
Abstract
Studies about diversification of Neotropical open-habitat organisms are scarce, especially in frogs, in which almost all studied species from Cerrado, Chaco, and Caatinga are mainly associated with gallery forest. Here we tested hypotheses about diversification in Neotropical open habitats (savannas and grasslands) using as model a small treefrog associated with these environments and widely distributed throughout South America. We sampled 68 individuals from 43 localities throughout the geographic distribution of Scinax fuscomarginatus and used one mitochondrial and three nuclear DNA markers to define genetic clusters. Based on population assignments and previously proposed diversification hypotheses for the Cerrado, we designed different models to be tested by approximate Bayesian computation (ABC). We found four genetic clusters which reflect the current distribution of open habitats of the Central Brazilian plateau and of the Cerrado lowlands. Among the models tested with ABC, the best-supported model was a scenario of multiple refugia in which all populations experienced population size reduction (bottleneck) in a relatively recent time, with the most ancient split event surrounding the Last Interglacial (LIG). During interglacial periods, warmer and wetter climate would have favored the geographic expansion of forests, resulting in the retraction and fragmentation of open habitats like savannas and grasslands. Populations of species associated with open habitats, including S. fuscomarginatus, would have remained isolated in relatively small stable areas with reduced gene flow among them.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Open habitats (savannas and grasslands) of the Neotropics occupy, semi-continuously, several ecoregions, mainly Cerrado and Pampas, and dominate important extensions of the Humid Chaco (Van Els et al., 2021). These formations are also present, but as smaller enclaves, in Caatinga, Amazon forest, Atlantic forest (semideciduous), and lowlands of northern South America, like the Guianan Savanna (Salgado-Laboriau, 1997; Behling and Hooghiemstra, 2001).
Several vertebrate species are associated with open habitats in the Neotropics; however, the evolutionary history of only a few has been studied. In this context, birds are the most studied group (Van Els et al., 2021; Ritter et al., 2021; Lima-Rezende et al., 2022), followed by reptiles (Quijada-Mascareñas et al., 2007; Santos et al., 2014). In frogs, several species mainly associated with Cerrado, Chaco, or Caatinga, and some species from the Diagonal of Open Formations (combination of Caatinga-Cerrado-Chaco) have been studied (e.g., Prado et al., 2012; Thomé et al., 2016; Oliveira et al., 2018; Brusquetti et al., 2019; Vasconcellos et al., 2019). Of these, only Dermatonotus muelleri (Boettger, 1885) inhabit open habitats (Oliveira et al., 2018), and the other species are associated with gallery forests and/or with the borders of gallery forests of the Cerrado (Prado et al., 2012; Vasconcellos et al., 2019) or occur mainly in dry forests of the Caatinga and the Chaco (Thomé et al., 2016; Brusquetti et al., 2019).
The Cerrado ecoregion harbors the major and most continuous savanna-like environments in the Neotropics (Salgado-Labouriau, 1997). The compartmentalization of the Cerrado landscape between plateaus (above 500 m altitude) and depressions (below 500 m altitude) due to the uplift of the Central Brazilian Plateau (CBP, 2–7 Mya; Silva, 1997) is suggested as one of the most important vicariant events that could have influenced the diversification of Cerrado species (Nogueira et al., 2011; Werneck, 2011). This event has been suggested as responsible for the diversification of some frogs and lizards (Werneck et al., 2012a; Recoder et al., 2014; Guarnizo et al., 2016; Oliveira et al., 2018), but with different mechanisms, including vicariance and dispersal. For example, a vicariant event was evoked to explain the diversification of the frog D. muelleri (Oliveira et al., 2018), suggesting that the highlands of the CBP would have split a widely distributed ancestral population. In a scenario of dispersion, the so-called plateau-depression hypothesis (Santos et al., 2014) suggests that the ancient plateaus harbor older lineages than the recently formed depressions, which were formed by erosion during the Quaternary and hold younger lineages resulting from recent colonization events (Silva, 1997).
Another hypothesis that has been invoked to explain diversification in open habitats is based on the Pleistocene refugia hypothesis (Haffer, 1969). This hypothesis suggests that Pleistocene climatic fluctuations would have induced several changes in South America vegetation cover. For example, the Amazon forest, during glacial periods, would have been invaded by tracts of savanna, isolating the forest in patches that would serve as refuges for forest-dependent taxa, leading to allopatric speciation. In contrast, during interglacial periods, the forest would have expanded and reconnected, leading to the range expansion of newly diversified taxa (Haffer, 1969).
Pleistocene climatic changes probably also led to vegetation changes in the Cerrado and other ecoregions, like Humid Chaco and Pampas. The warmer and more humid climate during interglacial periods would have promoted the geographic expansion of forest species, causing the reduction and fragmentation of savanna and grassland formations (Mäder & Freitas, 2019; Giudicelli et al., 2021), thus isolating animals associated with these environments. This hypothesis, named “isolation by instability”, suggests diversification by reduced gene flow between populations isolated by unstable areas (Vasconcellos et al., 2019).
All these scenarios are known to leave genetic signatures related to split events and historical demography. In a vicariant scenario some stability in historical demography and main split events coincident with the mentioned event are expected (Raposo do Amaral et al., 2013), while under a scenario of dispersion, new populations are expected to show lower genetic diversity and demographic growth originating from a small size population (founder effect; Santos et al., 2014). In a scenario related to Pleistocene climatic changes, populations are expected to have recently split and to have indications of a bottleneck (Carnaval et al., 2009).
Through approximate Bayesian computation (ABC) it is possible to generate specific models corresponding to these scenarios and find the most likely to have occurred according to empirical data (Beaumont, 2019). ABC has become popular for analyzing complex problems in a variety of fields (Beaumont, 2019) because the method bypasses the exact likelihood calculations by using summary statistics and simulations, thus expanding the range of models for which statistical inference can be considered (Sunnåker et al., 2013). Briefly, ABC analysis consists of formulating a model, fitting the model to data (parameter estimation), and comparing it to other models (Csilléry et al., 2010; Gelman et al., 2003). Furthermore, the increase in the number of ABC studies has led to the development of methods that can improve the accuracy of model choice, like regression-adjustment and the choice of summary statistics (Beaumont, 2019).
Scinax fuscomarginatus (Lutz, 1925) is an interesting model to test hypotheses about diversification in Neotropical open habitats due to its wide geographic distribution, primarily associated with savannas and grasslands. The species is a small treefrog that inhabits South American open areas east of the Andes, being one of the most widely distributed Neotropical anuran species (Brusquetti et al., 2014). This species occupies open habitats of Humid Chaco, Cerrado, Pantanal, Chiquitano dry forest, Mato Grosso seasonal forest, and Guianan savanna (Brusquetti et al., 2014).
We used mitochondrial and nuclear DNA of a wide sampling of S. fuscomarginatus to estimate genetic clustering and, based on ABC, tested the diversification hypotheses that could be responsible for the current genetic pattern. We tested hypotheses related to the uplift of the CBP (vicariance and plateau-depression hypotheses) and to the climate changes through Pleistocene glacial cycles (isolation by instability hypothesis).
Materials and Methods
Sampling, Laboratory Procedures and Molecular Methods
We produced a multilocus dataset including one mitochondrial and three nuclear gene fragments: mtDNA consisted of 615 bp of the Cytochrome c oxidase subunit I gene (COI) and nuDNA of 395 bp of the Ribosomal protein L3 (intron 5) gene (RPL3), 380 bp of the B–fibrinogen (intron 7) gene (BFIB), and 346 bp of the Lactose Dehydrogenase Chain Beta (Intron 3) gene (MVZ 27–28).
We extracted total genomic DNA from samples preserved in 95–100% ethanol (muscle or liver), using the DNeasy extraction kit (Qiagen, Valencia, CA, USA), following the manufacturer’s protocol. We amplified gene fragments via polymerase chain reaction using published primers and a commercial kit (Master Mix, Fermentas). For the amplification of the mitochondrial gene we used a step-up reaction (UP) following Lyra et al. (2017). For the amplification of the nuclear gene fragments we used an initial denaturation step of 3 min at 94 °C, followed by 35 cycles (45 cycles for difficult samples) (30 s of denaturation at 95 °C, 30 s of annealing at 50–64.3 °C, and 45 s of extension at 72 °C), and a final extension step of 7 min at 72 °C (see Table S1 in Online Resource for details). We purified PCR products using ExoSAP (Fermentas) and sent them to Macrogen Inc. (Seoul, South Korea) for sequencing. We checked chromatograms and edited sequences in CODONCODE ALIGNER v. 3.5.4 (Codon Code Corporation).
We separated sequences of individuals with heterozygous indels with the algorithm Process Heterozygous Indels in CodonCode Aligner v. 3.5.4 (Codon Code Corporation) and used Phase 2.1 (Stephens et al., 2001) implemented in DnaSP 5.1 (Librado & Rozas, 2009) to resolve haplotypes of heterozygous individuals, discarding those with less than 0.60 of the posterior probability. We aligned sequences from each gene fragment separately with muscle (Edgar, 2004) in MEGA 6 (Tamura et al., 2013). Within nuclear loci, we tested for recombination with PhiTest implemented in Splitstree v. 4.2 (Huson & Bryant, 2006).
To diminish missing data, after Phase analyses, we kept samples with sequences for at least the mtDNA gene and two nuclear genes, except for two samples with one nuclear gene (from northeastern Santa Cruz, Bolivia and southern Mato Grosso do Sul, Brazil) which were kept in order to maintain geographic representation. The final matrix included 68 tissue samples of S. fuscomarginatus from 43 localities (Fig. 1), representing populations from Cerrado, Humid Chaco, Chiquitano dry forest, Pantanal, Guianan savanna, and Mato Grosso seasonal forest (see Appendix S1 in Online Resource).

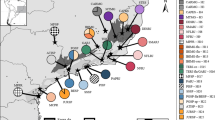
Sampled localities and population assignment of Scinax fuscomarginatus. Horizontal bars (right) correspond to each specimen analyzed in GENELAND, genetic clusters (groups) are represented by colors: group A = gray; group B = orange; group C = blue; group D = yellow. Numbers in the map and horizontal bars correspond to the map code in Appendix S1 in Online Resource. Blue lines represent main rivers, and black lines represent country borders. Ecoregions were modified from Olson et al. (2001)
Population Assignments
To assign individuals to genetic clusters we followed Carstens et al. (2013) including methods for the discovery of clusters followed by a validation step. Firstly, we tested for a correlation between genetic distance and geographical distance (isolation by distance, IBD). For this, we calculated genetic distance matrices for each nuclear gene in MEGA and geographic distance matrices using the dist function in R version 4.2.1 (R Core Team, 2019). We performed the Mantel test between both matrices for each nuclear gene fragment using the function mantel.rtest in the ‘ade4’ v. 1.7 R package (Dray & Dufour, 2007), employing 10,000 Monte Carlo permutations. For all fragments, we found a significant association between genetic and geographic distances (see Results section), indicating non-random-mating, which can affect the efficiency of methods of population assignment that do not incorporate the geographic location of individuals (e.g., STRUCTURE). In order to overcome the possible influence of isolation by distance, we used methods that consider the geographic distribution of individuals (GENELAND and BAPS) (see Thomé et al., 2016).
We ran GENELAND package v. 4.9.2 in R (Guillot et al., 2005a, b) for mitochondrial and nuclear datasets together, using ten independent runs and varying the number of populations from 1 to 15. Each run consisted of 200 iterations of burn-in, followed by 1,000,000 sampled iterations, sampled every 1,000 iterations. We assumed a spatially explicit model with the exact coordinates of samples and an uncorrelated model for allele frequencies.
We ran BAPS (Corander et al., 2008) combining nuclear and mitochondrial genes in a single dataset using the coordinates of samples. As the analysis of combined haploid and diploid sequence markers is technically not feasible in baps, we conducted a diploid analysis considering both phased haplotypes of nuDNA for each individual combined with its correspondent mtDNA sequence, which was duplicated in order to not allow a missing allele. We first ran the mixture model using a k range of 1–10, three times for each k. Then we used this result to run the admixture model (Corander & Marttinen, 2006) using 100 interactions, 200 reference individuals, and 10 interactions per individual. Admixed individuals were considered significant at P < 0.05.
In order to validate the different population assignments that resulted from GENELAND and BAPS, we used the multispecies coalescent model (Rannala & Yang, 2003; Edwards, 2009; Yang & Rannala, 2014) implemented in BPP v. 4.3 (Yang & Rannala, 2010) to estimation of the number of populations and topology of trees. This method has been successfully applied to species delimitation from multilocus datasets with the benefit that it accounts for gene tree conflict by modeling coalescent stochasticity when estimating species tree as a framework for species delimitation (Liu et al., 2009; Yang & Rannala, 2010). Moreover, this method has proven suitable for diagnoses population structure associated with population isolation but not species limits (Freudenstein et al., 2017). First we inferred a species tree using the multilocus coalescent model implemented in *BEAST (Heled & Drummond, 2010) on BEAST v. 2.4.3 (Bouckaert et al., 2014) for each population assignment (GENELAND and BAPS) to use as a guide tree for BPP analyses, considering the four loci. The nucleotide substitution model, range of the rate of heterogeneity, and proportion of invariant positions were inferred during the MCMC analysis with ‘bModelTest’ v. 1.2.1 package (Bouckaert, 2015) implemented in BEAST, with transition-transversion split option and empirical frequencies. For all loci we used strict clock model. We ran 100 million generations sampling every 10,000 with the Yule model tree prior and constant population size. To assess the stationarity of the Markov chains we examined the effective sample size (ESS) in the software TRACER v. 1.6 (Rambaut et al., 2013), ESS > 200 was expected at stationarity. The species tree was annotated with TREEANNOTATOR v. 2.4.3 (Drummond & Rambaut, 2007) as a maximum clade credibility tree and median heights as node ages; the first 2,000 trees (20% of the sampling) were discarded as burn-in. In BPP, as prior distributions of ancestral population size (theta) and root age (tau) can affect the posterior probabilities of the models (Yang & Rannala, 2010), we tested the resulting population assignments hypothesis using a range of prior combinations that included relatively large and small ancestral population sizes in combination with relatively deep and shallow divergence times, following Leaché and Fujita (2010). We performed A11 analyses (Yang & Rannala, 2014) for joint estimation of the number of populations and topology of trees using distinct combinations of theta and tau with the algorithms 0 and 1. The theta parameter (ancestral population size) varied from small (IG = 3, 0.02) to large (IG = 3, 0.2) and the tau parameter (divergence time) from deep (IG = 3, 0.01) to shallow (IG = 3, 0.001). We ran each analysis with automatic finetune for 100,000 generations, with 8,000 generations discarded as burn-in. For each prior combination we ran two replicates under the algorithms 0 and 1, totaling 16 analyses for each population assignment.
Genetic Diversity
For the mitochondrial gene fragment we calculated the basic statistics for each population assigned in the previous step: number of haplotypes, haplotype diversity, nucleotide diversity, and index of genetic differentiation Fst, and performed neutrality tests (Tajima’s D, R2 and Fu’s FS) in DnaSP 5.
Construction of Models to be Tested with Approximate Bayesian Computation (ABC)
All scenarios follow GENELAND assignment and the species tree topology inferred by *BEAST (see Results section and Online Resource), including four populations with geographic distribution limits that mainly follow boundaries between lowlands and plateaus of central Cerrado. Based on those population assignments we discarded the hypotheses of ancient divergence related to the center of CBP (Oliveira et al., 2018). We constructed four alternative diversification models representing three main scenarios: a simple vicariant scenario, a colonization scenario, and a recent refugia scenario (Fig. 2).
The vicariant model (model 1) considers constant population sizes after splits, with the first split corresponding to CBP uplift (7 − 2 Mya). The plateau-depression model (model 2), represents the colonization scenario, with stable populations in plateaus and small ancient populations in depressions (founder effect), with the first split also corresponding to CBP uplift. Two isolation by instability models (models 3 and 4) represent the refugia scenario. The first one with all populations experiencing size reductions, denoting small or micro-refuges (model 3), and the other with stable populations in highlands of the CBP, denoting a stable open environment area in central Brazil proposed for Cerrado trees (Collevatti et al., 2012; de Oliveira Buzatti et al., 2017; de Buzatti et al., 2018), which roughly corresponds to that region (model 4). In both latter models the most ancient split corresponds to Last Interglacial (LIG; 150,000 years ago).

Simulated scenarios of Scinax fuscomarginatus diversification. Genetic clusters (groups) are represented by colors. Times of divergences represented by t1, t2 and t3 and other priors are detailed in Materials and Methods. Best-supported model is highlighted in the red box
Model Testing with Approximate Bayesian Computation
We used the program ms (Hudson, 2002) to simulate genetic data under the defined demographic models. The simulations were conducted using the same loci lengths and sample sizes as the empirical data and run independently for each locus. The summary statistics computed for each marker were considered jointly in the model choice.
We conducted a preliminary analysis with 100,000 replicates to determine appropriate prior intervals for effective population size (Ne), θmit = Neµ and θnuc = 4Neµ. Firstly, we performed a cross-validation test to assess the performance of different methods of rejection [regular rejection (REJ), multinomial logistic regression (MNLOG) and neural networks (NN)], values of tolerance (0.001, 0.01, 0.05 and 0.1), and different combinations of summary statistics as vectors of three to five initially estimated statistics (pi, ss, D, thetaH, H). We applied the function cv4postpr from the R package ‘abc’ v. 2.1 (Csilléry et al., 2012; http://cran.r-project.org/web/packages/abc/index.html) which performs 10 simulations from the prior distribution of each model, used as PODs (pseudo-observed data). We considered as indicative of performance the ability to maximize the probability of choosing the simulated model over other models following Tsai and Carstens (2013). Then, we estimated new parameters with the function abc using the best performing method of rejection, value of tolerance and vector of summary statistics.
For this preliminary analysis we estimated a prior interval for the effective population size (Ne). Considering Ne = θ/µ, we used a mean COI mutation rate (µ) estimated for amphibians of 0.7% substitutions per lineage per million years based on the estimations of Freilich et al. (2014) and Meng et al. (2014) and theta-W calculated for COI in DnaSP (theta of COI = 3.88–28.86) resulting in Ne values from 900,000 to 6,000,000 that were used in a uniform distribution. For θ of mitochondrial and nuclear loci, we used broad intervals (0.01–150) drawn independently for each locus from a uniform distribution. We converted divergence times (t) in 4 N generations (coalescent time) using the formula: number of generations/Ne, assuming a generation time of one year (following Toledo and Haddad, 2005) and Ne previously estimated from COI. The prior for the time of the most ancient split event for models 1 and 2 (T3) represents the CBP, from mid-tertiary to early-quaternary (final plateau uplift), 7 to 2 Mya (Silva, 1997). To represent this time interval, we used a lognormal distribution, with a mean of 14.9 and standard deviation (SD) of 0.6, which in years corresponds to 1.1 to 7.9 Mya (95% HPD) with a median of 2.9 Mya. For models 3 and 4 the prior for T3 represents main interglacial periods of the Upper Pleistocene, the LIG and Sangamon interglacial period together (from 150 to 60 Kya). The Sangamon interglacial period represents the last interglacial period; some definitions localize this period between 130 and 70 Kya, but more recent definitions restrict this period between 130 and 115 Kya (Otvos, 2015; Gowan et al., 2021). However, after the Sangamon period, glacigenic deposits of the last Patagonian glaciation were dated from 60 Kya, although deposits of the Last Glacial Maximum (LGM) correspond to 25–20 Kya (Rutter et al., 2012). To represent this time interval, we used a lognormal distribution, with a mean of 12 and SD of 0.6, which in years corresponds to 60 to 437 Kya (95% HPD) with a median of 163 Kya. In all models subsequent split events (T2 and T1) were inserted between T3 and the present.
To draw the priors related to founder effect and bottleneck we followed Perez et al. (2016). The intensity of the the founder effect in the plateau-depression model (model 2) was sampled from a uniform distribution from 0 to 0.1 of the original population size. The exponential population growth rates were defined to ensure a size ratio of the founder and current population. To do this, we used a beta distribution with a mean of 1.0 and SD 15.0, concentrating this prior in very low values (median of 0.0452 and 95% HPD from 0.00169 to 0.218) to assure that the current population size sampled is several times larger than the founder population. The intensity of the bottleneck in the isolation by instability models (models 3 and 4) was sampled from a uniform distribution from 0.01 to 1 of the original population size. Statistics (pi, ss, D, thetaH, H) from empirical data were estimated with sample_stats.exe from a ms file generated in DnaSP.
For the final analysis, we adjusted priors for Ne and theta based on preliminary analysis results and kept the other parameters invariant. We conducted 500,000 replicates for each model and performed a new cross-validation test to compare the performance of the ABC analysis using the simulated data with and without transformation by Principal Component Analysis (PCA). In the transformed dataset, we replaced the summary statistics by the first six Principal Components, which comprise 90% of the information contained in the original dataset. We performed the ABC model selection using the algorithm, level of tolerance, dataset and summary statistics selected in both cross-validation steps.
After model selection, we checked whether the best-supported model provided a good fit to the data using the function gfit of the R package ‘abc’. To validate model selection results we performed a posterior predictive check (ppc). For this, we estimated parameters related to the best-supported model from simulations and from these parameters generated new summary statistics to check whether the selected model is able to reproduce observed values of summary statistics.
Results
Population Assignments
We found a significant association between genetic and geographic distances for all nuclear genes (MVZ 27–28: r = 0.66, P < 0.001; BFIB: r = 0.12, P < 0.05; RPL3: r = 0.15; P < 0.01). Different population assignment methods produced different results. In GENELAND, all replicates yielded k = 4, while BAPS yielded k = 9. However, the assignment of BAPS and GENELAND are equivalent in the sense that BAPS subdivided populations found by GENELAND (see Fig. 1 and Figure S1 in Online Resource).
BPP analysis with GENELAND population assignment recovered 100% of posterior probability. With BAPS population assignment, BPP results were less consistent, with posterior probability values from 96 to 99%. Among the nine populations, lower posterior probabilities were found in the groups with only one individual each (B3, C1, and C3). Considering BPP results and that BAPS population assignment is generally a subdivision of GENELAND groups, we based subsequent analyses on GENELAND results. The species trees inferred by BPP analyses were not reported because of low posterior probabilities, specially with BAPS assignments (0.0465 of posterior probability for best species tree, considering all runs). With GENELAND assignments, posterior probabilities were not so low, however the highest posterior probability was just 0.597, including, in all runs, three models in the 95% confidence interval.
In the GENELAND assignments (Figs. 1 and 3), group A clusters together samples from Cerrado lowlands, Mato Grosso seasonal forest, and Humid Chaco, from northeastern Brazil to northern Argentina; group B includes samples mainly from the highlands of the CBP, from central to southeastern Brazil; group C clusters the single sample from the Guianan savanna, with samples from western Cerrado and Chiquitano dry forest; and group D includes two samples from a single locality, from a transitional area between southern Cerrado and western Atlantic forest.
In all populations resulting from GENELAND analysis we found high levels of haplotype diversity (Hd) and low levels of nucleotide diversity (pi), which suggest recent population expansions, in concordance with the R2 index. Tajima’s D and Fu’s Fs were not significant (Table 1).

Sampled localities and population assignment of Scinax fuscomarginatus. Genetic clusters (groups) are represented by colors following GENELAND results: group A = grey; group B = orange; group C = blue; group D = yellow. (a) Geographical distribution of highlands (b) results of GENELAND population assignment analysis, colors represent the posterior probability of individuals membership, darker colors correspond to higher posterior probabilities. The sample from Venezuela (Group C) was not included in order to maintain visibility of the samples distribution
Model Testing with Approximate Bayesian Computation
The first cross-validation tests showed that the algorithm with best performance (higher probabilities of choosing the simulated models) was the MNLOG with tolerance of 0.05 and the vector of summary statistics containing ss, theta-H and H. The second cross-validation test, to compare the performance of the ABC analysis using the simulated data with and without transformation by PCA in final simulations, showed no significant differences in performances. Following this result, we used both datasets to choose the best model.
With both the transformed and untransformed datasets, model 3 resulted in the highest posterior probability (PP) (transformed dataset PP = 0.74 and untransformed dataset PP = 0.82) and Bayes Factor “substantial” relative to the best alternative model (model 4, isolation by instability with refuge in CBP) (Table 2). In gfit test, for both transformed and untransformed datasets, the best-supported model provided a good fit to the observed data (see Figure S2 in Online Resource). Posterior predictive checks (PPC) also supported the adequacy of the results showing the values of the observed data within the values of the simulated data (see Figure S3 in Online Resource).
ABC supports a scenario of multiple refuges, both in plateaus and lowlands (model 3: isolation by instability hypothesis), in which all populations experienced population size reduction (bottleneck effect) after divergence in a recent time (Pleistocene).
Estimated parameters show that all populations have decreased by about a third compared to ancestral population size (Table 3). The median of the most ancient split event (median for T3 = 173.05 Kya) and of the subsequent events (median for T2 = 75.6 Kya; median for T1 = 29.9 Kya) range from Middle to the Upper Pleistocene (Table 3).
Discussion
We found different population assignments with different methods, according to GENELAND, S. fuscomarginatus is genetically structured in four geographically associated lineages, while BAPS estimations resulted in nine. In several studies where more than one method of population assignment was used, an overestimation of genetic clusters by BAPS was observed (Latch et al., 2006; Zachos et al., 2016; Pinho et al., 2019; Terrones et al., 2022). Tests with simulated and real datasets suggested that the overestimation of K observed in BAPS is more frequent in populations poorly differentiated (Latch et al., 2006) and in populations with low number of individuals (Terrones et al., 2022); with a tendency to identify clusters with low number of individuals (Zachos et al., 2016) or spurious designation of individuals to clusters of only one individual (singleton clusters) (Pinho et al., 2019). In our BAPS analysis we found clusters from over ten to just one individual, and in all cases corresponding to subdivisions of clusters assigned by GENELAND. With BPP we found that GENELAND individual assignments were fully supported. Based on consistency of BPP results and on the fact that BAPS subdivided the clusters estimated by GENELAND we considered the results of BAPS an overestimation.
According to GENELAND assignments, S. fuscomarginatus is genetically structured in four geographically associated lineages, which follow the current distribution of open habitats of highlands of the CBP (groups B and D) and of the Cerrado lowlands, Mato Grosso seasonal forest, Humid Chaco, Chiquitano dry forest, and Guianan savanna (groups A and C) (Figs. 1 and 3). Among the models tested with ABC, the most likely to have occurred is the “isolation by instability” model, a scenario of multiple refugia in which all populations experienced population size reduction (bottleneck) in a relatively recent time, with the most ancient split event surrounding the LIG (about 150 Kya). Bottleneck events are also supported by basic statistics of the mitochondrial fragment. High haplotype diversity combined with low nucleotide diversity might be associated with recent population expansion (Eizirik et al., 2001; Althoff and Pellmyr, 2002; Joseph et al., 2002; Stamatis et al., 2004). This pattern is probably a consequence of an accumulation of mutations which results in a high number of closely related haplotypes, that is, many recently evolved haplotypes. We hypothesized that during interglacial periods, warmer and wetter climate would have favored the geographic expansion of forests, resulting in the retraction and fragmentation of open habitats like savannas and grasslands. Populations of S. fuscomarginatus, as well as other species associated with open habitats, would have remained isolated in relatively small stable areas with reduced gene flow among them.
Connections Between Open Areas North and South of the Amazonia
Although our sampling efforts were focused on open areas south of the Amazonia and testing connections through the Amazon forest is beyond our goals, we consider that the clustering of samples of populations from both sides of it deserves some discussion. With the exception of the disconnected Guianan savanna, which is part of the northern Amazonian savannas (Daly & Mitchell, 2000), the area occupied by samples of genetic groups A and C is a large and continuous savanna-like environment. A similar distribution has been described for birds (Mittermeier et al., 2010; Lima-Rezende et al., 2019, 2022; Ritter et al., 2021), snakes (Wüster et al., 2005; Quijada-Mascareñas et al., 2007), and plants (de Buzatti et al., 2018; Resende-Moreira et al., 2019). Rather than long-distance dispersal, recent connections through corridors in Amazon forest are suggested to explain this pattern (Lima-Rezende et al., 2019). Four corridors have been proposed: by Atlantic coast (Silva, 1995; Werneck et al., 2012b), by central Amazon forest (Haffer, 1967; Ledo & Colli, 2017; Ribeiro et al., 2016), and two by western Amazon forest: one along the base of the Andes mountain range (Silva, 1995; Webb, 1978; Werneck et al., 2012b) and another through the Madeira River basin (Ribeiro et al., 2016). However, palynological studies (Colinvaux et al., 1996, 2000; Bush et al., 2004) and past model reconstructions (Cowling et al., 2001; Claussen et al., 2013; O’ishi & Abe-Ouchi, 2013; Hopcroft & Valdes, 2015) have contradicted the connections of open habitats through central Amazon forest, showing a history of forest stability in the region.
Considering the geographic distribution of samples clustered in Group C (Fig. 1) and the above-mentioned evidence against Central Amazonian corridor, a plausible route for S. fuscomarginatus connections is by western Amazonia. Lima-Rezende et al. (2022) found high migration levels between Cerrado and Guianan savanna populations by the West Amazonia corridor in four bird species. Currently, this region is a continuum of open formations from Cerrado to the Madeira River basin, which are connected by Chiquitano dry forest and Beni Savanna (Fig. 1). A study of soil carbon isotopes suggested expansion of savanna vegetation in this area from early to middle Holocene (de Freitas et al., 2001) and grass and herb pollen records indicate predominance of tropical grasses and drier climate during Last Glacial Maximum (LGM) (Van Der Hammen & Hooghiemstra, 2000). However, Arruda et al. (2018) found that savanna expansions were restricted only to ecotonal areas and the forest was not replaced by savanna, but by another type of forest. Furthermore, other studies support stability in Amazon forest during LGM (Costa et al., 2018; Kern et al., 2022) and even an increase in precipitation during the last glacial cycles (Wang et al., 2004, 2017; Cheng et al., 2013).
As mentioned, grass pollen records have been interpreted as direct evidence of dry conditions (Van Der Hammen & Hooghiemstra, 2000); however, grass can occur in a variety of environments, especially in floodplains (Kirschner & Hoorn, 2020). The fluvial system of central Amazonia constituted by continuous areas of Terra Firme forest (non-flooded forests) and well defined river channels was only recently established, between 250 and 45 Kya, replacing widespread floodplains (Pupim et al., 2019; Hoorn et al., 2010). Central Amazonia would have been dominated by a broad Várzea-like environment with fewer areas of non-flooded forests than today (Pupim et al., 2019). These widespread flooded habitats could be considered a suitable environment for species such as S. fuscomarginatus associated with grass and herbaceous vegetation in floodplains (Prado et al., 2005). The predominance of Várzae-like environment and past dynamics of expansions and retractions of floodplains during the late Quaternary could have promoted connections through the Amazon forest. However, it is clear that a denser sampling, mainly in Guianan savanna, is required to adequately test this hypothesis or any other related to gene flow among populations both sides of the Amazon forest.
Interglacial periods as main diversification promoters in Scinax fuscomarginatus
Our ABC results support a more important influence of Pleistocene interglacial periods than old Pliocene vicariant or dispersal events related to the final uplift of the CBP. However, the geographic distribution of the genetic structure suggests an influence of the highlands of CBP, once the geographic distribution of the most widely distributed groups (A and B) corresponds with its limits (Fig. 3). During late Miocene, the CBP compartmentalization may have acted as the main driver of genetic structure of some widely distributed frogs, like D. muelleri and Physalaemus cuvieri Fitzinger, 1826 (Oliveira et al., 2018; Miranda et al., 2019). In other frog species as Boana lundii (Burmeister, 1856) and Boana raniceps (Cope, 1862), and several plant species, although genetic clustering is in concordance with boundaries of the CBP, diversification processes are more recent, related to Pleistocene events (Vasconcellos et al., 2019; Camurugi et al., 2021; de Buzatti et al., 2018). de Buzatti et al. (2018) suggested that the pattern observed in many phylogenetically distant plant species indicates a vicariant event at this region. However, in our ABC analysis the vicariant model (model 1) with the older split event in concordance with the CBP shows the lowest posterior probability (Table 2). The confidence of phylogeographic inferences based on demographic models is challenged by the appropriate model selection and also by misspecification of the models (see Carstens et al., 2022). Besides the mentioned issues and independently of the methods used in phylogeographic inferences, it is important to take into account that genetic signatures of ancient events may have been erased by recent events (Avise, 2000). Also, based on geographic distribution of main genetic populations (groups A and B), we suggest that current topography of the Cerrado may be acting on the genetic diversity of S. fuscomarginatus. The final uplift of the CBP (late-Tertiary to early-Quaternary) was accompanied by the subsidence of peripheral areas, forming depressions that progressively expanded during Quaternary cycles of erosion (Cole, 1986; Brasil & Alvarenga, 1989). In addition to increasing topographic complexity, these depressions harbor seasonally dry forests or gallery forests (Santos et al., 2014), reinforcing the compartmentalization of savanna and grassland habitats. Hence, as suggested for B. raniceps (Camurugi et al., 2021), although the initial diversification of S. fuscomarginatus may not have been influenced by the CBP uplift itself, the genetic structure within the species seems to be maintained by the current landscape of the Cerrado.
The most ancient genetic divergence (T3) of our best-supported model (model 3) is of about 160 Kya, corresponding to the LIG. We hypothesized that during the LIG, savanna and grassland like environments were fragmented, isolating S. fuscomarginatus populations and promoting genetic differentiation. Other open-habitat frogs with similar geographic distribution [Boana albopunctata (Spix, 1824), B. lundii, and B. raniceps] also showed an influence of climatic oscillations during the Pleistocene. However, divergence between genetic populations within these species was suggested to have occurred during cold and dry periods (glacial phases) from early to middle Pleistocene (Prado et al., 2012; Vasconcellos et al., 2019; Camurugi et al., 2021) and not in warm and humid periods (interglacial phases) as we propose for S. fuscomarginatus. We suggest that the differences in times of diversification between these open-habitat species and S. fuscomarginatus could be due to specific habitat associations. The Cerrado treefrogs B. lundii and B. albopunctata are associated with gallery forests (Prado et al., 2012; Vasconcellos et al., 2019), and B. raniceps, that although more widespread, is also mainly associated with gallery forests (Vaz-Silva et al., 2020). Expansions of forest formations during interglacial phases probably fragmented and isolated populations of S. fuscomarginatus, a species associated with savannas and grassland habitats (Toledo & Haddad, 2005; Brusquetti et al., 2014; Pupin et al., 2020), and, on the other hand, promoted expansions of the other mentioned species that are mainly associated with gallery forests.
Studies on population dynamics of plants associated with Neotropical savannas and grassland environments support range expansions in glacial phases, with a clear pattern in southern ecoregions, like Pampas and Humid Chaco (Mäder & Freitas, 2019; Giudicelli et al., 2021). Palynological records also support cycles of expansion of forest plant species in southeastern Brazil during the LIG (de Oliveira et al., 2020), and expansions of savannas from south as far as 20º latitude during the last glacial period (Behling, 2002). On the other hand, niche model studies based on tree species distribution (Caryocar brasiliense, Qualea multiflora, Q. parviflora, and Q. grandiflora) show evidence of geographic retraction during glacial phases (Ramos et al., 2007; Collevatti et al., 2012; de Oliveira Buzatti et al., 2017, 2018); however, these species are widely distributed in all phytophysionogmies of the Cerrado, not only those related to open habitats. The Cerrado ecoregion harbors a mosaic of formations, like semideciduous and gallery forests, and savanna vegetation proper (Salgado-Labouriau, 1997; Behling & Hooghiemstra, 2001; Bueno et al., 2017). In turn, these savanna formations are dominated by grasses but with different levels of density and sizes of trees and shrubs, ranging from grasslands with few or no tall woody plants to dense forest with more or less closed canopy (Silva, 1997). As mentioned before, these tree species are widely distributed in all phytophisignomies of the Cerrado, except gallery forest and semideciduous forest (Oliveira-Filho & Ratter, 1995; Collevatti et al., 2012; de Oliveira Buzatti et al., 2017, 2018), this habitat extent may influence analyses based on niche preferences (Collevatti et al., 2013) and may also represent environments other than those where S. fuscomarginatus occurs.
Based on the evidence presented here, we suggest an important role of the climatic dynamic during the Pleistocene and the recent compartmentalization of Cerrado on the diversification of a species highly associated with Neotropical savannas and grasslands. However, we are aware that to go deeper on the evolutionary history of this species and specifically, to better understand the trans-Amazonian connections, future research could focus sampling efforts on populations from northern side of the Amazon and also, that a denser molecular sampling will increase the capacity to better study demographic history and gene flow dynamics.
Data Availability
All sequences were deposited in GenBank, accession numbers are available in Appendix S1 in Online Resource. Input files and scripts used in genetic analyses are deposited in https://github.com/franbrusquetti/Scinax_fuscomarginatus_ABC.
References
Althoff, D. M., & Pellmyr, O. (2002). Examining genetic structure in a bogus yucca moth: A sequential approach to phylogeography. Evolution, 56(8), 1632–1643. https://doi.org/10.1111/j.0014-3820.2002.tb01475.x.
Arruda, D. M., Schaefer, C. E. G. R., Fonseca, R. S., Solar, R. R. C., & Fernandes-Filho, E. I. (2018). Vegetation cover of Brazil in the last 21 ka: New insights into the amazonian refugia and pleistocenic arc hypotheses. Global Ecology and Biogeography, 27(1), 47–56. https://doi.org/10.1111/geb.12646.
Avise, J. C. (2000). Phylogeography: The history and formation of species. Harvard University Press.
Beaumont, M. A. (2019). Approximate Bayesian computation. Annual Review of Statistics and Its Application, 6, 379–403.
Behling, H. (2002). South and southeast Brazilian grasslands during late quaternary times: A synthesis. Palaeogeography Palaeoclimatology Palaeoecology, 177(1–2), 19–27. https://doi.org/10.1016/S0031-0182(01)00349-2.
Behling, H., & Hooghiemstra, H. (2001). Neotropical savanna environments in space and time: late Quaternary interhemispheric comparisons. In V. Markgraf (Ed.), Interhemispheric climate linkages (pp. 307–323). Academic press. https://doi.org/10.1016/B978-012472670-3/50021-5.
Boettger, O. (1885). Liste von Reptilien und Batrachiern aus Paraguay. Zeitschrift für Naturwissenschaften. Halle, 58, 213–248.
Bouckaert, R. (2015). bModelTest: Bayesian site model selection for nucleotide data. bioRxiv, 1, 20792.
Bouckaert, R. R., Heled, J., Kühnert, D., Vaughan, T., Wu, C., Xie, D., Suchard, M. A., Rambaut, A., & Drummond, A. J. (2014). Beast 2: A software platform for bayesian evolutionary analysis. PLoS Computational Biology, 10(4), e1003537. https://doi.org/10.1371/journal.pcbi.1003537.
Brasil, A. E., & Alvarenga, S. M. (1989). Relevo. In A. C. Duarte (Ed.), Geografia do Brasil Região Centro-Oeste (pp. 53–72). FIBGE-Diretoria de Geociências.
Brusquetti, F., Jansen, M., Barrio-Amorós, C., Segalla, M., & Haddad, C. F. B. (2014). Taxonomic review of Scinax fuscomarginatus (Lutz, 1925) and related species (Anura; Hylidae). Zoological Journal of the Linnean Society, 171(4), 783–821. https://doi.org/10.1111/zoj.12148.
Brusquetti, F., Netto, F., Baldo, D., & Haddad, C. F. B. (2019). The influence of Pleistocene glaciations on Chacoan fauna: Genetic structure and historical demography of an endemic frog of the South American Gran Chaco. Biological Journal of the Linnean Society, 126(3), 404–416. https://doi.org/10.1093/biolinnean/bly203.
Bueno, M. L., Pennington, R. T., Dexter, K. G., Kamino, L. H. Y., Pontara, V., Neves, D. M., Ratter, J. A., & de Oliveira-Filho, A. T. (2017). Effects of Quaternary climatic fluctuations on the distribution of Neotropical savanna tree species. Ecography, 40(3), 403–414. https://doi.org/10.1111/ecog.01860.
Burmeister, H. (1856). Erläuterungen zur Fauna Brasiliens, enthaltend Abbildungen und ausführliche Beschreibungen neuer oder ungenügend bekannter Thier-Arten. Berlin: Georg Reimer.
Bush, M. B., Silman, M. R., & Urrego, D. H. (2004). 48,000 years of climate and forest change in a biodiversity hotspot. Science, 303(5659), 827–829. https://doi.org/10.1126/science.1090795.
Camurugi, F., Gehara, M., Fonseca, E. M., Zamudio, K. R., Haddad, C. F., Colli, G. R., Thomé, M. T. C., Prado, C. P. A., Napoli, M. F., & Garda, A. A. (2021). Isolation by environment and recurrent gene flow shaped the evolutionary history of a continentally distributed Neotropical treefrog. Journal of Biogeography, 48(4), 760–772. https://doi.org/10.1111/jbi.14035.
Carnaval, A. C., Hickerson, M. J., Haddad, C. F. B., Rodrigues, M. T., & Moritz, C. (2009). Stability predicts genetic diversity in the Brazilian Atlantic forest hotspot. Science, 323(5915), 785–789. https://doi.org/10.1126/science.1166955.
Carstens, B. C., Pelletier, T. A., Reid, N. M., & Satler, J. D. (2013). How to fail at species delimitation. Molecular Ecology, 22(17), 4369–4383. https://doi.org/10.1111/mec.12413.
Carstens, B. C., Smith, M. L., Duckett, D. J., Fonseca, E. M., & Thomé, M. T. C. (2022). Assessing model adequacy leads to more robust phylogeographic inference. Trends in Ecology & Evolution, 37(5), 402–410. https://doi.org/10.1016/j.tree.2021.12.007.
Cheng, H., Sinha, A., Cruz, F. W., Wang, X., Edwards, R. L., d’Horta, F. M., Ribas, C. C., Vuille, M., Stott, L. D., & Auler, A. S. (2013). Climate change patterns in Amazonia and biodiversity. Nature Communications, 4(1), 1–6. https://doi.org/10.1038/ncomms2415.
Claussen, M., Selent, K., Brovkin, V., Raddatz, T., & Gayler, V. (2013). Impact of CO2 and climate on last glacial maximum vegetation – a factor separation. Biogeosciences, 10(6), 3593–3604. https://doi.org/10.5194/bg-10-3593-2013.
Cole, M. M. (1986). The Savannas: Biogeography and Geobotany. Academic Press.
Colinvaux, P. A., de Oliveira, P. E., Moreno, J. E., Miller, M. C., & Bush, M. B. (1996). A long pollen record from lowland Amazonia: Forest and cooling in glacial times. Science, 274(5284), 85–88. https://doi.org/10.1126/science.274.5284.85.
Colinvaux, P. A., de Oliveira, P. E., & Bush, M. B. (2000). Amazonian and Neotropical plant communities on glacial time-scales: The failure of the aridity and refuge hypotheses. Quaternary Science Reviews, 19(1–5), 141–169. https://doi.org/10.1016/S0277-3791(99)00059-1.
Collevatti, R. G., Ribeiro, M. S. L., Souza Neto, A. C., Franco, A. A., de Oliveira, G. D., & Terribile, L. C. (2012). Recovering the demographical history of a Brazilian Cerrado tree species Caryocar brasiliense: Coupling ecological niche modeling and coalescent analyses. Natureza e Conservação, 10, 169–176. https://doi.org/10.4322/natcon.2012.024.
Collevatti, R. G., Terribile, L. C., de Oliveira, G., Lima-Ribeiro, M. S., Nabout, J. C., Rangel, T. F., & Diniz-Filho, J. A. F (2013). Drawbacks to palaeodistribution modelling: The case of South American seasonally dry forests. Journal of Biogeography, 40(2), 345–358. https://doi.org/10.1111/jbi.12005.
Cope, E. D. (1862). Catalogues of the reptiles obtained during the explorations of the Parana, Paraguay, Vermejo and Uraguay Rivers, by Capt. Thos. J. Page, U.S.N.; and of those procured by Lieut. N. Michler, U.S. Top. Eng., Commander of the expedition conducting the survey of the Atrato River. Proceedings of the Academy of Natural Sciences of Philadelphia, 14, 346–359.
Corander, J., & Marttinen, P. (2006). Bayesian identification of admixture events using multilocus molecular markers. Molecular Ecology, 15(10), 2833–2843. https://doi.org/10.1111/j.1365-294X.2006.02994.x.
Corander, J., Sirén, J., & Arjas, E. (2008). Bayesian spatial modelling of genetic population structure. Computational Statistics, 23, 111–129. https://doi.org/10.1007/s00180-007-0072-x.
Costa, G. C., Hampe, A., Ledru, M. P., Martinez, P. A., Mazzochini, G. G., Shepard, D. B., Werneck, F. P., Moritz, C., & Carnaval, A. C. (2018). Biome stability in South America over the last 30 kyr: Inferences from long-term vegetation dynamics and habitat modelling. Global Ecology and Biogeography, 27(3), 285–297. https://doi.org/10.1111/geb.12694.
Cowling, S. A., Maslin, M. A., & Sykes, M. T. (2001). Paleovegetation simulations of lowland Amazonia and implications for Neotropical allopatry and speciation. Quaternary Research, 55(2), 140–149. https://doi.org/10.1006/qres.2000.2197.
Csilléry, K., Blum, M. G. B., Gaggiotti, O. E., & François, O. (2010). Approximate Bayesian computation (ABC) in practice. Trends in Ecology and Evolution, 25(7), 410–418. https://doi.org/10.1016/j.tree.2010.04.001.
Csilléry, K., François, O., & Blum, M. G. B. (2012). Abc: An R package for approximate Bayesian computation (ABC). Methods in Ecology and Evolution, 3(3), 475–479. https://doi.org/10.1111/j.2041-210X.2011.00179.x.
Daly, D. C., & Mitchell, J. D. (2000). Lowland vegetation of tropical South America: An overview. In D. Lentz (Ed.), Imperfect balance: Landscape Transformations in the pre-columbian Americas (pp. 391–454). Columbia University press.
de Oliveira Buzatti, R.S., Pfeilsticker, T. S., de Magalhães, R. F., Bueno, M. L., Lemos-Filho, J. P., & Lovato, M. B. (2018). Genetic and historical colonization analyses of an endemic savanna tree, Qualea grandiflora, reveal ancient connections between Amazonian savannas and Cerrado core. Frontiers in Plant Science, 9, 981. https://doi.org/10.3389/fpls.2018.00981.
de Freitas, H. A., Pessenda, L. C. R., Aravena, R., Gouveia, S. E. M., de Souza Ribeiro, A., & Boulet, R. (2001). Late quaternary vegetation dynamics in the southern Amazon Basin inferred from carbon isotopes in soil organic matter. Quaternary Research, 55(1), 39–46. https://doi.org/10.1006/qres.2000.2192.
de Oliveira, P. E., Raczka, M., McMichael, C. N., Pinaya, J. L., & Bush, M. B. (2020). Climate change and biogeographic connectivity across the Brazilian cerrado. Journal of Biogeography, 47(2), 396–407. https://doi.org/10.1111/jbi.13732.
de Oliveira Buzatti, R. S., Lemos-Filho, J. P., Bueno, M. L., & Lovato, M. B. (2017). Multiple pleistocene refugia in the Brazilian cerrado: Evidence from phylogeography and climatic niche modelling of two Qualea species (Vochysiaceae). Botanical Journal of the Linnean Society, 185(3), 307–320. https://doi.org/10.1093/botlinnean/box062.
Dray, S., & Dufour, A. B. (2007). The ade4 package: Implementing the duality diagram for ecologists. Journal of statistical software, 22, 1–20. https://doi.org/10.18637/jss.v022.i04.
Drummond, A. J., & Rambaut, A. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evolutionary Biology, 7, 214. https://doi.org/10.1186/1471-2148-7.
Edgar, R. C. (2004). MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32(5), 1792–1797. https://doi.org/10.1093/nar/gkh340.
Edwards, S. V. (2009). Is a new and general theory of molecular systematics emerging? Evolution, 63(1), 1–19. https://doi.org/10.1111/j.1558-5646.2008.00549.x.
Eizirik, E., Kim, J. H., Menotti-Raymond, M., Crawshaw Jr, P. G., O’Brien, S. J., & Johnson, W. E. (2001). Phylogeography, population history and conservation genetics of jaguars (Panthera onca, Mammalia, Felidae). Molecular Ecology, 10(1), 65–79. https://doi.org/10.1046/j.1365-294X.2001.01144.x.
Freilich, X., Tollis, M., & Boissinot, S. (2014). Hiding in the highlands: Evolution of a frog species complex of the genus Ptychadena in the Ethiopian highlands. Molecular Phylogenetics and Evolution, 71, 157–169. https://doi.org/10.1016/j.ympev.2013.11.015.
Freudenstein, J. V., Broe, M. B., Folk, R. A., & Sinn, B. T. (2017). Biodiversity and the species concept—lineages are not enough. Systematic Biology, 66(4), 644–656. https://doi.org/10.1093/sysbio/syw098.
Gelman, A., Carlin, J. B., Stern, H. S., & Rubin, D. B. (2003). Bayesian data analysis (2nd ed.). Chapman & Hall.
Giudicelli, G. C., Turchetto, C., Guzmán-Rodriguez, S., Teixeira, M. C., Petzold, E., Bombarely, A., & Freitas, L. B. (2021). Population genomics indicates micro-refuges and riverine barriers for a southern South American grassland nightshade. Journal of Biogeography, 49(1), 51–65. https://doi.org/10.1111/jbi.14277.
Gowan, E. J., Rovere, A., Ryan, D. D., Richiano, S., Montes, A., Pappalardo, M., & Aguirre, M. L. (2021). Last interglacial (MIS 5e) sea-level proxies in southeastern South America. Earth System Science Data, 13(1), 171–197. https://doi.org/10.5194/essd-13-171-2021.
Guarnizo, C. E., Werneck, F. P., Giugliano, L. G., Santos, M. G., Fenker, J., Sousa, L., D’Angiolella, A. B., dos Santos, A. R., Strüssmann, C., Rodrigues, M. T., Dorado-Rodrigues, T. F., Gamble, T., & Colli, G. R. (2016). Cryptic lineages and diversification of an endemic anole lizard (Squamata, Dactyloidae) of the Cerrado hotspot. Molecular Phylogenetics and Evolution, 94, 279–289. https://doi.org/10.1016/j.ympev.2015.09.005.
Guillot, G., Estoup, A., Mortier, F., & Cosson, J. F. (2005a). A spatial statistical model for landscape genetics. Genetics, 170(3), 1261–1280. https://doi.org/10.1534/genetics.104.033803.
Guillot, G., Mortier, F., & Estoup, A. (2005b). Geneland: A computer package for landscape genetics. Molecular Ecology Notes, 5(3), 712–715. https://doi.org/10.1111/j.1471-8286.2005.01031.x.
Haffer, J. (1967). Speciation of Colombian forest birds west of the Andes. American Museum Novitates, 2294.
Haffer, J. (1969). Speciation in Amazonian forest birds. Science, 165(3889), 131–137. https://doi.org/10.1126/science.165.3889.131.
Heled, J., & Drummond, A. J. (2010). Bayesian inference of species trees from multilocus data. Molecular Biology and Evolution, 27(3), 570–580. https://doi.org/10.1093/molbev/msp274.
Hoorn, C., Wesselingh, F. P., ter Steege, H., Bermudez, M. A., Mora, A., Sevink, J., Sanmartín, I., Sanchez-Meseguer, A., Anderson, C. L., Figueiredo, J. P., Jaramillo, C., Riff, D., Negri, F. R., Hooghiemstra, H., Lundberg, J., Stadler, T., Särkinen, T., & Antonelli, A. (2010). Amazonia through time: Andean uplift, climate change, landscape evolution, and biodiversity. Science, 330(6006), 927–931. https://doi.org/10.1126/science.1194585.
Hopcroft, P. O., & Valdes, P. J. (2015). Last glacial maximum constraints on the Earth System model HadGEM2-ES. Climate Dynamics, 45(5), 1657–1672. https://doi.org/10.1007/s00382-014-2421-0.
Hudson, R. R. (2002). Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics, 18(2), 337–338. https://doi.org/10.1093/bioinformatics/18.2.337.
Huson, D. H., & Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Molecular Biology and Evolution, 23(2), 254–267. https://doi.org/10.1093/molbev/msj030.
Joseph, L., Wilke, T., & Alpers, D. (2002). Reconciling genetic expectations from host specificity with historical population dynamics in an avian brood parasite, Horsfield’s bronze-cuckoo chalcites basalis of Australia. Molecular Ecology, 11(4), 829–837. https://doi.org/10.1046/j.1365-294X.2002.01481.x.
Kern, A., Akabane, T. K., Ferreira, J. Q., Chiessi, C. M., Willard, D. A., Ferreira, F., Sanders, A. O., Silva, C. G., Rigsby, C., Cruz, F. W., Dwyer, G. S., & Baker, P. A. (2022). 1.8 Million Year History of Amazonian Biomes. Available at SSRN 4131078. https://doi.org/10.2139/ssrn.4131078.
Kirschner, J. A., & Hoorn, C. (2020). The onset of grasses in the Amazon drainage basin, evidence from the fossil record. Frontiers of Biogeography, 12(2), e44827. https://doi.org/10.21425/F5FBG44827.
Latch, E. K., Dharmarajan, G., Glaubitz, J. C., & Rhodes, O. E. (2006). Relative performance of bayesian clustering software for inferring population substructure and individual assignment at low levels of population differentiation. Conservation genetics, 7, 295–302. https://doi.org/10.1007/s10592-005-9098-1.
Leaché, A., & Fujita, M. (2010). Bayesian species delimitation in West African forest geckos (Hemidactylus fasciatus). Proceedings of the Royal Society of London B, 277(1697), 3071–3077. https://doi.org/10.1098/rspb.2010.0662.
Ledo, R. M. D., & Colli, G. R. (2017). The historical connections between the Amazon and the Atlantic Forest revisited. Journal of Biogeography, 44(11), 2551–2563. https://doi.org/10.1111/jbi.13049.
Librado, P., & Rozas, J. (2009). DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics, 25(11), 1451–1452. https://doi.org/10.1093/bioinformatics/btp187.
Lima-Rezende, C. A., Rocha, A. V., Couto, A. F., de Souza Martins, É., Vasconcelos, V., & Caparroz, R. (2019). Late pleistocene climatic changes promoted demographic expansion and population reconnection of a Neotropical savanna-adapted bird, Neothraupis fasciata (Aves: Thraupidae). PLoS One, 14(3), 1–19. https://doi.org/10.1371/journal.pone.0212876.
Lima-Rezende, C. A., Cabanne, G. S., Rocha, A. V., Carboni, M., Zink, R. M., & Caparroz, R. (2022). A comparative phylogenomic analysis of birds reveals heterogeneous differentiation processes among Neotropical savannas. Molecular Ecology, 31(12), 3451–3467. https://doi.org/10.1111/mec.16487.
Liu, L., Yu, L., Kubatko, L., Pearl, D. K., & Edwards, S. V. (2009). Coalescent methods for estimating phylogenetic trees. Molecular phylogenetics and evolution, 53(1), 320–328.
Lyra, M. L., Haddad, C. F. B., Azeedo-Espin, A. M. L. (2017) Meeting the challenge of DNA barcoding Neotropical amphibians: polymerase chain reaction optimization and new COI primers. Molecular Ecology Resources, 17(5), 966–980. https://doi.org/10.1111/1755-0998.12648
Mäder, G., & Freitas, L. B. (2019). Biogeographical, ecological, and phylogenetic analyses clarifying the evolutionary history of Calibrachoa in South American grasslands. Molecular Phylogenetics and Evolution, 141, 106614. https://doi.org/10.1016/j.ympev.2019.106614.
Meng, H., Li, X., & Qiao, P. (2014). Population structure, historical biogeography and demographic history of the Alpine Toad Scutiger ningshanensis in the Tsinling Mountains of Central China. Plos One, 9(6), e100729. https://doi.org/10.1371/journal.pone.0100729.
Miranda, N. E. O., Maciel, N. M., Lima-Ribeiro, M. S., Colli, G. R., Haddad, C. F. B., & Collevatti, R. G. (2019). Diversification of the widespread Neotropical frog Physalaemus cuvieri in response to Neogene-Quaternary geological events and climate dynamics. Molecular Phylogenetics and Evolution, 132, 67–80. https://doi.org/10.1016/j.ympev.2018.11.003.
Mittermeier, J. C., Zyskowski, K., Stowe, E. S., & Lai, J. E. (2010). Avifauna of the Sipaliwini savanna (Suriname) with insights into its biogeographic affinities. Bulletin of the Peabody Museum of Natural History, 51(1), 97–122. https://doi.org/10.3374/014.051.0101.
Nogueira, C., Ribeiro, S., Costa, G. C., & Colli, G. C. (2011). Vicariance and endemism in a Neotropical savanna hotspot: Distribution patterns of Cerrado squamate reptiles. Journal of Biogeography, 38(10), 1907–1922. https://doi.org/10.1111/j.1365-2699.2011.02538.x.
O’ishi, R., & Abe-Ouchi, A. (2013). Influence of dynamic vegetation on climate change and terrestrial carbon storage in the last glacial Maximum. Climate of the Past, 9(4), 1571–1587. https://doi.org/10.5194/cp-9-1571-2013.
Oliveira, E. F., Gehara, M., São-Pedro, V. A., Costa, G. C., Burbrink, F. T., Colli, G. R., Rodrigues, M. T., & Garda, A. A. (2018). Phylogeography of Muller’s termite frog suggests the vicariant role of the Central Brazilian Plateau. Journal of Biogeography, 45(11), 2508–2519. https://doi.org/10.1111/jbi.13427.
Oliveira-Filho, A. T., & Ratter, J. A. (1995). A study of the origin of central Brazilian forests by the analysis of plant species distribution patterns. Edinburgh Journal of Botany, 52(2), 141–194. https://doi.org/10.1017/S0960428600000949.
Olson, D. M., Dinerstein, E., Wikramanayake, E. D., Burgess, N. D., Powell, G. V. N., Underwood, E. C., D’amico, J. A., Itoua, I., Strand, H. E., Morrison, J. C., Loucks, C. J., Allnutt, T. F., Ricketts, T. H., Kura, Y., Lamoreux, J. F., Wettengel, W. W., Hedao, P., & Kassem, K. R. (2001). Terrestrial ecoregions of the world: A new map of life on earth: A new global map of terrestrial ecoregions provides an innovative tool for conserving biodiversity. BioScience, 51(11), 933–938. https://doi.org/10.1641/0006-3568(2001)051[0933:TEOTWA]2.0.CO;2.
Otvos, E. G. (2015). The last interglacial stage: Definitions and marine highstand, North America and Eurasia. Quaternary International, 383, 158–173. https://doi.org/10.1016/j.quaint.2014.05.010.
Perez, M. F., Bonatelli, I. A. S., Moraes, E. M., & Carstens, B. C. (2016). Model-based analysis supports interglacial refugia over long-dispersal events in the diversification of two South American cactus species. Heredity, 116(6), 550–557. https://doi.org/10.1038/hdy.2016.17.
Pinho, C., Cardoso, V., & Hey, J. (2019). A population genetic assessment of taxonomic species: The case of Lake Malawi cichlid fishes. Molecular Ecology Resources, 19(5), 1164–1180. https://doi.org/10.1111/1755-0998.13027.
Prado, C. P., Uetanabaro, M., & Haddad, C. F. B. (2005). Breeding activity patterns, reproductive modes, and habitat use by anurans (Amphibia) in a seasonal environment in the Pantanal, Brazil. Amphibia-Reptilia, 26(2), 211–221. https://doi.org/10.1163/1568538054253375.
Prado, C. P., Haddad, C. F. B., & Zamudio, K. R. (2012). Cryptic lineages and pleistocene population expansion in a Brazilian Cerrado frog. Molecular Ecology, 21(4), 921–941. https://doi.org/10.1111/j.1365-294X.2011.05409.x.
Pupim, F. N., Sawakuchi, A. O., Almeida, R. P. D., Ribas, C. C., Kern, A. K., Hartmann, G. A., Chiessi, C. M., Tamura, L. N., Mineli, T. D., Savian, J. F., Grohmann, C. H., Stern, B. Jr., Cruz, A. G., F. W., & Cracraft, J. (2019). Chronology of Terra Firme formation in Amazonian lowlands reveals a dynamic quaternary landscape. Quaternary Science Reviews, 210, 154–163. https://doi.org/10.1016/j.quascirev.2019.03.008.
Pupin, N. C., Brusquetti, F., & Haddad, C. F. B. (2020). Seasonality drives body size variation in a widely distributed Neotropical treefrog. Journal of Zoology, 312(2), 85–93. https://doi.org/10.1111/jzo.12787.
Quijada-Mascareñas, J. A., Ferguson, J. E., Pook, C. E., Salomao, M. D. G., Thorpe, R. S., & Wüster, W. (2007). Phylogeographic patterns of trans‐Amazonian vicariants and Amazonian biogeography: The Neotropical rattlesnake (Crotalus durissus complex) as an example. Journal of Biogeography, 34(8), 1296–1312. https://doi.org/10.1111/j.1365-2699.2007.01707.x.
R Core Team. (2019). R: A language and environment for statistical computing (version 4.2.1). R Foundation for Statistical Computing. https://www.R?project.org/.
Rambaut, A., Suchard, M. A., Xie, D., & Drummond, A. J. (2013). MCMC Trace Analysis Tool. Version v1. 6.0.
Ramos, A. C. S., Lemos-Filho, J. P., Ribeiro, R. A., Santos, F. R., & Lovato, M. B. (2007). Phylogeography of the tree Hymenaea stigonocarpa (Fabaceae: Caesalpinioideae) and the influence of quaternary climate changes in the Brazilian Cerrado. Annals of Botany, 100(6), 1219–1228. https://doi.org/10.1093/aob/mcm221.
Rannala, B., & Yang, Z. (2003). Bayes estimation of species divergence times and ancestral population sizes using DNA sequences from multiple loci. Genetics, 164(4), 1645–1656. https://doi.org/10.1093/genetics/164.4.1645.
Raposo do Amaral, F., Albers, P. K., Edwards, S. V., & Miyaki, C. Y. (2013). Multilocus tests of pleistocene refugia and ancient divergence in a pair of Atlantic Forest antbirds (Myrmeciza). Molecular Ecology, 22(15), 3996–4013. https://doi.org/10.1111/mec.12361.
Recoder, R. S., Werneck, F. P., Teixeira, M., Colli, G. R., Sites, J. W. Jr., & Rodrigues, M. T. (2014). Geographic variation and systematic review of the lizard genus Vanzosaura (Squamata, Gymnophthalmidae), with the description of a new species. Zoological Journal of the Linnean Society, 171(1), 206–225. https://doi.org/10.1111/zoj.12128.
Resende-Moreira, L. C., Knowles, L. L., Thomaz, A. T., Prado, J. R., Souto, A. P., Lemos‐Filho, J. P., & Lovato, M. B. (2019). Evolving in isolation: Genetic tests reject recent connections of Amazonian savannas with the central Cerrado. Journal of Biogeography, 46(1), 196–211. https://doi.org/10.1111/jbi.13468.
Ribeiro, P. C., Lemos-Filho, J. P., de Oliveira Buzatti, R. S., Lovato, M. B., & Heuertz, M. (2016). Species-specific phylogeographical patterns and Pleistocene east–west divergence in Annona (Annonaceae) in the Brazilian Cerrado. Botanical Journal of the Linnean Society, 181(1), 21–36. https://doi.org/10.1111/boj.12394.
Ritter, C. D., Coelho, L. A., Capurucho, J. M., Borges, S. H., Cornelius, C., & Ribas, C. C. (2021). Sister species, different histories: Comparative phylogeography of two bird species associated with Amazonian open vegetation. Biological Journal of the Linnean Society, 132(1), 161–173. https://doi.org/10.1093/biolinnean/blaa167.
Rutter, N., Coronato, A., Helmens, K., Rabassa, J., & Zárate, M. (2012). Glaciations in North and South America from the Miocene to the last glacial maximum: Comparisons, linkages and uncertainties. Springer. https://doi.org/10.1007/978-94-007-4399-1.
Salgado-Labouriau, M. L. (1997). Late quaternary paleoclimate in the savannas of South America. Journal of Quaternary Science, 12(5), 371–379. https://doi.org/10.1002/(SICI)1099-1417(199709/10)12:5%3C371::AID-JQS320%3E3.0.CO;2-3.
Santos, M. G., Nogueira, C., Giugliano, L. G., & Colli, G. R. (2014). Landscape evolution and phylogeography of Micrablepharus atticolus (Squamata, Gymnophthalmidae), an endemic lizard of the Brazilian Cerrado. Journal of Biogeography, 41(8), 1506–1519. https://doi.org/10.1111/jbi.12291.
Silva, J. M. C. (1995). Biogeographic analysis of the South American Cerrado avifauna. Steenstrupia, 21, 49–67.
Silva, J. M. C. (1997). Endemic bird species and conservation in the Cerrado Region, South America. Biodiversity and Conservation, 6, 435–450. https://doi.org/10.1023/A:1018368809116.
Stamatis, C., Triantafyllidis, A., Moutou, K. A., & Mamuris, Z. (2004). Mitochondrial DNA variation in Northeast Atlantic and Mediterranean populations of Norway lobster, Nephrops norvegicus. Molecular Ecology, 13(6), 1377–1390. https://doi.org/10.1111/j.1365-294X.2004.02165.x.
Stephens, M., Smith, N. J., & Donnelly, P. (2001). A new statistical method for haplotype reconstruction from population data. American Journal of Human Genetics, 68(4), 978–989. https://doi.org/10.1086/319501.
Sunnåker, M., Busetto, A. G., Numminen, E., Corander, J., Foll, M., & Dessimoz, C. (2013). Approximate Bayesian computation. PLoS Computational Biology, 9(1), e1002803. https://doi.org/10.1371/journal.pcbi.1002803.
Tamura, K., Stecher, G., Peterson, D., Filipski, A., & Kumar, S. (2013). MEGA 6: Molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution, 30(12), 2725–2729. https://doi.org/10.1093/molbev/mst197.
Terrones, A., van der Bank, M., Moreno, J., & Juan, A. (2022). DNA barcodes and microsatellites: How they complement for species identification in the complex genus Tamarix (Tamaricaceae). Journal of Systematics and Evolution, 60(5), 1140–1157. https://doi.org/10.1111/jse.12830.
Thomé, M. T. C., Sequeira, F., Brusquetti, F., Carstens, B., Haddad, C. F. B., Rodrigues, M. T., & Alexandrino, J. (2016). Recurrent connections between Amazon and Atlantic forests shaped diversity in Caatinga four-eyed frogs. Journal of Biogeography, 43(5), 1045–1056. https://doi.org/10.1111/jbi.12685.
Toledo, F., & Haddad, C. F. B. (2005). Reproductive biology of Scinax fuscomarginatus (Anura, Hylidae) in south-eastern Brazil. Journal of Natural History, 39(32), 3029–3037. https://doi.org/10.1080/00222930500221403.
Tsai, Y. H. E., & Carstens, B. C. (2013). Assessing model fit in phylogeographical investigations: An example from the North American sandbar willow Salix melanopsis. Journal of Biogeography, 40(1), 131–141. https://doi.org/10.1111/j.1365-2699.2012.02775.x.
Van Der Hammen, T., & Hooghiemstra, H. (2000). Neogene and quaternary history of vegetation, climate, and plant diversity in Amazonia. Quaternary Science Reviews, 19(8), 725–742. https://doi.org/10.1016/S0277-3791(99)00024-4.
Van Els, P., Zarza, E., Rocha Moreira, L., Gómez-Bahamón, V., Santana, A., Aleixo, A., Ribas, C. C., Sena do Rêgo, Dantas Santos, P., Zyskowski, M. P., Prum, K., R. O., & Berv, J. (2021). Recent divergence and lack of shared phylogeographic history characterize the diversification of Neotropical savanna birds. Journal of Biogeography, 48(5), 1124–1137. https://doi.org/10.1111/jbi.14065.
Vasconcellos, M. M., Colli, G. R., Weber, J. N., Ortiz, E. M., Rodrigues, M. T., & Cannatella, D. C. (2019). Isolation by instability: Historical climate change shapes population structure and genomic divergence of treefrogs in the Neotropical cerrado savanna. Molecular Ecology, 28(7), 1748–1764. https://doi.org/10.1111/mec.15045.
Vaz-Silva, W., Maciel, N. M., Nomura, F., Morais, A. R. D., Batista, V. G., Santos, D. L., Andrade, S. P., de Oliveira, A. A. B., Brandão, R. A., & Bastos, R. P. (2020). Guia de identificação das espécies de anfíbios (Anura e Gymnophiona) do estado de Goiás e do Distrito Federal, Brasil Central. Sociedade Brasileira de Zoologia. https://doi.org/10.7476/9786587590011.
Wang, X. F., Auler, A. S., Edwards, R. L., Cheng, H., Cristalli, P. S., Smart, P. L., Richards, D. A., & Shen, C. C. (2004). Wet periods in northeastern Brazil over the past 210 kyr linked to distant climate anomalies. Nature, 432(7018), 740–743. https://doi.org/10.1038/nature03067.
Wang, X., Edwards, R. L., Auler, A. S., Cheng, H., Kong, X., Wang, Y., Cruz, F. W., Dorale, J. A., & Chiang, H. W. (2017). Hydroclimate changes across the Amazon lowlands over the past 45,000 years. Nature, 541(7636), 204–207. https://doi.org/10.1038/nature20787.
Webb, S. D. (1978). A history of savanna vertebrates in the new world. Part II: South America and the great interchange. Annual Review of Ecology and Systematics, 9(1), 393–426. https://doi.org/10.1146/annurev.es.09.110178.002141.
Werneck, F. P. (2011). The diversification of eastern South American open vegetation biomes: Historical biogeography and perspectives. Quaternary Science Reviews, 30(13–14), 1630–1648. https://doi.org/10.1016/j.quascirev.2011.03.009.
Werneck, F. P., Gamble, T., Colli, G. R., Rodrigues, M. T., & Sites, J. W. Jr. (2012a). Deep diversification and long-term persistence in the South American ‘dry diagonal’: Integrating continent‐wide phylogeography and distribution modeling of geckos. Evolution: International Journal of Organic Evolution, 66(10), 3014–3034. https://doi.org/10.1111/j.1558-5646.2012.01682.x.
Werneck, F. P., Nogueira, C., Colli, G. R., Sites Jr, J. W., & Costa, G. C. (2012b). Climatic stability in the Brazilian Cerrado: Implications for biogeographical connections of South American savannas, species richness and conservation in a biodiversity hotspot. Journal of Biogeography, 39(9), 1695–1706. https://doi.org/10.1111/j.1365-2699.2012.02715.x.
Wüster, W., Ferguson, J. E., Quijada-Mascareñas, J. A., Pook, C. E., Salomão, M. G., & Thorpe, R. S. (2005). Tracing an invasion: Landbridges, refugia, and the phylogeography of the Neotropical rattlesnake (Serpentes: Viperidae: Crotalus durissus). Molecular Ecology, 14(4), 1095–1108. https://doi.org/10.1111/j.1365-294X.2005.02471.x.
Yang, Z., & Rannala, B. (2010). Bayesian species delimitation using multilocus sequence data. Pnas, 107(20), 9264–9269. https://doi.org/10.1073/pnas.0913022107.
Yang, Z., & Rannala, B. (2014). Unguided species delimitation using DNA sequence data from multiple loci. Molecular Biology and Evolution, 31(12), 3125–3135. https://doi.org/10.1093/molbev/msu279.
Zachos, F. E., Frantz, A. C., Kuehn, R., Bertouille, S., Colyn, M., Niedziałkowska, M., Pérez_González, J., Skog, A., Sprĕm, N., & Flamand, M. C. (2016). Genetic structure and effective population sizes in European red deer (Cervus elaphus) at a continental scale: Insights from microsatellite DNA. Journal of Heredity, 107(4), 318–326. https://doi.org/10.1093/jhered/esw011.
Acknowledgements
For donation of tissue samples, we thank César Barrio-Amorós (CAB), Guarino Colli (CHUNB), Natan Medeiros Maciel (ZUFG), Martin Jansen (MNKA), Diego Baldo (LGE), Franco L. Souza (ZUFMS), Taran Grant (CTMZ) and Marcos Andrés de Carvalho (UFMT). We also thank Manolo Perez for help in ABC analyses and Robert Owen for reviewing English text. The following agencies provided financial support: Consejo Nacional de Ciencia y Tecnologıa (CONACYT, Paraguay) through Programa Nacional de Incentivo a Investigadores (PRONII, fellowship to F.B.), Programa Paraguayo para el Desarrollo de la Ciencia y Tecnología (PROCIENCIA, grant PINV15-684 to F.B.), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPQ) for a research fellowship to C.F.B.H., and Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP Proc. #2018/03428-5 and FAPESP Proc. #2021/10639-5).
Author information
Authors and Affiliations
Contributions
F.B and N.C.P. conceived the ideas; F.B. collected the data; C.F.B.H. provided the resources, logistical support and funding; F.B and N.C.P. analyzed the data and led the writing. All authors contributed with manuscript review and editing.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Brusquetti, F., Pupin, N.C. & Haddad, C.F.B. Model-Based Analyses Suggest Pleistocene Refugia over Ancient Divergence as Main Diversification Driver for a Neotropical Open-Habitat Treefrog. Evol Biol 50, 432–446 (2023). https://doi.org/10.1007/s11692-023-09614-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11692-023-09614-3