
Abstract
POMA/Cellulose and POEA/Cellulose nanocomposites were successfully synthesized based on interfacial polymerization over cellulose fibers extracted from the Amazon P. pellucida. The triclinic structure of poly(o-methoxyaniline) (POMA) and poly(o-ethoxyaniline) (POEA) was maintained after polymerization over cellulose fibers. However, possible chemical interactions between polymers and cellulose chains resulted in the increase of the average crystallite size, suggesting an oriented polymerization as well as the cellulose surface modification. The average crystallite size of POMA changed from (32 ± 2) Å (for pure POMA) to (41 ± 2) Å (for POMA in the nanocomposite form). The average crystallite size of POEA changed from (32 ± 2) Å (for pure POEA) to (44 ± 2) Å (for POEA in the nanocomposite form). Initially, the extracted cellulose presented average crystallite size of (29 ± 2) Å, while the cellulose crystallites in the nanocomposites were found around (59 ± 2) Å (POMA/Cellulose) and (92 ± 2) Å (POEA/Cellulose). The morphology of POMA/Cellulose was significantly different from that observed in the pure as-synthesized POMA: globular vesicular shape was formed during the polymerization over the cellulose surface. In the POEA/Cellulose nanocomposite, the cellulose nanofibrils were also completely recovered by POEA consisting of well-defined nanometric spheres. These results were correlated with the thermal stability of the developed nanocomposites by Thermogravimetric Analysis (TG/dTG). Thus, the interfacial synthesis of POMA and POEA over a cellulose matrix was reported here, contributing to a better understanding of the thermal, structural and morphological properties of these resulting nanocomposites.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The search for friendly materials has made cellulose one of the most studied natural polymers because it can be obtained from a variety of sources and crystal structures (Jonoobi et al. 2015; Manzato et al. 2017). Peperomia pellucida (Linn.) Humb. Bonpl. & Kunth (Piperaceae) is known as “erva-de-jabuti” in the Amazon State, Brazil. The local folk medicine describes this species as emollient, diuretic and useful against cough and throat inflammation (Da Silva et al. 1999). P. pellucida is rich in cellulose, representing a potential source to obtain this natural polymer, as well as to prepare nanocomposites, increasing its commercial value besides proposing a technological application for this Amazon species.
Cellulose has been used in the preparation of nanocomposite materials together with conjugated polymers, aiming at improvement of their physicochemical properties (Zheng et al. 2017). There are numerous composites based on cellulose and bio/synthetic polymers that have been frequently used for different purposes. The hydroxyl groups covering the cellulose fibers (Jie et al. 2019; Svenningsson et al. 2019), as well as the regular arrangement of their molecules allow the interaction with a variety of conjugated polymers (Sharifi et al. 2018; Fei et al. 2019). Polyaniline derivatives such as POMA and POEA have received considerable attention due to their controllable chemical and electrical properties, easy methods of preparation, low cost of monomers and good environmental stability. Cellulose/conjugated polymer nanocomposites have attracted considerable attention due to their application in antimicrobial surface (Hanif et al. 2019), targeted drug delivery (Dai and Si 2018) and heavy metal absorption (Jahan et al. 2018). Nanosized polymer particles can form a fine and homogenous distributed network along the cellulose matrix. The nanosized reinforcement particles can occupy substantially greater number of sites of the cellulose’s molecular structure. For this reason, POMA and POEA were synthesized by in situ interfacial polymerization in the presence of the extracted cellulose.
A novel methodology for in situ interfacial polymerization of POMA and POEA over cellulose fibers was described here. A systematic structural, morphological, and thermal investigation was proposed: X-ray Diffraction technique (XRD) was applied to evaluate the long-range order achieved as a consequence of very short-range interactions. The Le Bail method (Le Bail 2005) was performed using the previous XRD patterns to refine cell parameters and estimate crystallite size. Peak broadening based on linear combinations of spherical harmonics was useful to evaluate the anisotropic crystallite shape. Scanning Electron Microscopy (SEM) was useful to assess the material morphology, as well as to verify the recovering of the cellulose fibers. Then, these results were correlated with the thermal stability of the developed nanocomposites using Thermogravimetric Analysis (TG/dTG).
Experimental
Materials
Chemicals
Acetone (CH3(CO)CH3) and ethyl alcohol (C2H6O) were purchased from Biotec®; sodium hydroxide (NaOH) was purchased from Dinamica®; hydrochloric acid (HCl) and glacial acetic acid (CH3COOH) were purchased from Labsynth® and sodium chlorite (NaClO2), chloroform (CHCl3), ammonium persulfate (APS, (NH4)2S2O8), o-anisidine (C7H9NO) and o-phenetidine (C8H11NO) were purchased from Sigma Aldrich®.
Plant material
Peperomia pellucida (Linn.) Humb. Bonpl. & Kunth (SisGen n° A26CD5E) was collected at Federal University of Amazonas (UFAM), Manaus/AM—Brazil (3° 5′21″ S; 59° 57′51″ W) and dried under controlled humidity (50% R.H.) at 27 °C. Botanical identification (voucher specimen HUAM-10759) was carried out at UFAM following the established protocol.
Methods
Cellulose extraction
The stems from P. pellucida were separated from leaves, cut into pieces (about 3 cm) and washed with distilled water under constant stirring at (70 ± 2) °C for 60 min. Then, the stems were dried in oven for 24 h at (60 ± 2) °C.
The specific chemical treatments employed for extraction of cellulose was performed in the following order: extractives, alkalinization and bleaching. For removing the extractives (waxes and oils), the stems were subjected to a solvent mixture of ethanol/acetone (95:5, v/v) for 20 min at (70 ± 2) °C. After reach the room temperature, the system was filtered, and the stems were washed using distilled water and dried in oven for 4 h at (60 ± 2) °C.
Alkalization treatment was applied to swell raw fibers. Then, fibers were treated with 5 wt% NaOH solution for 1 h at (60 ± 2) °C to reduce hemicellulose and lignin contents. The resulting lignocellulosic fibers were washed using distilled water until reach pH 6.
Bleaching treatment was performed to extract cellulose fibers. A solution was prepared using distilled water (120 mL), glacial acetic acid (1 mL) and sodium chlorite (2.5 g). Then, lignocellulosic fibers (3 g) were added to this solution under constant stirring for 1 h at (70 ± 2) °C. An additional portion of glacial acetic acid (1 mL) and sodium chlorite (2.5 g) were added to the system under constant stirring for 2 h. This procedure was repeated, and the system was maintained at constant stirring for 1 h. After reaching room temperature, the system was filtered and washed using distilled water until reach pH 6 for the obtainment of the bleached fibers.
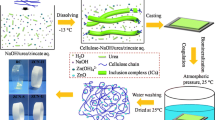
Interfacial polymerization and nanocomposite preparation
Interfacial polymerization of o-anisidine and o-phenetidine (heterogeneous biphasic system) was performed at 27 °C and based on previous report (Ferreira and Sanches 2017) with marginal modifications.
An amount of 2.16 g of o-anisidine was added to 100 mL of chloroform to form the organic phase. On the other hand, 3.42 g of APS was dissolved in HCl (100 mL, 1 M) to form the aqueous phase. Then, the aqueous phase was added drop by drop to the organic phase. The system remained without any stirring for 24 h (interfacial polymerization). The dark precipitate (POMA) was filtered, washed with acetone, and dried in a desiccator until achieve constant weight. The same methodology was performed using o-phenetidine to prepare POEA.
For the preparation of nanocomposites, the cellulose extracted previously from the P. pellucida stems was added to the organic phase described above at ratio of (1:0.25, w/w) monomer/cellulose allowing the in situ interfacial polymerization. Thus, the nanocomposites POMA/Cellulose and POEA/Cellulose were obtained.
XRD analysis
XRD data were obtained at the Laboratório de Materiais (LabMat—UFAM) in a Panalytical diffractometer, Empyrean model, operating with CuKα, 40 kV and 40 mA. XRD patterns were collected in the range 2θ = 5–70°, step of 0.013° and 5 s/step.
Le Bail method
The software package Fullprof (Rodríguez-Carvajal 2002) was used to perform the Le Bail method (Le Bail 2005). The least squares method (Pawley 1981) was applied to refine cell parameters. Peak profile was adjusted according to the pseudo-Voigt function modified by Thompson–Cox–Hastings (Thompson et al. 1987). A LaB6 crystal standard was employed to obtain the Instrumental Resolution Function (IRF) parameters. Crystal data from the end-capped tetrameter of aniline (Evain et al. 2002) were considered as initial parameters for POMA and POEA refinement (triclinic, P\(\stackrel{-}{1}\); a = 5.7328 Å; b = 8.8866 Å; c = 22.6889 Å; α = 82.7481°; β = 84.5281° and γ = 88.4739°). Crystal data of cellulose polymorphs Iα (triclinic; a = 6.717 Å; b = 5.962 Å; c = 10.400 Å; α = 118.080°; β = 114.800° and γ = 80.370°) and Iβ (monoclinic; a = 7.784 Å; b = 8.201 Å; c = 10.380 Å; α = 90°; β = 90° and γ = 96.550°) were considered as initial parameters for the extracted cellulose refinement (French 2014). Crystallite anisotropy was evaluated using linear combination of spherical harmonics (SHP) (Popa 1998).
Thermal analysis
TG/dTG analyses were carried out on a TA Instruments model STD Q600 at the Laboratório de Materiais da Amazônia e Compósitos (LaMAC-FT/UFAM). Measurements were carried out using 10–12 mg of samples in alumina crucibles, N2 atmosphere (flow of 10 mL/min) at heating rate of 10 °C/min, from 25 to 700 °C.
SEM analysis
SEM experiments were performed on a Carl Zeiss equipment model Supra 35, using 1.0 kV at 25 °C. Powder samples were placed on a carbon tape and recovered with a thin gold layer.
Results and discussion
XRD analysis
The semi-crystalline XRD pattern of the extracted cellulose is shown in Fig. 1. Table 1 shows the angular peak positions (2θ) of the obtained XRD patterns. Well-defined diffraction peaks were observed at 2θ = 16.0°, 2θ = 22.4° and 2θ = 35.0° as a result of some regular alignment of the cellulose chains. This parallel, regular alignment allowed the formation of the so-called crystallites, which are embedded in the regions where the polymer chains are randomly oriented. The observed broad peaks are due to the nanosized crystallites.

Semi-crystalline XRD patterns of the a extracted cellulose, POMA and POMA/Cellulose nanocomposite, and b extracted cellulose, POEA and POEA/Cellulose nanocomposite
Cellulose polymorphism has been associated with the source and extraction methodology (Abdul Khalil et al. 2012; French 2014). According to the obtained XRD pattern, the extracted cellulose could be described as type Iα (triclinic) or Iβ (monoclinic) (French 2014) since these both polymorphs presented diffraction peaks at similar angular positions. For this reason, the Le Bail refinement (Le Bail 2005) was performed to determine the extracted cellulose polymorph.
The semi-crystalline XRD patterns of the as-synthesized POMA and POEA are shown in Fig. 1a, b, respectively. To the best of our knowledge, no reports on structural characterization of POMA or POEA prepared by interfacial polymerization have been found in the scientific literature. Previous work reported the conventional chemical polymerization of POMA at different time of synthesis. Diffraction peaks were found at 2θ = 7.9°, 12.5°, 17.5°, 24.8° and 26.8° (Sanches et al. 2013). Similarly, the angular peak positions of the chemically synthesized POEA were found at 2θ = 4°, 8°, 12°, 16°, 24°, 26°, 38°, 44° and 52° (Leite et al. 2008).
Although the polymerization methodology reported in the present work is different from the above-mentioned reports, the angular peak positions were almost similar. However, peak displacements to smaller angular positions were observed in the XRD pattern of POEA when compared to that of POMA. These angular displacements are related to different unit cell parameters and, consequently, to the different size of the side group located in the aromatic rings at the ortho position: POMA and POEA present o-OCH3 and o-OCH2CH3 side groups, respectively. For this reason, larger lattice parameters are required to accommodate the POEA molecules in its unit cell (Sanches et al. 2013; Silva et al. 2014).
The nanocomposite POMA/Cellulose (Fig. 1a) presented diffraction peaks at similar angular positions from those found in the as-synthesized POMA. This fact indicates that the interfacial polymerization in the presence of cellulose was successfully performed. Peak displacements to larger angular positions were observed when compared to the XRD pattern of the pure POMA. On the other hand, peak displacement to lower angular positions was verified when compared to the XRD pattern of the extracted cellulose. These results are significantly related to decrease and increase of the lattice parameters of POMA and cellulose, respectively, in the nanocomposite form. Chemical interaction between the conjugated polymer and cellulose during the in situ interfacial polymerization was suggested.
The nanocomposite POEA/Cellulose (Fig. 1b) presented smaller number of diffraction peaks when compared to the as-synthesized POEA. Particularly between 2θ = 20°–30°, the most intense diffraction contribution was related to cellulose due to the more intense diffraction peak at 2θ = 22.3°. This result may be related to the less effective polymerization of POEA in the presence of cellulose when compared to in situ interfacial polymerization of POMA due to the different molecular structure of both monomers.
Le Bail method
The extracted cellulose presented diffraction pattern possibly described as type Iα or Iβ (French 2014). For this reason, the Le Bail method was performed to confirm the extracted cellulose polymorph. This tool has been useful to propose unit cell parameters for semi-crystalline materials, as well as to assess crystallite size and shape. Table 2 shows the refined parameters.
The structural refinement of the extracted cellulose considering the polymorphs Iα and Iβ were consistent with the triclinic and monoclinic crystal structures, respectively. However, the refined crystallite size values along the [100] and [001] directions were found, respectively, around 5 Å and 7 Å (considering the monoclinic structure, Iβ), which are physically inconsistent with the refined cell parameters a and b, respectively. For this reason, the crystal structure of the extracted cellulose is described only as the polymorph Iα.
According to Table 2, the largest crystallite size was found along the [010] direction. In contrast, the smallest size was expected to be found along the larger lattice parameter, [001]. The crystallite average size was found around 29 Å. The reflection planes (hkl) of major contribution in the diffraction peaks of the extracted cellulose were (− 101), (0-11) and (010) for the peak located at 2θ = 16°; (002), (1-10) and (110) for the peak located at 2θ = 22.3°; and (020) and (201) for the peak at 2θ = 35°. Crystallite size and shape projections of the extracted cellulose along [100], [010] and [001] directions are shown in Fig. 2. A crystallite prolate-like shape was found in the extracted Iα cellulose, as clearly observed along [001] × [010] directions (Fig. 2a).

Crystallite size and shape projections of the extracted Iα cellulose along [100], [010] and [001] directions
The crystal structure of the as-synthesized POMA and POEA was consistent with the triclinic system. The c refined parameters were found around 18.76–18.86 Å, suggesting that POMA and POEA presented four repetitive units per unit cell (tetrameric molecules) arranged along the [001] direction. An increase of the refined unit cell parameter b was observed in POEA when compared to that of POMA (Table 3), which is related to the larger o-CH2CH3 side group of POEA. For this reason, the chain alignment along the [010] direction was considerably affected (Silva et al. 2014). As a result, the unit cell volume of POEA (1752 Å3) was higher than that of POMA (1434 Å3). These results agreed with the XRD data highlighting the displacements of the angular peak positions. According to Table 3, the largest crystallite size was found along the [100] direction. In contrast, the smallest size was found along the [010] direction probably due to the side group presented in the aromatic rings of the polymers, which impairs the regular alignment of polymer chains along this direction. Although both polymers presented similar average crystallite size (~ 32 Å), POEA presented higher crystallite growth along the [100] direction (~ 52 Å). This fact can be explained due to the larger lateral group, increasing the difficulty of alignment.
The triclinic crystal system of POMA and POEA was maintained after the in situ interfacial polymerization over cellulose surface to form, respectively, the nanocomposites POMA/Cellulose and POEA/Cellulose. A decrease of the unit cell volume from 1434 Å3 (pure POMA) to 1411 Å3 (POMA in the nanocomposite form) was observed. These results were expected since slight displacements of the angular peak positions to larger Bragg angles were observed. On the other hand, an increase of the unit cell volume from 298 Å3 (pure extracted cellulose) to 327 Å3 (cellulose in the nanocomposite form) was also observed. This result was also expected since slight displacements of the angular peak positions to larger Bragg angles were observed.
The structural parameters of the nanocomposite POEA/Cellulose were similar to those of POMA/Cellulose. The triclinic crystal system was also maintained after the in situ interfacial polymerization of POEA over the cellulose surface. A decrease of the unit cell volume was observed from 1752 Å3 (for pure POEA) to 1715 Å3 (for POEA in the nanocomposite form). On the other hand, an increase of the unit cell volume of cellulose was also increased from 298 Å3 (for pure extracted cellulose) to 332 Å3 (for the cellulose in the nanocomposite form), corroborating with the observed angular displacements of the peak positions of both POEA and cellulose phases.
These results suggested a structural modification of the cellulose fibers by the in situ polymerization of POMA or POEA. The cellulose chains have three hydroxyl groups that form the intermolecular and intramolecular hydrogen bonds (Moon et al. 2011; Jonoobi et al. 2015). Possible chemical interactions between the o-OCH3 or o-OCH2CH3 groups (from POMA and POEA, respectively) and the cellulose chains resulted in the increase of the unit cell volume of the nanocomposites. This interaction may also be the reason for the increase of the average crystallite size of both conjugated polymers and cellulose in the nanocomposite form, indicating that the in situ polymerization over the cellulose surface may result in an oriented polymerization in nanocomposites when compared to the pure polymers.
Crystallite size and shape projections of the as-synthesized POMA and POEA along [100], [010] and [001] directions are shown in Fig. 3a–f. Due to the different anisotropic growths of crystallites, their shapes were slightly different, but in general tending to prolate-like shape.

Crystallite size and shape projections of the as-synthesized POMA and POEA along [100], [010] and [001] directions
Figure 4 shows the crystallite shape projections along [100], [010] and [001] directions of the nanocomposites. Despite observing structural changes in the nanocomposite’s refinement when compared to the pure polymers, no significant changes in the crystallite shape were observed.

Crystallite shape projections along [100], [010] and [001] directions for the phases of the nanocomposites
The polymerization of POMA and POEA over cellulose surface considerably influenced the crystallite size. The in situ polymerization clearly favored the crystallite growth. The average crystallite size of POMA changed from 32 Å (for pure POMA) to 41 Å (for POMA in the nanocomposite form). The average crystallite size of POEA changed from 32 Å (for pure POEA) to 44 Å (for POEA in the nanocomposite form). The cellulose crystallites were also found larger: initially the extracted cellulose presented average crystallite size of 29 Å, while the cellulose crystallites in the nanocomposites were found around 59 Å (POMA/Cellulose) and 92 Å (POEA/Cellulose). The structural refinement suggests interactions between reinforcement and matrix, resulting in ordered polymer chains and larger average crystallite sizes.
Thermal analysis
Figure 5 shows the TG/dTG curves of the in natura stems of P. pellucida, POMA, POEA and nanocomposites (POMA/Cellulose and POEA/Cellulose).

TG/dTG curves of the a in natura stems of P. pellucida, b POMA and POMA/Cellulose nanocomposite and c POEA and POEA/Cellulose nanocomposite
The first thermal event identified in the in natura stems (Fig. 5a) was observed from 25 to 150 °C, which was assigned to the moisture release, resulting in 11.8% of mass loss. The second event was observed from 210 to 360 °C, resulting in 38.9% of mass loss. This wide range of temperature was related to the degradation of hemicellulose, lignin and cellulose (Chen et al. 2011; Abdul Khalil et al. 2012; Zhao et al. 2017; Yu et al. 2017). The first possibly degraded compound was hemicellulose, since its thermal degradation has been reported from 200 °C and depend on the lignocellulosic source; cellulose presents degradation temperature from 280 to 350 °C; lignin presents the highest degradation temperature, ranging from 200 to approximately 600 °C. However, the thermal degradation of lignin may be extended up to 900 °C (Peng and Wu 2010; Werner et al. 2014; Zhao et al. 2017; Yu et al. 2017). The third thermal event represents a set of decomposition reactions occurring simultaneously from 400 to 700 °C. These reactions may be related to the remaining lignin from the second thermal event, as well as to the gradual decomposition process of lignin intermediates (Brebu and Vasile 2010; Stefanidis et al. 2014; Zhao et al. 2017; Yu et al. 2017). The in natura stems of P. pellucida presented residual mass about 20.9% corresponding to the ashes resulting from the thermal degradation process, which also may be constituted of inorganic compounds (Llorente et al. 2006).
The first thermal event of the extracted cellulose (Fig. 5a) was observed from 25 to 100 °C and was assigned to the moisture release. The second event was observed from 270 to 350 °C and was assigned to the break of the main chain bonds (Jonoobi et al. 2015). The extracted cellulose presented higher thermal stability than the in natura stems due to the removal of components contributing to the degradation process, such as oils, greases and hemicellulose (C.S. et al. 2016). The third thermal event was observed from 380 to 530 °C and was related to the lignin decomposition, which probably was not totally removed previously (Chen et al. 2011; Abdul Khalil et al. 2014). The extracted cellulose presented 3.2% of residual mass, suggesting that the components from the in natura stems were efficiently removed (Spinacé et al. 2009; Manzato et al. 2017).
Figure 5b shows the TG/dTG curves of the as-synthesized POMA and POMA/Cellulose nanocomposite. The first thermal event observed for POMA was verified from 25 to 132 °C and assigned to the moisture release (Sreedhar et al. 2006; Alves et al. 2010). Two competing reactions were observed from 132 to 270 °C: the first reaction presented maximum dTG at 166 °C and was assigned to the release of hydrated water, while the second reaction presented maximum dTG at 199 °C (starting before the previous reaction was completed) and was assigned to the release of counterions (Alves et al. 2010; Ferreira and Sanches 2017). The fourth and fifth thermal events presented maximum temperatures of 367 °C and 535 °C, respectively, and were associated to the chemical reactions resulted from the polymer degradation (Kulkarni et al. 1989; Sreedhar et al. 2006).
The TG/dTG curves of the POMA/Cellulose presented the first thermal event from 25 to 141 °C assigned to the moisture release (Kulkarni et al. 1989; Raghunathan et al. 2017). Three competing reactions were observed from 141 to 300 °C: the first one was represented by a shoulder observed from 141 to 202 °C and was attributed to the release of hydration water; the second one was observed from 202 to 231 °C (starting before the previous reaction was completed) and was assigned to the release of counterions (Lee et al. 2012; He et al. 2016; Raghunathan et al. 2017); the third one was observed from 231 to 300 °C and was assigned to the cellulose degradation. This peak was observed in lower temperatures when compared to the extracted cellulose (Fig. 5a) and can be explained by the influence of the counterions on the degradation process. The fourth event was observed from 300 to 400 °C and may be related to polymer degradation (Lee et al. 2012; He et al. 2016). This event was also observed in lower temperatures when compared to the as-synthesized POMA and can be explained by the interactions between matrix and reinforcement, which can influence on the degradation process. The fifth thermal event was observed from 400 °C, and was related to the prolongation of the polymer chains degradation.
The first thermal event of POEA (from 25 to 150 °C, Fig. 5c) was associated to the moisture release. Two competing reactions were observed from 150 to 300 °C: the first one presented maximum dTG at 193 °C and was assigned to the release of hydration water; the second one (a shoulder presenting maximum dTG at approximately 216 °C) started before the previous reaction was completed, and was assigned to the release of counterions (Kulkarni et al. 1989; Sreedhar et al. 2006). The fourth event (from 300 to 480 °C) resulted from the breakdown of the POEA chains (Sreedhar et al. 2006; Alves et al. 2010), which was extended over 700 °C.
The TG/dTG curves of the POEA/Cellulose nanocomposite are similar to previous reports on cellulose matrix composites (Yan and Xu 2015; He et al. 2016). Five main thermal events were observed: the first one (from 25 to 150 °C) was assigned to the moisture related. The second event was observed from 150 to 230 °C and was assigned to the release of hydration water. For this event, the maximum peak observed on the dTG curve was found at 192 °C. Two competing reactions were observed from 230 to 300 °C: the first one presented maximum dTG temperature of 275 °C and was related to the release of counterions; the second one was represented by a shoulder starting at 300 °C and assigned to the cellulose degradation (starting before the previous reaction was completed). The release of counterions was observed at higher temperatures when compared to the as-synthesized POEA probably due to the interactions between matrix and reinforcement. Some reports attribute this stage to the start of the polymer degradation in association to the cellulose chain degradation. Then, the fifth thermal event started from 440 °C and was assigned to the polymer chain degradation.
Some reports have shown an improvement of the thermal stability of cellulose in the nanocomposite form, attributing to the conducting polymer a retarding action of mass loss (Lee et al. 2012; He et al. 2016; Raghunathan et al. 2017). However, these results were not observed here, since the thermal stability of the samples was evaluated as a function of the temperature at which the degradation of the polymer chains was started. For this reason, we suggest the lower stability of the nanocomposites due to the presence of counterions in the polymer chains: these counterions may have contributed to accelerate the degradation process with the increase of temperature.
Morphological analysis
Figure 6a shows the SEM image of the in natura stems of P. pellucida, highlighting its rough surface composed of several filaments. Roughness was reduced—Fig. 6b—after chemical treatment, revealing a crumpled paper-like morphology. Figure 6c, d reveals several nanofibrils, which may be a result of the dissolution of the plant components during the alkaline treatment and bleaching processes, resulting in the separation of the cellulose filaments presented in the cell wall layers.

a SEM image of the in natura stems of P. pellucida; b crumpled paper-like morphology of the extracted cellulose; c, d nanofibrils of the extracted cellulose
The morphology of the as-synthesized conjugated polymers is shown in Fig. 7. Randomly dispersed microplates and granules were verified in POMA morphology (Fig. 7a, b), indicating a competitive growth between these morphologies (multimorphology) during the polymerization process. This result may be related to the synthesis methodology. Sanches et al. (2013) reported the conventional chemical synthesis of POMA and verified vesicle globular morphology. Siva and Sathiyanarayanan (2016) obtained chemically synthesized POMA nanospheres in the presence of cationic surfactant in aqueous medium. Zhang et al. (2009) verified the influence of temperature and reaction time on the formation of POMA microspheres, revealing that the kinetics polymerization plays an important role on the amount of fully formed microspheres. The POEA morphology was more uniform than that observed on POMA images, presenting micrometric granules constituted of nanosized structures similar to nanofibers (Fig. 7b, c). Unlike POMA, POEA did not present multimorphology. This fact was probably due to the larger size of the side group (O–CH2CH3) of POEA (which may have influenced the fashion at which the polymer chains were arranged), as well as to the interfacial polymerization methodology.

SEM images of the as-synthesized (a, b) POMA and (c, d) POEA
Figure 8 shows the morphology of POMA/Cellulose and POEA/Cellulose nanocomposites. The morphology of the nanocomposites presented different characteristics when compared to those observed in the pure polymers. In addition, the conjugated polymers presented a tendency of polymerization on the surface of the fibers, acting as a polymeric reinforcement phase in the nanocomposites. Furthermore, this characteristic may be related to a chemical interaction between matrix (cellulose) and reinforcement (POMA or POEA).

SEM images of the as-synthesized a, b POMA/Cellulose and c, d POEA/Cellulose nanocomposites
The SEM images of the POMA/Cellulose (Fig. 8a, b) showed more uniform morphology than that observed in pure POMA (Fig. 7a, b). This fact was also probably due to a possible chemical interaction between cellulose fibers and POMA: the cellulose fiber may have acted as a pathway for polymerization, probably orienting the polymer growth on its surface due to the interaction between both phases. Furthermore, the polymer morphology on the nanocomposite form was significantly different from that observed in the pure POMA: globular vesicular morphology was formed during the polymerization over the cellulose fibers. A total recovery of an individual cellulose fiber, as well as the formation of polymer vesicles is shown in Fig. 8b.
Regarding the POEA/Cellulose nanocomposite (Fig. 8c, d), the cellulose fibers were also completely recovered by POEA in the form of granule-like nanostructures. The morphology of POEA in the nanocomposite form was also different from that observed in the pure POEA. The in situ interfacial polymerization promoted the interaction between both phases, modifying the surface of the cellulose fibers. The complete recovery of the cellulose fibers also suggested a chemical interaction between matrix and reinforcement. Cellulose fibers acted as a pathway for polymerization, probably orienting the polymer growth on its surface. This fact significantly influenced the polymer morphology: in the nanocomposite form, POEA consisted of well-defined nanometric spheres. In conventional polymerization, similar results were obtained by Stejskal et al. (2008) using polyaniline as reinforcement.
Conclusions
POMA/Cellulose and POEA/Cellulose were successfully synthesized based on the in situ polymerization over cellulose fibers extracted from the Amazon P. pellucida. The methodology applied to obtain cellulose promoted the removal of a significant content of their constituents, such as lignin and hemicellulose, promoting a better thermal stability of the extracted cellulose when compared to the in natura stems. The Le Bail method represents an important tool to characterize semi-crystalline materials and allowed the identification of the extracted cellulose as type Iα. In addition, this tool was also useful to determine the cell parameters of extracted cellulose, polymers, as well as the individual phases of nanocomposites. The crystalline structure of POMA and POEA were significantly influenced by the presence of their respective side groups, directly influencing on lattice parameters of the unit cells, as well as on the crystallite size. However, the crystallites (both polymeric and cellulose) of the nanocomposites were larger when compared to the crystallites of the individual phases. This fact occurred probably because cellulose acted as a pathway for polymerization, orienting the growth of the polymers on its surface through a possible chemical interaction that favored the alignment of the chains. The possible chemical interaction between matrix and reinforcement also influenced the polymer morphology. Finally, we suggest the lower stability of the nanocomposites due to the presence of counterions in the polymer chains.
References
Abdul Khalil HPS, Bhat AH, Ireana Yusra AF (2012) Green composites from sustainable cellulose nanofibrils: a review. Carbohydr Polym 87:963–979. https://doi.org/10.1016/j.carbpol.2011.08.078
Abdul Khalil HPS, Davoudpour Y, Islam MN et al (2014) Production and modification of nanofibrillated cellulose using various mechanical processes: a review. Carbohydr Polym 99:649–665. https://doi.org/10.1016/j.carbpol.2013.08.069
Alves WF, Venancio EC, Leite FL et al (2010) Thermo-analyses of polyaniline and its derivatives. Thermochim Acta 502:43–46. https://doi.org/10.1016/j.tca.2010.02.003
Brebu M, Vasile C (2010) Thermal degradation of lignin—a review. Cell Chem Technol 44:353–363
Chen W, Yu H, Liu Y et al (2011) Isolation and characterization of cellulose nanofibers from four plant cellulose fibers using a chemical-ultrasonic process. Cellulose 18:433–442. https://doi.org/10.1007/s10570-011-9497-z
CS JC, George N, Narayanankutty SK (2016) Isolation and characterization of cellulose nanofibrils from arecanut husk fibre. Carbohydr Polym 142:158–166. https://doi.org/10.1016/j.carbpol.2016.01.015
Da Silva MHL, Zoghbi MDGB, Andrade EHA, Maia JGS (1999) The essential oils of Peperomia pellucida Kunth and P. circinnata Link var. circinnata. Flavour Fragr J 14:312–314. https://doi.org/10.1002/(SICI)1099-1026(199909/10)14:5%3c312::AID-FFJ835%3e3.0.CO;2-B
Dai L, Si C (2018) Recent advances on cellulose-based nano-drug delivery systems: design of prodrugs and nanoparticles. Curr Med Chem 26:2410–2429. https://doi.org/10.2174/0929867324666170711131353
Evain M, Quillard S, Corraze B et al (2002) A phenyl-end-capped tetramer of aniline. Acta Crystallogr Sect E Struct Rep Online 58:o343–o344. https://doi.org/10.1107/S1600536802002532
Fei G, Wang Y, Wang H et al (2019) Fabrication of bacterial cellulose/polyaniline nanocomposite paper with excellent conductivity, strength, and flexibility. ACS Sustain Chem Eng 7:8215–8225. https://doi.org/10.1021/acssuschemeng.8b06306
Ferreira AA, Sanches EA (2017) Multimorphologies of hydrochloride polyaniline synthesized by conventional and interfacial polymerization. J Mol Struct 1143:294–305. https://doi.org/10.1016/j.molstruc.2017.04.104
French AD (2014) Idealized powder diffraction patterns for cellulose polymorphs. Cellulose 21:885–896. https://doi.org/10.1007/s10570-013-0030-4
Hanif Z, Khan ZA, Siddiqui MF et al (2019) Polypyrrole-based conducting and antibacterial hybrid cellulose membranes: a study on the effect of UV exposure on the conductivity and formation of silver nanoparticles. Sensors Mater 31:1927–1938. https://doi.org/10.18494/SAM.2019.2310
He W, Tian J, Li J et al (2016) Characterization and properties of cellulose nanofiber/polyaniline film composites synthesized through in situ polymerization. BioResources 11:8535–8547. https://doi.org/10.15376/biores.11.4.8535-8547
Jahan K, Kumar N, Verma V (2018) Removal of hexavalent chromium from potable drinking using a polyaniline-coated bacterial cellulose mat. Environ Sci Water Res Technol 4:1589–1603. https://doi.org/10.1039/c8ew00255j
Jie LIU, Huaifang W, Lin Z (2019) Preparation, structure and performances of cross-linked regenerated cellulose fibers. Wuhan Univ J Nat Sci 24:1–7
Jonoobi M, Oladi R, Davoudpour Y et al (2015) Different preparation methods and properties of nanostructured cellulose from various natural resources and residues: a review. Cellulose 22:935–969. https://doi.org/10.1007/s10570-015-0551-0
Kulkarni VG, Campbell LD, Mathew WR (1989) Thermal stability of polyaniline. Synth Met 30:321–325. https://doi.org/10.1016/0379-6779(89)90654-1
Le Bail A (2005) Whole powder pattern decomposition methods and applications: a retrospection. Powder Diffr 20:316–326. https://doi.org/10.1154/1.2135315
Lee BH, Kim HJ, Yang HS (2012) Polymerization of aniline on bacterial cellulose and characterization of bacterial cellulose/polyaniline nanocomposite films. Curr Appl Phys 12:75–80. https://doi.org/10.1016/j.cap.2011.04.045
Leite FL, Alves WF, Mir M et al (2008) TEM, XRD and AFM study of poly(o-ethoxyaniline) films: new evidence for the formation of conducting islands. Appl Phys A Mater Sci Process 93:537–542. https://doi.org/10.1007/s00339-008-4686-9
Llorente M, Laplaza J, Cuadrado R, García J (2006) Ash behaviour of lignocellulosic biomass in bubbling fluidised bed combustion. Fuel 85:1157–1165. https://doi.org/10.1016/j.fuel.2005.11.019
Manzato L, Rabelo LCA, de Souza SM et al (2017) New approach for extraction of cellulose from tucumã’s endocarp and its structural characterization. J Mol Struct 1143:229–234. https://doi.org/10.1016/j.molstruc.2017.04.088
Moon RJ, Martini A, Nairn J et al (2011) Cellulose nanomaterials review: structure, properties and nanocomposites. Chem Soc Rev 40:3941. https://doi.org/10.1039/c0cs00108b
Pawley GS (1981) Unit-cell refinement from powder diffraction scans. J Appl Crystallogr 14:357–361. https://doi.org/10.1107/S0021889881009618
Peng Y, Wu S (2010) The structural and thermal characteristics of wheat straw hemicellulose. J Anal Appl Pyrolysis 88:134–139. https://doi.org/10.1016/j.jaap.2010.03.006
Popa NC (1998) The (hkl) dependence of diffraction-line broadening caused by strain and size for all Laue groups in rietveld refinement. J Appl Crystallogr 31:176–180. https://doi.org/10.1107/S0021889897009795
Raghunathan SP, Narayanan S, Poulose AC, Joseph R (2017) Flexible regenerated cellulose/polypyrrole composite films with enhanced dielectric properties. Carbohydr Polym 157:1024–1032. https://doi.org/10.1016/j.carbpol.2016.10.065
Rodríguez-Carvajal J (2002) An introduction to the program (Version July 2001). Lab Leon Brillouin 1–139
Sanches EA, Soares JC, Mafud AC et al (2013) Structural and morphological characterization of chloride salt of conducting poly(o-methoxyaniline) obtained at different time synthesis. J Mol Struct 1039:167–173. https://doi.org/10.1016/j.molstruc.2012.12.025
Sharifi H, Zabihzadeh M, Ghorbani M (2018) The application of response surface methodology on the synthesis of conductive polyaniline/cellulosic fiber nanocomposites. Carbohydr Polym 194:384–394. https://doi.org/10.1016/j.carbpol.2018.04.083
Silva ADS, Soares JC, Mafud AC et al (2014) Structural and morphological characterization of poly(o-ethoxyaniline) emeraldine-salt form using FTIR, XRD, Le Bail method and SEM. J Mol Struct 1071:1–5. https://doi.org/10.1016/j.molstruc.2014.04.039
Siva T, Sathiyanarayanan S (2016) Cationic surfactant assisted synthesis of poly o-methoxy aniline (PoMA) hollow spheres and their self healing performance. RSC Adv 6:2944–2950. https://doi.org/10.1039/C5RA23090J
Spinacé MAS, Lambert CS, Fermoselli KKG, De Paoli MA (2009) Characterization of lignocellulosic curaua fibres. Carbohydr Polym 77:47–53. https://doi.org/10.1016/j.carbpol.2008.12.005
Sreedhar B, Sairam M, Chattopadhyay DK et al (2006) Thermal and XPS studies on polyaniline salts prepared by inverted emulsion polymerization. J Appl Polym Sci 101:499–508. https://doi.org/10.1002/app.23301
Stefanidis SD, Kalogiannis KG, Iliopoulou EF et al (2014) A study of lignocellulosic biomass pyrolysis via the pyrolysis of cellulose, hemicellulose and lignin. J Anal Appl Pyrolysis 105:143–150. https://doi.org/10.1016/j.jaap.2013.10.013
Stejskal J, Trchová M, Kovářová J et al (2008) Polyaniline-coated cellulose fibers decorated with silver nanoparticles. Chem Pap 62:181–186. https://doi.org/10.2478/s11696-008-0009-z
Svenningsson L, Lin Y, Karlsson M et al (2019) Molecular orientation distribution of regenerated cellulose fibers investigated with polarized raman spectroscopy. Macromolecules 52:3918–3924. https://doi.org/10.1021/acs.macromol.9b00520
Thompson P, Cox D, Hastings J (1987) Rietveld refinement of Debye-Scherrer synchrotron X-ray data from Al2O3. J Appl Crystallogr 20:79–83. https://doi.org/10.1107/s0021889887087090
Werner K, Pommer L, Broström M (2014) Thermal decomposition of hemicelluloses. J Anal Appl Pyrolysis 110:130–137. https://doi.org/10.1016/j.jaap.2014.08.013
Yan J, Xu R (2015) Reinforced conductive polyaniline-paper composites. BioResources 10:4065–4076. https://doi.org/10.15376/biores.10.3.4065-4076
Yu J, Paterson N, Blamey J, Millan M (2017) Cellulose, xylan and lignin interactions during pyrolysis of lignocellulosic biomass. Fuel 191:140–149. https://doi.org/10.1016/j.fuel.2016.11.057
Zhang L, Peng H, Sui J et al (2009) Self-assembly of poly(o-methoxyaniline) hollow microspheres. J Phys Chem C 113:9128–9134. https://doi.org/10.1021/jp900267t
Zhao C, Jiang E, Chen A (2017) Volatile production from pyrolysis of cellulose, hemicellulose and lignin. J Energy Inst 90:902–913. https://doi.org/10.1016/j.joei.2016.08.004
Zheng W, Lv R, Na B et al (2017) Nanocellulose-mediated hybrid polyaniline electrodes for high performance flexible supercapacitors. J Mater Chem A 5:12969–12976. https://doi.org/10.1039/C7TA01990D
Acknowledgements
This work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq—Grant number 305161/2017-2), Fundação de Amparo à Pesquisa do Estado do Amazonas (FAPEAM—Processo 062.00121/2019), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and Laboratório de Materiais (LabMat e UFAM) for the X-ray difraction measurements.
Author information
Authors and Affiliations
Contributions
BAF carried out the experiment, verified the analytical methods and performed the computations. ALFR, SXL, LMO and MMB contributed to the interpretation of the results. EAS conceived the original idea, supervised the findings of this work and wrote the manuscript with support from PHC. All the authors discussed the results and contributed to the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
de A. Feitosa, B., Rocha, A.L.F., Lima, S.X. et al. Nanocomposites based on the cellulose extracted from the Amazon Peperomia pellucida and polyaniline derivatives: structural and thermal properties. Chem. Pap. 75, 1809–1821 (2021). https://doi.org/10.1007/s11696-020-01435-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-020-01435-4