower molecular mobility.
Non-Isothermal Crystallization
To generate non-isothermal conditions, the temperature was raised in a stepwise function once crystallization had begun. Initially crystallization was conducted at 10 °C before raising the temperature to 22 °C at two different times, one early in the plateau and the other late (Fig. 2). The aim was to see the effect of melting α crystals on the overall crystallization process. A comparison of Fig. 2a with the isothermal runs shows that β′ crystallization took place much more quickly when α crystals were formed first and subsequently destabilized by heating. The same effect has been observed for cocoa butter [16] and milk fat [3]. These results were attributed to the presence of β′ nuclei in the non-isothermal case but the source of these nuclei has not been explained.

Non-isothermal crystallization curves with temperature stepped from 10 °C to 22 °C at 5 min (a) and 20 min (b)
Foubert et al. [14] claimed that α to β′ transformation occurs in the solid state on the basis of their time-resolved X-ray diffraction work, which explains the formation of β′ seed crystals from destabilized α crystals. However, this is incompatible with the deep dip observed in the SFC curve of Fig. 2a, which indicated that the α crystals were changed to a form that was included in the liquid component of the FID signal. A second objection against the concept of solid-state transformation is that milk fat is a comparatively heterogeneous mix of triglycerides and hence the α crystals are more mixed than the β′ [17]. Solid state transformation would not provide an opportunity for the crystals to demix (i.e. migration apart of non-isomorphic molecules).
The results reported here indicate that for fresh α crystals the β′ transformation is melt mediated for milk fat. The most likely explanation for the accelerated β′ crystallization requires consideration of the nature of molten fats. Larsson [18] and Hernqvist [19] have described the molecules in molten fats as having dynamic lamellar structures. These lamellar units need to be rearranged to contain a series of adjacent isomorphic molecules to be able to crystallize. When fresh α crystals melt, they are broken down into lamellar units that already contain isomorphic molecules. The units are relatively large in size because the temperature is well below the β′ melting point, and these units are immediately available either to combine to form a β′ nuclei or to bind to an existing β′ crystal. By contrast, the crystallization of β′ crystals directly from the melt must wait for lamellar units to rearrange to contain isomorphic molecules. This explanation is supported by the work of Ueno et al. [20] who used time-resolved X-ray diffraction to demonstrate the formation of a liquid crystal phase when SOS was subjected to a similar non-isothermal crystallization cycle.
When the heating was performed towards the end of the first plateau, the minimum of the dip in SFC was much closer to the final value (Fig. 2b). There are two possible explanations for this; either the character of the α crystals had changed during the plateau, or there were more β′ crystals already present accelerating the incorporation of the lamellar units into crystals.
The most logical change to occur in the character of the α crystals would be demixing, which is favored thermodynamically. If the demixing was sufficient, it could result in α crystals that were pure enough to enable solid state β′ transformation to occur. The crystallization kinetics of the α modification are extremely fast and, hence, the demixing process is expected to be also reasonably quick. This explanation can be related with those of Foubert et al. [14] if one considers the time–temperature history experienced by the fat. In their experiment, the α crystals were held long enough to demix, which would have enabled solid mediated β′ transformation to occur.
The demixing and subsequent solid-state transformation of α crystals would also help explain the observation of Mazzanti et al. [21] who found that the appearance of β crystals in milk fat was linked to first obtaining α crystals. Presumably, if the α crystals were sufficiently demixed, they would be capable of transforming directly into β crystals. Finally, Ueno et al. [20] found solid-state transformation occurred when α crystals of SOS were tempered, although care should be taken when extrapolating results from single triglycerides to complex mixtures.
Crystallization in SSHE Plants
Because crystallization within the pin-workers of a crystallization plant occurs almost adiabatically, we attempted to simulate these conditions by raising the water bath temperature as crystallization progressed (Fig. 3). As expected from the non-isothermal trials, the heat released during the α crystallization phase served to destabilize these crystals so that they transformed to the β′ form and the resultant SFC reached a plateau 2–3 times faster than the isothermal case. Therefore, α polymorph crystals are metastable and some change needs to be made to their environment to destabilize them so that they transform to the β′ form. In the isothermal case, when the β′ crystals begin to form the composition of the liquid fat surrounding the α crystals changes thereby destabilizing them but this is a comparatively slow process. In the non-isothermal and adiabatic experiments, the α crystals were destabilized by increasing the temperature, which is more direct and rapid.

Simulated adiabatic crystallization curve starting at 10 °C
The difference between isothermal and adiabatic situations in combination with other factors can explain some of the variation in crystallization kinetics between laboratory measurements and in-plant experience. It is interesting to surmise how the findings of this paper with regard to isothermal and non-isothermal crystallization can be applied in practice. For instance, operators of crystallization plants avoid certain fat blends because they are either fast or slow crystallizing.
In the case of a slow crystallizing blend, crystallization should be accelerated by destabilizing the α crystals that form in the first cooling stages of the crystallization plant. The obvious way to destabilize these crystals is by heating but directly heating the product reduces thermal efficiency. Alternatively, a small proportion of the molten feed material could be bypassed around the first cooling stage and blended in prior to the pin-working stage. The majority of the feed material would pass through the first cooling stage forming α crystals, which would then be destabilized into β′ crystals at the feed end of the pin-worker.
In the case of a fast crystallizing blend, approximating isothermal rather than adiabatic conditions within the crystallization plant could reduce the crystallization speed. This requires a configuration with the individual heat exchanger barrels and pin-workers to be interspersed along the length on the plant rather than being grouped together as is the current practice.
References
Mulder H, Walstra P (1974) The milk fat globule pudoc. Wageningen, The Netherlands
van Beresteyn ECH (1972) Polymorphism in milk fat in relation to the solid/liquid ratio. Neth Milk Dairy J 26:117–130
Ten Grotenhuis E, van Aken GA, van Malssen KF, Schenk H (1999) Polymorphism of milk fat studied by differential scanning calorimetry and real-time X-ray powder diffraction. J Am Oil Chem Soc 76:1031–1039
Timms RE (1980) The phase behaviour and polymorphism of milk fat, milk fat fractions and fully hardened milk fat. Aust J Dairy Technol 35:47–53
Sato K, Kuroda T (1987) Kinetics of melt crystallization and transformation of tripalmitin polymorphs. J Am Oil Chem Soc 64:124–127
Lopez C, Lavigne F, Lesieur P, Bourgaux C, Ollivon M (2001) Thermal and structural behavior of milk fat. 1. Unstable species of anhydrous milk fat. J Dairy Sci 84:756–766
Lopez C, Lavigne F, Lesieur P, Keller G, Ollivon M (2001) Thermal and structural behavior of anhydrous milk fat. 2. Crystalline forms obtained by slow cooling. J Dairy Sci 84:2402–2412
Lopez C, Lesieur P, Bourgaux C, Ollivon M (2005) Thermal and structural behavior of milk fat. 3. Influence of cooling rate. J Dairy Sci 88:511–526
Gribnau MCM (1992) Determination of solid/liquid ratios of fats and oils by low-resolution pulsed NMR. Trends Food Sci Technol 3:186–190
van Aken GA, Visser KA (2000) Firmness and crystallization of milk fat in relation to processing conditions. J Dairy Sci 83:1919–1932
Rao CS, Hartel RW (2006) Scraped surface heat exchangers. Crit Rev Food Sci Nutr 46:207–219
Mersmann A (1995) Crystallization technology handbook. Marcel Dekker, New York
Mulder H (1953) Melting and solidification of milk fat. Neth Milk Dairy J 7:149–174
Foubert I, Dewettinck K, Janssen G, Vanrolleghem PA (2005) Modelling two-step isothermal fat crystallization. J Food Eng 75:551–559
Hagemann JW (1988) Thermal behaviour and polymorphism of acylglycerides. In: Garti N, Sato K (eds) Crystallization and polymorphism of fats and fatty acids. Marcel Dekker, New York, pp 9–95
van Malssen K, Peschar R, Schenk H (1996) Real-time X-ray powder diffraction investigations on cocoa butter. I. Temperature-dependent crystallization behavior. J Am Oil Chem Soc 73:1209–1215
Wesdorp LH (1990) Liquid-multiple solid phase equilibria in fats—theory and experiments. Ph.D. thesis, Delft University of Technology, Delft, The Netherlands
Larsson K (1972) Molecular Arrangement in Glycerides. Fette Seiffen Anstrichm 74:136–142
Hernqvist L (1988) Crystal structures of fats and fatty acids. In: Garti N, Sato K (eds) Crystallization and polymorphism of fats and fatty acids. Marcel Dekker, New York, pp 97–137
Ueno S, Minato A, Seto H, Amemiya Y, Sato K (1997) Synchrotron radiation X-ray diffraction study of liquid crystal formation and polymorphic crystallization of SOS (sn-1,3-Distearoyl-2-oleoyl Glycerol) J Phys Chem B 101:6847–6854
Mazzanti G, Guthrie SE, Sirota EB, Marangoni AG, Idziak SHJ (2004) Effect of minor components and temperature profiles on polymorphism in milk fat. Crystal Growth Des 4:1303–1309
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Janssen, P.W.M., MacGibbon, A.K.H. Non-Isothermal Crystallization of Bovine Milk Fat. J Am Oil Chem Soc 84, 871–875 (2007). https://doi.org/10.1007/s11746-007-1105-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11746-007-1105-x