
Abstract
To further elucidate the molecular mechanism involved in peach (Prunus persica cv. Xiacui) fruit ripening, a differential proteomic analysis was conducted using iTRAQ based strategy for studying peach fruit ripening from day 15 before commercial harvesting (D0) to 15-day storage period at 25 °C. 64 differentially expressed proteins (≥1.5-fold, p ≤ 0.05) were successfully identified. Lipoxygenase (LOX) family members exposed the status of causing disorder metabolism and senescence by increased expressions. Some well-known antioxidant enzymes did not show significant expressions especially in later storage time. Nevertheless, proteins involved in ABA metabolism were up-regulated, demonstrating the important role of ABA in adaptation of plants to various environmental stresses. Glutathione S-transferase, cysteine proteinase inhibitor, transcription factor bHLH135 all showed great potential in response to ROS or other stresses. Proteins associated with flavonoid biosynthesis and amino acid metabolism also revealed critical roles in metabolism during storage.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
As one of the most important temperate stone fruit crops in the word, peach fruits received considerable attention due to its high nutritional and esthetic value. Some critical components such as pigments, flavor, aroma and other compounds are influenced by the ripening process. Therefore, unraveling the molecular and biochemical bases underlying peach fruit ripening has biological and dietary significance, which direct impacts not only in general fruit quality traits, but also in consumer acceptability, and post-harvest disease incidence (El-Sharkawy et al. 2016).
ROS are well-known as a family of free radicals including superoxide (O ·−2 ), hydroxyl (OH·), and hydrogen peroxide (H2O2), which can cause oxidative damage to biological macromolecules. Additionally, other biotic and abiotic stresses can also lead to remarkable metabolic disorders during fruit ripening. For response to internal and external stresses, fruits have derived its own cellular enzymatic and non-enzymatic mechanisms (Singh et al. 2012). The enzymatic antioxidant defense in plants includes enzymes such as superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPX) and peroxidase (POD). The non-enzymatic antioxidant system consists of compounds such as ascorbate and glutathione, lipid-soluble compounds such as carotenoids (Apel and Hirt 2004).
In the past few years, proteomics have triggered a scientific burst in all biological sciences, and have been widely used in studying various fruit ripening and senescence, such as apple, tomato, grape, and so on (Palma et al. 2011). Jiang et al. (2014) studied peach fruit during ripening upon post-harvest heat combined with 1-MCP treatment, and they found that most identified proteins were involved in stress response and energy metabolism. Zhang et al. (2010) found that proteins related to phenolic compounds were repressed in peach fruits stored at 0 °C with regard to storing at 5 °C.
Two-dimensional electrophoresis (2-DE) was the widely used method in proteomic analysis during past years, and extensive 2-DE research has been conducted to investigate protein expressions and accumulation in fruits (Jiang et al. 2014; Shi et al. 2014; Yu et al. 2015; Zhang et al. 2011). However, studies declared 2-DE has several limitations such as low reproducibility and difficult separation of hydrophobic proteins, low identification rate of proteins as well as low solubility of extremely large/small or acidic/basic proteins (Cheng et al. 2016). As a new and promising approach for proteomics research, isobaric tags for relative and absolute quantification (iTRAQ) have aroused considerable attention of researchers (Li et al. 2016). It showed an obvious advantage of simultaneously identifying and quantifying proteins from multiple samples, retaining important post-translational modification information, which are not completed by the 2-DE technique (Zieske 2006). Hereby, iTRAQ technique is used to identify and quantify differential proteins from peach fruit in this experiment to assess the biological characteristics of ripening. In total, 1384 proteins and unique 9816 peptides were identified, among which 64 proteins were successfully identified to show increased or decreased abundance (≥1.5-fold, p ≤ 0.05), involving in multiple pathways. Our research can provide an important molecular basis and reference information for future research.
Materials and methods
Plant materials and treatments
Field sampling and assays were carried out with peach fruits grown in the Jiangsu Academy of Agricultural Sciences orchard, Nanjing, Jiangsu Province, China. We defined peach fruits at commercial maturity after 120 days after florescence. Picking processes were divided to three times during the ripening stages: the optimal date of commercially mature (designated as D0), 15 and 7 days prior to D0 (D-15 and D-7), respectively. On the optimal date of harvest (D0), peach fruits with uniform sizes and without visual defects were picked and held at 25 ± 2 °C for 7 and 15 days (D7 and D15), maintaining the relative humidity between 80 and 90%. Samples (30 fruits) collected at each time points were immediately frozen in liquid nitrogen and stored at −20 °C until further analyses. There were three biological replicates for each measurement in our experiment.
Respiratory rate, color and firmness determination
Respiratory rate, as indicated by CO2 production, was measured with a portable infrared CO2 analyzer (CheckMate 3, Dansensor, Denmark) by placing samples (4–5 fruits) in a vessel for 1 h.
Ten peach fruits were evaluated for tristimulus color using a Minolta colorimeter (Konica Minolta CR-400, Co. Ltd., Osaka, Japan) on the basis of the CIELAB color system (L, a and b). Ten fruit were used to determine firmness using Fruit Hardness Tester (FHM-5, Japan), and firmness of peel and pulp were both measured in this experiment.
Malondialdehyde (MDA), H2O2 and O ·−2 content determination
MDA was measured by 2-thiobarbituric acid reaction as described by Wu et al. (2016). The amount of MDA was estimated as μmol/g FW.
H2O2 determination measurement was conducted using Hydrogen Peroxide Assay Kit (Jiancheng, China). Manipulated in accordance with the instructions of the kit. H2O2 content was expressed as μmol/g FW.
The O ·−2 content was measured according to the method as stated in previous research (Wu et al. 2016). A standard curve was used to calculate the O ·−2 concentration with NaNO2 as a standard. O ·−2 concentration was expressed as μmol/g FW.
Protein preparation
5 g peach tissue was ground into powder in liquid nitrogen, added 10 mL Lysis buffer (7 M Urea, 2 M Thiourea, 4% CHAPS, 40 mM Tris–HCl, pH 8.5) containing 1 mM PMSF and 2 mM ethylene diamine tetraacetic acid (EDTA, final concentration). After 5 min, added 10 mM dl-dithiothreitol (DTT, final concentration) to the samples. The suspension was sonicated at 200 W for 15 min, then centrifuged at 4 °C, 30,000g for 15 min. The supernatant was collected and added with 5 volume of chilled acetone containing 10% (v/v) TCA, and then the sample incubated at −20 °C over night. The next day, the mixture was centrifuged at 4 °C, 30,000g and discarded the supernatant. The precipitate was washed with chilled 100% acetone for three times. The pellet was air-dried and dissolved in lysis buffer (7 M urea, 2 M thiourea, 4% NP40, 20 mM Tris–HCl, pH 8.5). Sonicated the suspension at 200 W for 15 min, then centrifuged at 4 °C, 30,000g for 15 min. Transferred the supernatant to another tube. 10 mM DTT (final concentration) was added and incubated at 56 °C for 1 h to reduce disulfide bonds in proteins of the supernatant. Subsequently, 55 mM IAM (final concentration) was added to block the cysteines and incubated for 1 h in the darkroom. Added 5 volume of chilled acetone to the supernatant and mixed well for 2 h at −20 °C to precipitate proteins. After centrifugation at 4 °C, 30,000g, the supernatant was discarded. The pellet was air-dried for 5 min, then dissolved in 500 μL 0.5 M borane-triethylamine complex (TEAB, Applied Biosystems, Milan, Italy), and sonicated at 200 W for 15 min. Finally, samples were centrifuged at 4 °C, 30,000g for 15 min and transferred the supernatant to a new tube. Protein concentrations in the extracts were determined by the method of Bradford (1976), using bovine serum albumin (BSA) as a standard. The proteins in the supernatant were kept at −80 °C for further analysis. There were three biological replicates in each sample.
iTRAQ labeling and SCX fractionation
Total protein (100 μg) was taken out of each biological replicate, then digested the protein with Trypsin Gold (Promega, Madison, WI, USA) at 37 °C for 16 h (protein:trypsin 30:1). After trypsin digestion, peptides were dried by vacuum centrifugation. Peptides were reconstituted in 200 μL 0.5 M TEAB and processed according to the manufacture’s protocol for 8-plex iTRAQ reagent (Applied Biosystems). Briefly, one unit of iTRAQ reagent was thawed and reconstituted in 24 μL isopropanol. Samples were labeled with the iTRAQ tags as follow: Sample D-15 (113 tag), Sample D-7 (114 tag), Sample D0 (115 tag), Sample D7 (116 tag), Sample D15 (117 tag). After labeled with the isobaric tags, the peptides were incubated at room temperature for 2 h. The labeled peptide mixtures were collected and dried by vacuum centrifugation.
SCX chromatography was performed with a LC-20 AB HPLC pump system (Shimadzu, Kyoto, Japan). The iTRAQ labeled peptide mixtures were reconstituted with 4 mL buffer A (25 mM NaH2PO4 in 25% ACN, pH 2.7) and loaded onto a 4.6 × 250 mm Ultremex SCX column containing 5 μm particles (Phenomenex, CA, USA). Eluted the peptides at a flow rate of 1 mL/min with a gradient of buffer A for 10 min, 5–60% buffer B (25 mM NaH2PO4, 1 M KCl in 25% ACN, pH 2.7) for 27 min, 60–100% buffer B for 1 min. Prior to the next injection, the system was then maintained at 100% buffer B for 1 min before equilibrating with buffer A for 10 min. Elution was monitored by measuring the absorbance at 214 nm. Collected fractions every 1 min. The eluted peptides were pooled into 20 fractions, desalted with a Strata XC18 column (Phenomenex) and vacuum dried.
LC–ESI–MS/MS analysis based on Triple TOF 5600
Each fraction was resuspended in buffer A (5% ACN, 0.1% FA) and centrifuged at 20,000g for 10 min. 10 μL supernatant was loaded on a LC-20 AD nano HPLC (Shimadzu, Kyoto, Japan) by the autosampler onto a 2 cm C18 trap column. Then, the peptides were eluted onto a 10 cm analytical C18 column (inner diameter 75 μm) packed in-house. The samples were loaded at 8 μL/min for 4 min, then the 35 min gradient was run at 300 nL/min starting from 2 to 35% B (95% ACN, 0.1% FA), followed by 5 min linear gradient to 60%, then, followed by 2 min linear gradient to 80%, and maintenance at 80% B for 4 min, and finally return to 5% in 1 min. Data acquisition was performed with a Triple TOF 5600 System (AB SCIEX, Concord, ON) fitted with a Nanospray III source (AB SCIEX, Concord, ON) and a pulled quartz tip as the emitter (New Objectives, Woburn, MA). Data was acquired using an ion spray voltage of 2.5 kV, curtain gas of 30 psi, nebulizer gas of 15 psi, and an interface heater temperature of 150 °C. The MS was operated with a resolving power of 30,000 (full width at half maximum, FWHM) for TOF MS scans. For IDA, survey scans were acquired in 250 ms and as many as 30 product ion scans were collected if exceeding a threshold of 120 counts per second (counts/s) and with a 2+ to 5+ charge-state. Total cycle time was fixed to 3.3 s. Q2 transmission window was 100 Da for 100%. Four time bins were summed for each scan at a pulser frequency value of 11 kHz through monitoring of the 40 GHz multichannel TDC detector with four-anode channel detect ion. A sweeping collision energy setting of 35 ± 5 eV coupled with iTRAQ adjust rolling collision energy was applied to all precursor ions for collision-induced dissociation. Dynamic exclusion was set for 1/2 of peak width (15 s), and then the precursor was refreshed off the exclusion list.
Quantification and database search
Protein identification was performed using Mascot search engine (Matrix Science, London, UK; version 2.3.02). For protein identification, a mass tolerance of 0.05 Da was permitted for intact peptide masses and 0.1 Da was permitted for fragmented ions, with allowance for one missed cleavage in the trypsin digests. Settings included Gln->pyro-Glu (N-term Q), oxidation (M), deamidated (NQ) as the potential variable modifications, and carbamidomethyl (C), iTRAQ 8 plex (N-term), iTRAQ 8 plex (K) as fixed modifications. The charge states of peptides were set to +2 and +3. Specifically, an automatic decoy database search was performed in Mascot by choosing the decoy checkbox in which a random sequence of database is generated and tested for raw spectra as well as the real database. To reduce the probability of false peptide identification, only peptides with significance scores (≥20) at the 99% confidence interval by a Mascot probability analysis greater than “identity” were counted as identified. And each confident protein identification requires at least one unique peptide.
iTRAQ 8-plex was chosen for protein quantification during the search, proteins containing at least two unique spectra were selected for quantification analysis. The quantitative protein ratios were weighted and normalized by the median ratio in Mascot. All proteins data with fold-change ≥1.5 and p ≤ 0.05 were considered significantly expressed in this study. Proteins were identified by searching from Genome Database for Rosaceae (http://www.rosaceae.org/node/355), The kegg database (http://www.genome.jp/kegg/) and the COG database (http://www.ncbi.nlm.nih.gov/COG/) were used to classify these identified proteins.
Data analysis
Proteins were grouped according to their fold change compared to D0 through a two-way hierarchical clustering methodology using the software Cluster 3.0 and Treeview. Value was defaulted to 1 when the fold change compared to D0 was not significant (“ns” in Table 1). Rows were mean centered, and each row named with spot number corresponding to Table 1. Principal component analysis (PCA) of proteins was applied to the correlation matrix to reduce its dimensionality using Simca-P 11.5 software. Using un-rotated principal component (PC) scores, and the relation between the different time points and different proteins were showed in loading plot maps and score plot maps. Statistical analysis of respiratory rate, O .−2 content, H2O2 content, MDA content, color and firmness between two different groups were performed with the SPSS 18.0 software, using a one-way ANOVA by LSD test (p ≤ 0.05). In our results, we focus on comparison of D0 and other groups.
Results and discussion
Morphological and physiological changes of peach fruits
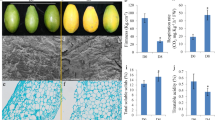
As shown in Fig. 1, the color and firmness changed gradually during storage time, and respiration rate increased quickly and reached a maximum at D15. Fruit respiration produce energy in the presence of an oxygen substrate and is a major factor contributing to ripening and senescence. As productions of ROS, O ·−2 and H2O2 occurs inevitably when there is a serious imbalance in cell compartment between production of ROS and antioxidant defense, and the content of MDA level can reflect the degree of cell membrane lipid peroxidation. In our study, the rate of CO2 production, O ·−2 , H2O2 and MDA contents of peaches all increased during storage period (Fig. 1), consistent with published studies (Huan et al. 2016), which indicated that oxidative stress happened during ripening and senescence in peach fruit. It’s also worth noting that changes of MDA, O ·−2 and H2O2 contents were roughly the same as that of the respiration rate, suggesting that ROS content was greatly influenced by respiratory activity.

a CO2 production rate, b O ·−2 , H2O2 and MDA contents, c firmness, d color, e representative photos of peaches during storage period. Statistical analyses of these indexes between two different groups were conducted using a one-way ANOVA. p values ≤0.05 by LSD test (SPSS v.18 package). In our results, we focus on comparison of D0 and other groups. As shown in Fig. 1. Double asterisks means p ≤ 0.01; asterisk means 0.01 < p ≤ 0.05 (compared to D0)
Protein classification and analysis
The distribution and classification of differentially abundant proteins in each sample are shown in Fig. 2, including those associated with energy metabolism (17%), carbohydrate metabolism (9%), response to stress (14%), lipid metabolism (5%), nucleotide metabolism (3%), amino acid metabolism (14%), flavone, and flavonol biosynthesis (3%), others (27%) and unknown (8%).

a Distribution of differentially abundant proteins in each sample. b Classification of differentially expressed proteins (fold change ≥1.5, p ≤ 0.05) into functional categories based on COG database (http://www.ncbi.nlm.nih.gov/COG/)
Cluster analysis of all proteins is shown in Fig. 3, and proteins with similar trend gathered together and were distinguished in a intuitive map between pre- and post-harvest stage. PCA analysis showed that using these components in a 2-D (plotting PC1 and PC2) representation allows to effectively separate samples into original groups (Fig. 4a) and respective proteins (Fig. 4b). The 64 differentially accumulated proteins were centralized into two PCs, of which PC1 and PC2 explained 42.75 and 29.00% of the variance, respectively. Proteins with similar change will gather together, and proteins with greater change will stay far away from zero on the axis.


Representation of the samples based on main principal components found after PCA. 2-D plot of main principal components (PC1 and PC2). a Proteins of samples (loading plot), b spot maps (score plot). The small triangles show the distribution of the differentially abundant proteins, and numbers beside the triangles are in correspondence with Table 1
Proteins related with energy production and carbohydrate metabolism
Photosynthesis is a complex metabolic process enabling photosynthetic organisms to use solar energy to convert carbon dioxide into biomass, which is an essential step in the biosynthesis of organic compounds. Chlorophyll a–b binding protein CP26 (CBP, protein 3), Oxygen-evolving enhancer protein 1 (OEEP1, protein 2), oxygen-evolving enhancer protein 2-2 (OEEP2, spot 1) and ferredoxin-NADP reductase (FNADP, spot 4) are proteins that belongs to photosystem. Moreover, ribulose bisphosphate carboxylase/oxygenase (Rubisco1, protein 7 and Rubisco 2, protein 8), ribulose bisphosphate carboxylase/oxygenase activase B (Rubisco activase B, protein 9), and ribulose bisphosphate carboxylase/oxygenase activase (Rubisco activase, protein 10) are involved in CO2 fixation in photosynthesis. In this study, the abundance of these proteins were higher in D-15 than other groups, which indicated that not only in the leaves, photosynthesis was still active in green fruits. The decreased profiles of these proteins would impair the capacity of the fruit cells to generate energy and reducing power through photosynthesis. This would lead to decomposition of carbohydrate to produce energy.
Only two proteins related to ATP production in this experiment, including NADH dehydrogenase, FAD-containing subunit (NADHF, protein 5), NADH dehydrogenase (ubiquinone) 1 alpha subcomplex subunit 2 (NADH1, protein 6), which means that the energy metabolism including the pathway and intensity had not changed greatly during peach fruit ripening period. However, the generally decreased expression of NADH dehydrogenase and NADH1 indicates that the ATP generation capacity and/or utilization is reduced. Similar results were also obtained in previous report (Jiang et al. 2014), which confirms fruit ripening is a continuous and gradual changing process, and NADHF 1.88-fold increase on D15 suggested that NADHF play important role in energy metabolism during late stage of fruit ripening.
Cellulose is a central component in plant cell walls and highly abundant in the secondary walls, which keeps fruit firmness by making up the primary cell wall combined with pectin and hemicelluloses (Bu et al. 2013; Kim et al. 2013). Pectate lyase act by depolymerizing cell wall, thus destroying the integrity of the plant tissues (Collmer and Keen 1986). In this research, pectate lyase 18 (PEL 18, protein 13) and pectate lyase 20 (PEL 20, protein 14) were identified to increase in the later stages of storage time, inducing the decrease of pectin content and causing of fruit softening in later ripening stages. Similar result about pectate lyase enzyme activity during banana fruit ripening was also mentioned (Payasi and Sanwal 2003). Endoglucanase 6 (EG6, protein 12) mainly possesses the function of depolymerization of cellulose, and cellulose synthase (CS, protein 16) is also considered important enzyme in cellulose metabolism. It is interesting to note in this context that high level of EG6 in D-15 were observed. One possibility is that turnover of the wall material is being differentially regulated during fruit development (Lunn et al. 2013). It is of particular interest that the expression patterns of CS may induce increase or maintain normal trend of cellulose content and consistent level of cellulose throughout ripening were also reported in tomato fruit (Lunn et al. 2013). Based on above results, we hypothesized that the degradation of the cell wall was mainly due to hydrolysis of pectin, which is consistent with established literature in kiwifruit (Bauchot et al. 1999), but the mechanism inside that need to be further studied.
Proteins associated with response to stress
Fruit ripening is an aerobic metabolic process generating reactive oxygen species (ROS) which can cause damage to fruit tissue, and the internal and environmental stresses can also lead to remarkable metabolic disorders. Fruits have derived its own cellular enzymatic and non-enzymatic mechanisms for response to these stresses. Studies have stated that UDP-glycosyltransferase (UGT, protein 21) play important role not only in the detoxification of xenobiotics and in the regulation of endobiotics, but also in the biosynthesis, storage and transport of secondary metabolites (Ahn et al. 2014). What’s more, Glutathione S-transferases (GST, protein 18), cysteine proteinase inhibitor (CPI, protein 23) and transcription factor bHLH135 (bHLH, protein 20) have been suggested to play a significant role in plant development and defense (Dong et al. 2014; Solomon et al. 1999). Contrary to above proteins, catalase isozyme 1 (CAT, protein 22) was decreased in abundance during ripening, showing the highest expression at D-15, similar to published report (Wu et al. 2016). Figure 1 shows the increase trend of ROS contents, and UGT, GST, CPI and bHLH were all increased in abundance during later stages of ripening, which can be helpful to removal of toxins. Study also showed that cysteine protease could lead to the acceleration of programmed cell death, and the corresponding up-regulated CPI can significantly act against the proteolytic cysteine protease (Solomon et al. 1999).
Abscisic acid (ABA) serves as an endogenous hormone that plays a key role in adaptation of plants to various environmental stresses (Zeevaart and Creelman 1988). 9-cis-epoxycarotenoid dioxygenase (NCED) catalyze the rate-limiting step in ABA biosynthetic pathway. Abscisic acid receptors (ABAR, protein 24) function as a bona fide receptor for ABA that constitute the core ABA component and connect all aspects of ABA signaling networks in plants (Miyakawa et al. 2013). In our study, the abundance of 9-cis-epoxycarotenoid dioxygenase NCED1 (NCED1, protein 19) and ABAR all increased during fruit ripening, showing a preference for the synthesis and signal transduction of ABA, which is conducive to response to various to environmental cues.
Proteins involved in lipid metabolism
Acetyl-CoA carboxylase (ACCase, protein 29) catalyzes the committed and rate-limiting step of the fatty acid biosynthesis pathway by converting acetyl-CoA to malonyl-CoA. In our study, ACCase 1 was identified increased to 1.59-fold at D15, promoting the synthesis of fatty acids at the last stage of ripening. Similar result of fatty acid content was also found in P. lentiscus L. fruits (Trabelsi et al. 2012). Fatty acids can serve as metabolic intermediate for conversion of various substances in plant, and production of aroma volatiles are mainly derived from fatty acids (Wu et al. 2011). The increased expression of ACCase 1 in our result may be helpful for aroma synthesis during ripening.
Membrane lipid peroxidation has been suggested as a characteristic of senescing system, which is initiated by either enzymatic activities or ROS (Shewfelt and del Rosario 2000). In our results, linoleate 9S-lipoxygenase 5 (L9s-LOX5, protein 27) and linoleate 13S-lipoxygenase 2-1(L13s-LOX2, protein 28) belong to LOXs family and catalyze the oxidation of linoleate into 9S-HPODE and 13-HPODE, respectively. Whereas a contrary tendency of these proteins’ expressions were observed. The abundance of L13s-LOX2 was higher at D-15 than that in other days, consistent with the result in sweet cherry and peach research, which was more abundant in the unripe fruits and decreased in ripe fruits. It is possible that the profile of L13s-LOX2 reflected the influences of light exposure and the lessening of chloroplastic functionality during ripening (Prinsi et al. 2016). However, L9s-LOX5 significantly increased in abundance at D7 and D15, consistent with the result of kiwifruit, reflecting the degradation of membrane lipids that might be caused by the increased L9s-LOX5 activity. Additionally, L9s-LOX5 might be core members for aroma volatile biosynthesis during later stage of ripening (Zhang et al. 2009).
Nucleotide and amino acid metabolism related
Nudix hydrolases (Nudx) are a family of proteins which can catalyze the hydrolysis of a variety of nucleoside diphosphate derivatives, such as ADP-ribose, NADH, dATP and NADPH (Kang et al. 2003; Ogawa et al. 2008). Serine carboxypeptidases (SCPs) belong to the hydrolase family that catalyze the reaction of C-terminal amino acids removal from peptides, and serine carboxypeptidase-like proteins (SCPLs) share high levels of similarities to SCPs, comprise a large family of proteins. Studies found that Nudx and SCPs/SCPLs could eliminate excess toxic metabolites (Yoshimura and Shigeoka 2015). In our result, the protein expression of nudix hydrolase 2 (Nudx 2, protein 30) was found to showed 2.08-fold at D15, and serine carboxypeptidase-like 52 (SCP, protein 32) and serine carboxypeptidase II-2 (SCPII, protein 33) were all increased during later stage of ripening. Thus, it can be speculated that the imbalance between production and elimination of toxins can induce up-regulation of these proteins. Moreover, the up-regulated expressions of SCP and SCPII might be helpful to resistance to programmed cell death in senescence (Schaller 2004).
Aspartic protease (AP, protein 34) is a member of a class of endopeptidases and shows preferential specificity for cleavage at peptide bonds between hydrophobic amino acid residues (Wang et al. 2008). Studies about the functions of Aps in plant are limited in the past years. However, it has been suggested that AP might be involved in the degradation of plant proteins in response to pathogens (Rodrigo et al. 1989, 1991). Alkaline elastase (AE, protein 35) produced by alkalophilic Bacillus YaB was a new type of proteinase which had a very high optimum pH and high elastolytic activity (Tsai et al. 1986). In our study, AP and AE showed 1.78 and 1.68-fold at D-15, but no significant expressions were showed in later ripening, and the internal mechanism need to be further studied.
Secondary metabolism related
Within the plant, secondary metabolites such as phenols play important roles in aroma and can increase the resistance to oxidative stress. In this view, it is interesting to highlight the identifications of anthocyanidin 3-O-glucosyltransferase (3GT, protein 42) and leucoanthocyanidin dioxygenase (LDOX, protein 41). They are all key enzymes for the synthesis and stability of anthocyanidins (Gollop et al. 2001). In our result, 3GT and LDOX were detected at the normal level in early stages of ripening, but showed 1.77-fold and 2.52-fold at D15, respectively. 3GT expression was consistent with the gene expression profile in grape tissue (Kobayashi et al. 2001). It needs to note that the color of both peel and pulp change significantly during ripening, as shown in Fig. 1. Thus, increased expression of 3GT and LODX are helpful for anthocyanin synthesis and stability, which are the fundamental factors in color change of peach in late ripening stages. Proteins link secondary metabolism and other pathways are shown in Fig. 5.

Overview of the main metabolic pathway and possible roles in up- or down-regulated identified proteins of peach fruits. Protein numbers are in correspondence with Table 1, and numbers in green present down-regulated proteins; numbers in red present up-regulated proteins. EMP Embden–Meyerhof–Parnas pathway, HMP hexose monophosphate pathway, OAA oxaloacetate, GSSG oxidized glutathione, GSH reduced glutathione
Conclusion
In summary, majority of energy production and carbohydrate metabolism related proteins showed decreased tendency during storage, but NADHF showed high level at D15, indicating its critical role in maintaining energy metabolism. Among successfully identified three lipoxygenases (LOXs) family members LOX2 and LOX5, LOX5 showed the greatest expression at D15 contrary to LOX2, exposing the status of causing disorder metabolism and senescence.
Data obtained in this experiment suggested that the altered metabolism may occur in peach fruit due to LOX and ROS during storage period. Expressions of GST, UTG and CPI were obvious increased at later stage, effectively removed toxins generated from internal or external stimuli. Proteins involved in ABA metabolism such as NCED1 and ABAR increased to maximum at D15, suggesting key role of ABA in adaptation of plants to various environmental stress. As a novel protein, bLHL and SCP showed great potential capacity in response to external stress. Proteins associated with flavonoid biosynthesis revealed critical roles in color change of peach in ripening stages. The identification of all proteins provides a good starting point to further dissect the mechanism of ripening by combining genetic, transcriptional, and other approaches.
Author contribution statement
Author ZY designed the experiment, and XW conducted the experiment and wrote this paper. AMM, MY and RM were participated in part of this experiment and provided some guidance. ZY helped with further revision to the manuscript. All authors read and approved the final version of the manuscript.
References
Ahn SJ, Dermauw W, Wybouw N, Heckel DG, Van Leeuwen T (2014) Bacterial origin of a diverse family of UDP-glycosyltransferase genes in the Tetranychus urticae genome. Insect Biochem Mol 50:43–57
Apel K, Hirt H (2004) Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annu Rev Plant Biol 55:373–399
Bauchot AD, Hallett IC, Redgwell RJ, Lallu N (1999) Cell wall properties of kiwifruit affected by low temperature breakdown. Postharvest Biol Technol 16:245–255
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Bu JW, Yu YC, Aisikaer G, Ying TJ (2013) Postharvest UV-C irradiation inhibits the production of ethylene and the activity of cell wall-degrading enzymes during softening of tomato (Lycopersicon esculentum L.) fruit. Postharvest Biol Technol 86:337–345
Cheng X, Deng G, Su Y, Liu JJ, Yang Y, Du GH, Chen ZY, Liu FH (2016) Protein mechanisms in response to NaCl-stress of salt-tolerant and salt-sensitive industrial hemp based on iTRAQ technology. Ind Crop Prod 83:444–452
Collmer A, Keen NT (1986) The role of pectic enzymes in plant pathogenesis. Annu Rev Phytopathol 24:383–409
Dong Y, Wang CP, Han X, Tang S, Liu S, Xia XL, Yin WL (2014) A novel bHLH transcription factor PebHLH35 from Populus euphratica confers drought tolerance through regulating stomatal development, photosynthesis and growth in Arabidopsis. Biochem Biophys Res Commun 450:453–458
El-Sharkawy I, Sherif S, Qubbaj T, Sullivan AJ, Jayasankar S (2016) Stimulated auxin levels enhance plum fruit ripening, but limit shelf-life characteristics. Postharvest Biol Technol 112:215–223
Gollop R, Farhi S, Perl A (2001) Regulation of the leucoanthocyanidin dioxygenase gene expression in Vitis vinifera. Plant Sci 161:579–588
Huan C, Jiang L, An X, Yu M, Xu Y, Ma R, Yu Z (2016) Potential role of reactive oxygen species and antioxidant genes in the regulation of peach fruit development and ripening. Plant Physiol Biochem 104:294–303
Jiang L, Zhang L, Shi Y, Lu Z, Yu Z (2014) Proteomic analysis of peach fruit during ripening upon post-harvest heat combined with 1-MCP treatment. J Proteom 98:31–43
Kang LW, Gabelli SB, Cunningham JE, O’Handley SF, Amzel LM (2003) Structure and mechanism of MT-ADPRase, a nudix hydrolase from Mycobacterium tuberculosis. Structure 11:1015–1023
Kim WC, Kim JY, Ko JH, Kim J, Han KH (2013) Transcription factor MYB46 is an obligate component of the transcriptional regulatory complex for functional expression of secondary wall-associated cellulose synthases in Arabidopsis thaliana. J Plant Physiol 170:1374–1378
Kobayashi S, Ishimaru M, Ding CK, Yakushiji H, Goto N (2001) Comparison of UDP-glucose: flavonoid 3-O-glucosyltransferase (UFGT) gene sequences between white grapes (Vitis vinifera) and their sports with red skin. Plant Sci 160:543–550
Li JJ, Ding XL, Han SH, He TT, Zhang H, Yang LS, Yang SP, Gai JY (2016) Differential proteomics analysis to identify proteins and pathways associated with male sterility of soybean using iTRAQ-based strategy. J Proteom 138:72–82
Lunn D, Phan TD, Tucker GA, Lycett GW (2013) Cell wall composition of tomato fruit changes during development and inhibition of vesicle trafficking is associated with reduced pectin levels and reduced softening. Plant Physiol Biochem 66:91–97
Miyakawa T, Fujita Y, Yamaguchi-Shinozaki K, Tanokura M (2013) Structure and function of abscisic acid receptors. Trends Plant Sci 18:259–266
Ogawa T, Yoshimura K, Miyake H, Ishikawa K, Ito D, Tanabe N, Shigeoka S (2008) Molecular characterization of organelle-type nudix hydrolases in Arabidopsis. Plant Physiol 148:1412–1424
Palma JM, Corpas FJ, del Rio LA (2011) Proteomics as an approach to the understanding of the molecular physiology of fruit development and ripening. J Proteom 74:1230–1243
Payasi A, Sanwal GG (2003) Pectate lyase activity during ripening of banana fruit. Phytochemistry 63:243–248
Prinsi B, Negri AS, Espen L, Piagnani MC (2016) Proteomic comparison of fruit ripening between ‘Hedelfinger’ sweet cherry (Prunus avium L.) and its somaclonal variant ‘HS’. J Agric Food Chem 64:4171–4181
Rodrigo I, Vera P, Conejero V (1989) Degradation of tomato pathogenesis-related proteins by an endogenous 37-kDa aspartyl endoproteinase. Eur J Biochem 184:663–669
Rodrigo I, Vera P, Vanloon LC, Conejero V (1991) Degradation of tobacco pathogenesis-related proteins—evidence for conserved mechanisms of degradation of pathogenesis-related proteins in plants. Plant Physiol 95:616–622
Schaller A (2004) A cut above the rest: the regulatory function of plant proteases. Planta 220:183–197
Shewfelt RL, del Rosario BA (2000) The role of lipid peroxidation in storage disorders of fresh fruits and vegetables. HortScience 35:575–579
Shi Y, Jiang L, Zhang L, Kang R, Yu Z (2014) Dynamic changes in proteins during apple (Malus × domestica) fruit ripening and storage. Hortic Res 1:6
Singh SP, Singh Z, Swinny EE (2012) Climacteric level during fruit ripening influences lipid peroxidation and enzymatic and non-enzymatic antioxidative systems in Japanese plums (Prunus salicina Lindell). Postharvest Biol Technol 65:22–32
Solomon M, Belenghi B, Delledonne M, Menachem E, Levine A (1999) The involvement of cysteine proteases and protease inhibitor genes in the regulation of programmed cell death in plants. Plant Cell 11:431–443
Trabelsi H, Cherif OA, Sakouhi F, Villeneuve P, Renaud J, Barouh N, Boukhchina S, Mayer P (2012) Total lipid content, fatty acids and 4-desmethylsterols accumulation in developing fruit of Pistacia lentiscus L. growing wild in Tunisia. Food Chem 131:434–440
Tsai YC, Lin SF, Li YF, Yamasaki M, Tamura G (1986) Characterization of an alkaline elastase from alkalophilic Bacillus Ya-B. Biochim Biophys Acta 883:439–447
Wang LZ, Wang M, Li QH, Cai TY, Jiang WB (2008) Partial properties of an aspartic protease in bitter gourd (Momordica charantia L.) fruit and its activation by heating. Food Chem 108:496–502
Wu MC, Hu HT, Yang L, Yang L (2011) Proteomic analysis of up-accumulated proteins associated with fruit quality during autumn olive (Elaeagnus umbellata) fruit ripening. J Agric Food Chem 59:577–583
Wu X, Jiang L, Yu M, An X, Ma R, Yu Z (2016) Proteomic analysis of changes in mitochondrial protein expression during peach fruit ripening and senescence. J Proteom 147:197–211
Yoshimura K, Shigeoka S (2015) Versatile physiological functions of the nudix hydrolase family in Arabidopsis. Biosci Biotechnol Biochem 79:354–366
Yu F, Shao XF, Yu LN, Xu F, Wang HF (2015) Proteomic analysis of postharvest peach fruit subjected to chilling stress or non-chilling stress temperatures during storage. Sci Hortic (Amsterdam) 197:72–89
Zeevaart JAD, Creelman RA (1988) Metabolism and physiology of abscisic-acid. Annu Rev Plant Phys 39:439–473
Zhang B, Yin XR, Li X, Yang SL, Ferguson IB, Chen KS (2009) Lipoxygenase gene expression in ripening kiwifruit in relation to ethylene and aroma production. J Agric Food Chem 57:2875–2881
Zhang CF, Ding ZS, Xu XB, Wang Q, Qin GZ, Tian SP (2010) Crucial roles of membrane stability and its related proteins in the tolerance of peach fruit to chilling injury. Amino Acids 39:181–194
Zhang L, Yu Z, Jiang L, Jiang J, Luo H, Fu L (2011) Effect of post-harvest heat treatment on proteome change of peach fruit during ripening. J Proteom 74:1135–1149
Zieske LR (2006) A perspective on the use of iTRAQ (TM) reagent technology for protein complex and profiling studies. J Exp Bot 57:1501–1508
Acknowledgements
This investigation was financially supported by the following projects: The Innovation Project of Jiangsu Agricultural Science [CX(15)1020], the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) and Jiangsu key laboratory for horticultural crop genetic improvement (2015022).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by W. Wang.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wu, X., Mason, A.M., Yu, M. et al. Quantitative proteomic analysis of pre- and post-harvest peach fruit ripening based on iTRAQ technique. Acta Physiol Plant 39, 181 (2017). https://doi.org/10.1007/s11738-017-2478-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11738-017-2478-6