
Abstract
Huntington’s Disease (HD) is an autosomal dominant disease that occurs as a result of expansion of the trinucleotide repeat CAG (glutamine) on the HTT gene. HD patients exhibit various forms of mitochondrial dysfunction within neurons and peripheral tissues. Cardiolipin (Ptd2Gro) is a polyglycerophospholipid found exclusively in mitochondria and is important for maintaining mitochondrial function. We examined if altered Ptd2Gro metabolism was involved in the mitochondrial dysfunction associated with HD. Mitochondrial basal respiration, spare respiratory capacity, ATP coupling efficiency and rate of glycolysis were markedly diminished in Epstein-Barr virus transformed HD lymphoblasts compared to controls (CTRL). Mitochondrial supercomplex formation and Complex I activity within these supercomplexes did not vary between HD patients with different length of CAG repeats and appeared unaltered compared to CTRL. In contrast, in vitro Complex I enzyme activity in mitochondrial enriched samples was reduced in HD lymphoblasts compared to CTRL. The total cellular pool size of Ptd2Gro and its synthesis/remodeling from [3H]acetate/[14C]oleate were unaltered in HD lymphoblasts compared to CTRL. In addition, the molecular species of Ptd2Gro were essentially unaltered in HD lymphoblasts compared to CTRL. We conclude that compared to CTRL lymphoblasts, HD lymphoblasts display impaired mitochondrial basal respiration, spare respiratory capacity, ATP coupling efficiency and rate of glycolysis with any pathological CAG repeat length, but this is not due to alterations in Ptd2Gro metabolism. We suggest that HD patient lymphoblasts may be a useful model to study defective energy metabolism that does not involve alterations in Ptd2Gro metabolism.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Phospholipids are important structural and functional components of the cell membrane [1]. Cardiolipin (Ptd2Gro) is a major phospholipid and is found exclusively in the mitochondria and comprises approximately 3–15 % of the entire phospholipid phosphorus mass of the mitochondria [2–4]. In rat neurons and human lymphoblasts Ptd2Gro comprises approximately 5.7 and 5.4 %, respectively, of the total phospholipid composition [1, 5]. Ptd2Gro is required for modulation of the activity of enzymes involved in the generation of ATP in mitochondria (reviewed in [4]). In fact, Ptd2Gro is thought to be the “glue” that holds the mitochondrial respiratory complexes together [6]. Thus, maintenance of the appropriate content and fatty acid composition of Ptd2Gro in mitochondria is essential for cellular function and ATP generation.
Huntington’s Disease (HD) is a neurodegenerative genetic disorder and is the most common genetic cause of repetitive abnormal movements called chorea (reviewed in [7, 8]). The prevalence of HD is 7 in 100,000 in the Caucasian population [9, 10]. Physical symptoms begin at any age, although the mean is 35–44 years of age [11]. HD is an autosomal dominant disease that occurs as a result of the expansion of the trinucleotide repeat CAG (glutamine) [12]. The HTT gene, which codes for the Huntingtin protein (HTT), is found on chromosome 4p16.3 [11, 12]. Under normal circumstances, 10–35 CAG repeats can be found on this gene. However, if a certain threshold is achieved (>40 CAG repeats), full penetrance of the disease will take place [13]. The greater the amount of CAG repeats present in the HTT gene, the earlier the onset of the disease [14]. Mitochondrial dysfunction is manifest in HD neurons and in peripheral tissues of animal models of HD and in HD patients (reviewed in [15]). The cytotoxicity induced by mutant HTT is mediated, in part, by an alteration in normal mitochondrial function and dynamics (reviewed in [15, 16]). The mitochondrial dysfunction in HD includes deficits in mitochondrial protein import [17], calcium-handling and ATP production (reviewed in [15, 16, 18]). Mitochondrial fission/fusion dynamics and mitochondrial trafficking are also aberrant in HD (reviewed in [18, 19]) and might be key to mitochondrial dysfunction. This is suggested by studies showing that inhibition of the mitochondrial fission protein, dynamin-related protein-1, decreased mitochondrial dysfunction, improved HD cell survival in vitro and ameliorated motor dysfunction in a HD transgenic model [20]. A link between the mitochondrial fission/fusion machinery, Bcl-2 family members and mitochondrial outer-membrane permeabilization is beginning to emerge (reviewed in [21]). Ptd2Gro is involved in this link as Ptd2Gro interacts with Bcl-2 family members and Ptd2Gro metabolism is involved in mitochondrial fusion [22–24]. Previously, we showed that expression of the mitochondrial fusion protein, mitofusion-2, slows down de novo Ptd2Gro biosynthesis immediately post mitochondrial fusion [25]. Recently, it was observed that CTRL and mutant HTT bind to large unilamellar vesicles containing Ptd2Gro [26]. However, the biological significance of this is unknown as Ptd2Gro will bind to a number of proteins due to its multivalent properties.
Since Ptd2Gro is abundant in mitochondria and plays a key role in energy production, alterations in Ptd2Gro metabolism could potentially be involved in the pathogenic mechanisms leading to the development of the mitochondrial dysfunction observed in HD. In this study, we examined mitochondrial function in HD lymphoblasts and examined if alterations in Ptd2Gro content, synthesis and remodeling, mitochondrial respiratory supercomplex formation and Complex I activity within these supercomplexes contributed to the mitochondrial defect observed in this disease. We show that HD lymphoblasts exhibit extensive mitochondrial dysfunction but that this is not due to alterations in Ptd2Gro metabolism. We further show that HD lymphoblasts do not appear to exhibit altered mitochondrial respiratory supercomplex assembly compared to CTRL cells and that no correlation exists between the number of CAG repeats and the degree of mitochondrial dysfunction in these cells.
Materials and Methods
The Epstein-Barr virus transformed control (CTRL) and HD lymphoblasts used in this study are indicated in Table 1 and were obtained from Coreill Institute for Medical Research, Camden, New Jersey. Mitochondrial stress test and glycolysis stress equipment and test reagents were obtained from Seahorse Bioscience (North Billerica, MA, USA). [3H]Acetate and [1-14C]oleic acid were obtained from Dupont, Mississauga, Ontario, Canada. Dulbecco’s Modified Eagle’s Medium and fetal bovine serum were products of Canadian Life Technologies (GIBCO), Burlington, Ontario, Canada. Lipid standards were obtained from Serdary Research Laboratories, Englewood Cliffs, New Jersey, USA. Thin layer chromatographic plates (silica gel G, 0.25 mm thickness) were obtained from Fisher Scientific, Winnipeg, Canada. Ecolite scintillant was obtained from ICN Biochemicals, Montreal, Quebec, Canada. Mitochondrial Isolation Kit for Profiling Cultured Cells was obtained from abcam, Cambridge, MA. Bradford protein assay kit was from Bio-Rad laboratories, Mississauga, ON, Canada. Blue Native Polyacrylamide Gel Electrophoresis (BN-PAGE) analysis system was obtained from Life Technologies (Thermo Fischer Scientific brand). In-gel activity assay components were obtained from Sigma. All other chemicals were certified ACS grade or better and obtained from Sigma Chemical Company, St. Louis, USA or Fisher Scientific, Winnipeg, Canada.
Assessment of Mitochondrial Function and Rate of Glycolysis
Mitochondrial function and rate of glycolysis were determined using a Seahorse XF24 analyzer (Seahorse Biosciences, North Billerica, MA, USA). Lymphoblasts were grown in suspension in RPMI-1640 medium containing 10 % fetal bovine serum until reaching a concentration of 106 cells/ml. The medium was replaced with XF assay medium supplemented with 1 mM pyruvate, 25 mM glucose (pH = 7.4) for mitochondrial stress test. Cells were then plated on an XF analyzer 24-well plate (200,000 cells per well) that was coated with Cell Tak (2 µg per well) to attach the cells. The plate was then centrifuged at 1100 rpm for 10 min at room temperature. The cells were then maintained at 37 °C in a carbon dioxide-free incubator for 1 h prior to assay. A mitochondrial stress test was performed to assess cellular mitochondrial function. The test was done by first measuring the baseline oxygen consumption rate (OCR). This was then followed by sequential OCR measurements after injection of oligomycin (1 µM), Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (1 µM) and Rotenone + Antimycin A (1 µM), respectively, to obtain the key parameters of mitochondrial function that includes basal respiration, coupling efficiency, and spare respiratory capacity. The rate of glycolysis was determined by measuring the extracellular acidification rate (ECAR). Cells were grown as above. The growth media was replaced with XF assay media (unbuffered Dulbecco’s Modified Eagle’s Medium, 0 mM glucose, pH = 7.35) supplemented with l-glutamine (final concentration = 2 mM). To each sample well of a Seahorse XF analyzer 24-well plate (coated with Cell Tak as above), 200,000 cells were added. The plate was centrifuged at 1100 rpm for 10 min at room temperature. The cells were then incubated at 37 °C in a carbon dioxide-free incubator for 1 h. The rate of glycolysis was measured by detecting the change in ECAR after injecting glucose into each well (final concentration = 10 mM).
Radiolabeling of Ptd2Gro and Pool Size Determination
CTRL and HD lymphoblasts were grown as above and the medium was removed and incubated for 24 h with 1 µCi of [3H]acetate or 0.1 mM[1-14C]oleic acid (bound to bovine serum albumin in a 1:1 molar ratio). Cells were then harvested and radioactivity incorporated into Ptd2Gro determined as described [27]. Phospholipids were separated by two-dimensional thin layer chromatography and the phospholipid phosphorus content of Ptd2Gro determined as previously described [2, 27] (Suppl. Figure 1). Ptd2Gro fatty acyl molecular species mass were quantified using electrospray ionization-mass spectrometry coupled to high-performance liquid chromatography as previously described [28].
Measurement of Mitochondrial Supercomplex Assembly and Complex I Activity
Cellular mitochondrial fractions were isolated using the abcam Mitochondrial Isolation Kit for Cultured Cells (catalogue #: ab110170) as described in the kit’s protocol. All isolation procedures were performed at 4 °C. Briefly, cells were washed by re-suspension in ice-cold phosphate buffered saline, centrifuged at 600×g for 10 min and the supernatant was removed. The cell pellet was resuspended in Reagent A and then incubated on ice for 10 min. This was followed by homogenization by 30 strokes of a tight fitting Dounce A homogenizer to damage at least 50 % of the cells. Cellular damage was assessed by staining an aliquot with Trypan blue and observed under a phase contrast microscope. The homogenate was then centrifuged at 1000×g for 10 min to remove cellular debris. The supernatant was then transferred to a new tube while the cell pellet was re-suspended in Reagent B and the homogenizing and centrifugation steps were repeated. The two resulting supernatant volumes were then combined followed by centrifugation at 12,000×g for 15 min at 4 °C. The resulting pellet was the mitochondrial fraction and was re-suspended in Reagent C supplemented with protease inhibitors. Mitochondrial protein (65 µg) was treated with 50 µL of 0.2 % n-dodecyl β-d-maltoside (DDM) and placed on ice for 15 min. After this time, the samples were centrifuged at 20,000×g for 30 min at 4 °C. The supernatant was collected and stored at −80 °C. To perform the BN-PAGE (previously described in [29]), 1 µL of Coomassie® G-250 additive (charge shift molecule) was added to 20 µL of the resulting supernatant and the mixture was then placed into corresponding wells of a 3–12 % gradient acrylamide bisacrylamide gel for identification of mitochondrial respiratory complexes and supercomplexes. An in-gel activity assay of Complex I, NADH dehydrogenase (ubiquinone), was performed on the BN-PAGE gel using NADH as the electron donor and nitroblue tetrazolium chloride as the electron acceptor as previously described [29]. The resulting purple color formed (formazan pigment) represents activity. Complex I in vitro activity in isolated mitochondrial fractions was determined as described [30].
Other Determinations
Protein concentration was determined by the method of Bradford [31]. All data were expressed as means ± SD. The differences between the CTRL and HD groups were evaluated by Student’s t test. All values with p < 0.05 were considered statistically significant.
Results
Basal Respiration, Coupling Efficiency, Spare Respiratory Capacity and Extracellular Acidification Rate Are Reduced in HD Lymphoblasts
It was unknown if the mitochondrial dysfunction in HD was due to alterations in Ptd2Gro metabolism. To determine if alteration in CL metabolism was involved in the mitochondrial dysfunction associated with HD, we initially examined mitochondrial function in CTRL and HD lymphoblasts. The oxygen consumption rate (OCR) was determined by performing a mitochondrial stress test using a Seahorse XF24 analyzer in confluent CTRL and HD lymphoblasts. OCR was lower in the four HD lymphoblast lines compared to the four lines of CTRL lymphoblasts (Fig. 1a). In addition, the results between individual CTRL and HD lymphoblasts did not differ by more than 15 %. The basal respiration rate was reduced by 60 % in HD cells compared to CTRL (Fig. 1b, p < 0.001). Coupling efficiency (determined by the ratio between oligomycin sensitive rate and basal rate) was reduced 35 % (p < 0.01) in HD lymphoblasts compared to CTRL (Fig. 1c). Spare respiratory capacity (determined by the difference between maximal respiration and basal respiration) was reduced 90 % (p < 0.001) in HD lymphoblasts compared to CTRL (Fig. 1d). We examined the extracellular acidification rate (ECAR), a measure of glycolysis, in the presence of glucose. The rate of glycolysis was reduced 55 % (p < 0.01) in HD lymphoblasts compared to CTRL (Fig. 1e). Thus, HD lymphoblasts exhibited a mitochondrial and glycolytic dysfunction at any pathological CAG length (from 46 to 250 CAG in the mutant HD allele in the cell lines analysed).

Bioenergetic profiles of control and HD lymphoblasts. a O2 consumption rates (OCR) of CTRL and HD lymphoblasts were measured in real time under baseline conditions and after injection of mitochondrial inhibitors as indicated. Oligo oligomycin, FCCP Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone, Rot. + AA Rotenone + Antimycin A. Basal respiration (b), spare respiratory capacity (c), coupling efficiency (d), extracellular acidification rate (ECAR) (e) and glycolytic rate (f) were determined as described in “Materials and Methods”. Data are represented as the means ± SD (n = 4). ***p < 0.001, **p < 0.01
Ptd2Gro Metabolism is Unaltered in HD Lymphoblasts
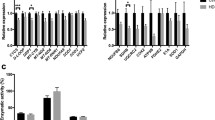
The pool size of Ptd2Gro was examined in CTRL and HD lymphoblasts. Total cellular Ptd2Gro levels were unaltered in HD lymphoblasts compared to CTRL (Fig. 2a). In addition, the total level of Ptd2Gro was unaltered in mitochondria prepared from HD lymphoblasts compared to CTRL (Table 2). The level of some Ptd2Gro species (16:1-18:1-18:1-18:2; 18:1-18:1-18:2-18:2; 18:1-18:1-18:2-20:4; 18:1-18:1-18:1-20:4) were elevated in HD lymphoblasts compared to CTRL. However, the level of tetralinoleoyl-Ptd2Gro (18:2-18:2-18:2-18:2), which plays a key role in mitochondrial respiratory chain supercomplex assembly, was unaltered in HD lymphoblasts compared to CTRL. To examine Ptd2Gro biosynthesis/remodeling, CTRL and HD lymphoblasts were incubated for 24 h with 0.1 µM [3H]acetate or 0.1 mM [1-14C]oleic acid bound to albumin (1:1 molar ratio) and radioactivity incorporated into Ptd2Gro determined. Incorporation of [3H]acetate into Ptd2Gro was unaltered in HD lymphoblasts compared to CTRL (Fig. 2b). In addition, incorporation of [1-14C]oleic acid into Ptd2Gro was unaltered in HD lymphoblasts compared to CTRL (Fig. 2c). Thus, Ptd2Gro metabolism is largely unaltered in HD lymphoblasts.

Ptd2Gro mass, synthesis and remodeling in HD lymphoblasts. a Mitochondrial fractions were prepared and Ptd2Gro content determined as described in “Materials and Methods”. b CTRL and HD lymphoblasts were incubated with [3H]acetate for 24 h and radioactivity incorporated into Ptd2Gro determined as described in the “Materials and Methods” section. c CTRL and HD lymphoblasts were incubated with [1-14C]oleate for 24 h and radioactivity incorporated into Ptd2Gro determined as described in “Materials and Methods”. Data represent the means ± SD (n = 4)
Mitochondrial Respiratory Supercomplex Assembly is Unaltered in HD Lymphoblasts
We examined if the reduction in mitochondrial function in HD lymphoblasts was due to alterations in mitochondrial supercomplex formation. Mitochondria were isolated from CTRL and HD lymphoblasts and 65 µg of mitochondrial protein subjected to BN-PAGE analysis. There were no alterations in mitochondrial supercomplex formation in all HD cell lines compared to all CTRL cells (Fig. 3a). We performed in-gel activity assays for Complex I on the BN-PAGE gel. No apparent alteration in Complex I activity within supercomplexes was observed in all HD cells compared to all CTRL cells (Fig. 3b, c). Complex I enzyme activity was then assayed in enriched mitochondrial fractions prepared from CTRL and HD lymphoblasts. Complex I in vitro activity was reduced 43 % (p < 0.05) from 28.07 ± 4.53 in CTRL to 16.12 ± 1.58 nmol/min/mg protein (n = 3). Thus, although Complex I activity appeared unaltered within mitochondrial respiratory supercomplexes it’s in vitro activity was reduced in mitochondria of HD lymphoblasts compared to CTRL cells.

Supercomplex assembly, Complex I activity and Complex I activity within supercomplexes in HD lymphoblasts. a Mitochondria were isolated from CTRL or HD lymphoblasts and 65 µg of mitochondrial protein subjected to BN-PAGE analysis as described in “Materials and Methods”. A representative gel of the four CTRL and HD lymphoblasts is depicted. Molecular mass marker ladder (L) is indicated on the left lane with molecular mass indicated in kDa. Mitochondrial respiratory complexes and supercomplexes are indicated on the right. CI Complex I, CII Complex II, CIII Complex III, CIV Complex IV, CV Complex V. b Complex I activity was determined in the above BN-PAGE gel using the in-gel activity assay described in “Materials and Methods”. Activity of Complex I is indicated on the left and Complex I containing supercomplexes are indicated on the right. Refer to Table 1 for cell identifier for each lane. c Relative levels of Complex I and Complex I containing supercomplexes in ascending order
Discussion
HD is caused by an expansion of a CAG trinucleotide sequence that encodes a polyglutamine tract in the huntingtin (HTT) protein [7, 8]. The cytotoxicity induced by mutant HTT proteins is mediated, in part, by an alteration in normal mitochondrial function and dynamics (reviewed in [15, 16]). In the current study we explored whether alteration in Ptd2Gro metabolism was involved in the mitochondrial dysfunction associated with HD. Ptd2Gro is required for optimal assembly of mitochondrial respiratory supercomplexes and reduction in tetralinoleoyl-Ptd2Gro is thought to be the cause of the profound mitochondrial dysfunction and mitochondrial supercomplex disassembly observed in Barth Syndrome [5, 29, 32–34]. Mitochondrial function and Ptd2Gro metabolism in lymphoblasts from four HD patients and four age-matched CTRL were examined. The major findings of this study were (1) HD lymphoblasts display impaired glycolytic rate and mitochondrial function including reduced basal respiration, spare respiratory capacity, ATP coupling efficiency, and in vitro Complex I activity compared to CTRL healthy cells, (2) there is no significant difference in total Ptd2Gro mass and Ptd2Gro biosynthesis/remodeling (measured via [3H]acetate or [14C]oleic acid incorporation into Ptd2Gro) between CTRL and HD lymphoblasts, and (3) mitochondrial respiratory supercomplex assembly does not appear altered between CTRL and HD patients.
The HD lymphoblasts used in our study exhibited profound glycolytic and mitochondrial dysfunction compared to CTRL cells but this did not appear to be related to the number of CAG repeats (from 46 to 250, in the cell lines used in this study), since the degree of glycolytic and mitochondrial dysfunction, as assessed by ECAR and OCR, respectively, did not differ between HD cells by more than 15 %. Analysis of a larger number of cell lines will be required to exclude correlation between the number of CAG repeats and the degree of glycolytic and mitochondrial dysfunction in HD lymphoblasts. The reduction in glycolytic rate was consistent with that observed in striatal neurons prepared from a transgenic rat model of HD [35]. Although Seahorse XF analysis indicated that HD cells exhibited reduced basal respiration, spare respiratory capacity and ATP coupling efficiency, the pool size of total cellular and mitochondrial Ptd2Gro and its biosynthesis/remodeling were unaltered in HD lymphoblasts compared to CTRL cells. These data indicate that the mitochondrial dysfunction in HD lymphoblasts is not due to alterations in Ptd2Gro metabolism.
BN-PAGE analysis revealed no apparent alteration in supercomplex formation from isolated mitochondria of HD lymphoblasts. Since tetralinoleoyl-Ptd2Gro has been implicated in mitochondrial supercomplex assembly, it was not surprising that assembly of these supercomplexes appeared unaltered in HD cells in which mitochondrial tetralinoleoyl-Ptd2Gro levels were unaltered. However, we observed a 48 % (p < 0.05) reduction in Complex I in vitro enzyme activity in mitochondria enriched samples isolated from HD lymphoblasts. This reduction correlated with the degree of mitochondrial dysfunction observed in the Seahorse analysis and suggests that Complex I enzyme activity may be impaired in HD lymphoblasts. A previous study had shown that Complex I activity was reduced in platelet mitochondria prepared from HD patients [36].
We did not observe any apparent difference in the level of Complex II in HD lymphoblasts, a complex that has been often implicated in HD, when equivalent amounts of mitochondrial fractions were separated by BN-PAGE. Inhibition of mitochondrial complex II causes selective degeneration of striatal neurons [37, 38], the most affected cells in HD [7]. Moreover, neuronal susceptibility to mitochondrial Complex II inhibition was shown to increase with age in animals and could be associated with the delayed onset of neurodegeneration observed in HD [39]. This is in contrast with previous studies where BN-PAGE analysis of mitochondria in striatal neurons of N171-82Q transgenic mice showed that the assembly of Complex II in mitochondria were altered at an early disease stage [40]. Complex II defects have also been reported in muscle from HD patients [41]. The differences across studies might be due to the type of tissue or cells examined, their energy requirements and the detergents used for supercomplex assembly analysis. In the current study, we utilized DDM for isolation of our mitochondrial supercomplexes. Some studies have used digitonin for isolation of supercomplexes in tissues. However, we found that digitonin was not ideal for Complex I separation in lymphoblasts. In fact, the milder digitonin treatment of WT and HD lymphoblasts resulted in only a single band upon BN-PAGE analysis (data not shown). Thus, it is possible that the much harsher DDM, which was required to isolate supercomplexes in lymphoblasts, may have affected supercomplex assembly in our preparations. In summary, we suggest that HD patient lymphoblasts may be a useful model to study defective energy metabolism that does not involve alterations in Ptd2Gro metabolism.
Abbreviations
- HD:
-
Huntington’s Disease (HD)
- Ptd2Gro:
-
Cardiolipin
- CTRL:
-
Control
- HTT:
-
Huntingtin protein
- BN-PAGE:
-
Blue native polyacrylamide gel electrophoresis
- ECAR:
-
Extracellular acidification rate
- NADH:
-
Nicotinamide adenine dinucleotide
- DDM:
-
N-Dodecyl β-d-maltoside
- OCR:
-
Oxygen consumption rate
- ATP:
-
Adenosine triphosphate
References
White DA (1973) Form and function of phospholipids. In: Ansell GB, Hawthorne JN, Dawson RM (eds) Phospholipids. Elsevier Biomedical, Amsterdam
Poorthuis BJ, Yazaki PJ, Hostetler KY (1976) An improved two dimensional thin-layer chromatography system for the separation of phosphatidylglycerol and its derivatives. J Lipid Res 17(4):433–437
Hostetler KY (1982) Polyglycerophospholipids: phosphatidylglycerol, diphosphatidylglycerol, and bis (monoacylglycero) phosphate. In: Hawthorne JN, Ansell GB (eds) Phospholipids. Elsevier Biomedical, Amsterdam
Hatch GM (2004) Cell biology of cardiac mitochondrial phospholipids. Biochem Cell Biol Biochim et Biol Cell 82(1):99–112
Schlame M et al (2003) Phospholipid abnormalities in children with Barth syndrome. J Am Coll Cardiol 42(11):1994–1999
Zhang M, Mileykovskaya E, Dowhan W (2002) Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J Biol Chem 277(46):43553–43556
Walker FO (2007) Huntington’s disease. Lancet 369(9557):218–228
Schneider SA, Walker RH, Bhatia KP (2007) The Huntington’s disease-like syndromes: what to consider in patients with a negative Huntington’s disease gene test. Nat Clin Pract Neurol 3(9):517–525
Harper PS (1992) The epidemiology of Huntington’s disease. Hum Genet 89(4):365–376
Pringsheim T et al (2012) The incidence and prevalence of Huntington’s disease: a systematic review and meta-analysis. Mov Disord 27(9):1083–1091
Myers RH (2004) Huntington’s disease genetics. NeuroRx 1(2):255–262
MacDonald ME et al (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 72(6):971–983
Kremer B et al (1994) A worldwide study of the Huntington’s disease mutation. The sensitivity and specificity of measuring CAG repeats. N Engl J Med 330(20):1401–1406
Andrew SE et al (1994) Huntington disease without CAG expansion: phenocopies or errors in assignment? Am J Hum Genet 54(5):852–863
Sassone J et al (2009) Huntington’s disease: the current state of research with peripheral tissues. Exp Neurol 219(2):385–397
Oliveira JM (2010) Nature and cause of mitochondrial dysfunction in Huntington’s disease: focusing on huntingtin and the striatum. J Neurochem 114(1):1–12
Yano H et al (2014) Inhibition of mitochondrial protein import by mutant huntingtin. Nat Neurosci 17(6):822–831
Costa V, Scorrano L (2012) Shaping the role of mitochondria in the pathogenesis of Huntington’s disease. EMBO J 31(8):1853–1864
Reddy PH (2014) Increased mitochondrial fission and neuronal dysfunction in Huntington’s disease: implications for molecular inhibitors of excessive mitochondrial fission. Drug Discov Today 19(7):951–955
Guo X et al (2013) Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration. J Clin Invest 123(12):5371–5388
Jourdain A, Martinou JC (2009) Mitochondrial outer-membrane permeabilization and remodelling in apoptosis. Int J Biochem Cell Biol 41(10):1884–1889
Kuwana T et al (2002) Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111(3):331–342
Gonzalvez F et al (2008) Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J Cell Biol 183(4):681–696
Choi SY et al (2006) A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat Cell Biol 8(11):1255–1262
Xu FY et al (2010) The dynamics of cardiolipin synthesis post-mitochondrial fusion. Biochim Biophys Acta 1798(8):1577–1585
Kegel KB et al (2009) Polyglutamine expansion in huntingtin alters its interaction with phospholipids. J Neurochem 110(5):1585–1597
Hatch GM, McClarty G (1996) Regulation of cardiolipin biosynthesis in H9c2 cardiac myoblasts by cytidine 5′-triphosphate. J Biol Chem 271(42):25810–25816
Sparagna GC et al (2005) Quantitation of cardiolipin molecular species in spontaneously hypertensive heart failure rats using electrospray ionization mass spectrometry. J Lipid Res 46(6):1196–1204
Mejia EM, Cole L, Hatch GM (2014) Cardiolipin metabolism and the role it plays in heart failure and mitochondrial supercomplex formation. Cardiovasc Haematol Disord Drug Targets 14(2):98–106
Zheng XX et al (1990) Evaluation of procedures for assaying oxidative phosphorylation enzyme activities in mitochondrial myopathy muscle biopsies. Biochim Biophys Acta 1019(1):1–10
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Schlame M et al (2002) Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Ann Neurol 51(5):634–637
McKenzie M et al (2006) Mitochondrial respiratory chain supercomplexes are destabilized in Barth syndrome patients. J Mol Biol 361(3):462–469
van Gestel RA et al (2010) The influence of the acyl chain composition of cardiolipin on the stability of mitochondrial complexes; an unexpected effect of cardiolipin in alpha-ketoglutarate dehydrogenase and prohibitin complexes. J Proteom 73(4):806–814
Gourane C et al (2013) Early deficits in glycolysis are specific to striatal neurons from a rat model of Huntington disease. PLoS One 8(11):e81528
Parker WD Jr et al (1990) Evidence for a defect in NADH: ubiquinone oxidoreductase (complex I) in Huntington’s disease. Neurology 40(8):1231–1234
Bazzett TJ et al (1993) Chronic intrastriatal dialytic administration of quinolinic acid produces selective neural degeneration. Exp Neurol 120(2):177–185
Novelli A et al (1988) Glutamate becomes neurotoxic via the N-methyl-d-aspartate receptor when intracellular energy levels are reduced. Brain Res 451(1–2):205–212
Brouillet E et al (1993) Age-dependent vulnerability of the striatum to the mitochondrial toxin 3-nitropropionic acid. J Neurochem 60(1):356–359
Damiano M et al (2013) A role of mitochondrial complex II defects in genetic models of Huntington’s disease expressing N-terminal fragments of mutant huntingtin. Hum Mol Genet 22(19):3869–3882
Turner C, Cooper JM, Schapira AH (2007) Clinical correlates of mitochondrial function in Huntington’s disease muscle. Mov Disord 22(12):1715–1721
Acknowledgments
We thank Dr. Fred Y. Xu for technical assistance. This work was supported by grants from the Huntington Society of Canada (S.S. & G.M.H.), Canadian Institutes of Health Research MOP 111219 (S.S.) and the Heart and Stroke Foundation of Canada G-14-0005708 (G.M.H.) E.M.M. is the recipient of a Children’s Hospital Research Institute of Manitoba—Research Manitoba Ph.D. studentship. S.S. is a Canada Research Chair in Neurobiology of Huntington Disease. G.M.H. is a Canada Research Chair in Molecular Cardiolipin Metabolism.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
About this article

Cite this article
Mejia, E.M., Chau, S., Sparagna, G.C. et al. Reduced Mitochondrial Function in Human Huntington Disease Lymphoblasts is Not Due to Alterations in Cardiolipin Metabolism or Mitochondrial Supercomplex Assembly. Lipids 51, 561–569 (2016). https://doi.org/10.1007/s11745-015-4110-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11745-015-4110-0