
Abstract
The purpose of this study is to investigate the effect of copper powder oxidation on the deposition efficiency, microstructure, wear and corrosion resistance. The gas-atomized copper powders in the as-received (Cu-Safina and Cu-FST) and oxidized states (Cu-treat, oxidized in air, 25 °C for 5 months; Cu-treat1, oxidized at 100 °C for 1 h; and Cu-treat2, oxidized at 200 °C for 1 h) were used to prepare the coatings by cold gas spray (CGS). XPS analysis detected Cu2O and CuO for all feedstock powders, increasing for oxidized ones. The deposition efficiency and thickness of the coatings followed the order: Cu-Safina > Cu-FST > Cu-treat1 > Cu-treat > Cu-treat2. For oxidized coatings, SEM images showed more defected microstructure, increase in pores, and microcracks. Cu-FST coating showed a sliding wear rate of (0.13 ± 0.01) × 10-4 mm3 N−1 m−1), and abrasive wear rate of (3.2 ± 0.2) × 10−4 mm3 N−1 m−1. Gas-atomized powder coatings showed a better corrosion resistance performance. The electrolyte did not reach the substrate/coating interface for t ≥ 700 h and the coatings resisted for 2000 h in salt fog tests. However, oxidized coatings showed low corrosion resistance due to the presence of cracks and defects, and the coating/substrate was severely damaged after ≈100 h in 3.5wt.%NaCl solution.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cold gas spray (CGS) is becoming a very promising technique for coating production, especially for materials susceptible to oxidation, such as copper (Ref 1,2,3,4). These coatings are mainly used in heating systems, conductive tracks, and flange parts due to their corrosion resistance and sliding wear resistance (Ref 1,2,3). The underlying principle of CGS is that the sprayed material is no longer heated up to a semi-molten or fully molten state to be stuck on the substrate surface. Instead, solid-state powders are accelerated in a high-pressure supersonic gas jet (in a nozzle with convergent-divergent geometry) that allows the particles to be plastically deformed during impact with the target to form interconnected splats adhered to the substrate, resulting in an overlay deposit (Ref 1). Unlike other thermal spraying techniques, CGS uses higher kinetic energy instead of heat energy for the deposition (Ref 2). The work temperature does not exceed 1000 °C; the particle speed during spraying can reach, depending on the gas or spray parameters, between 500 and 1200 m s−1 (Ref 3, 4).
The formation mechanism of CGS coatings is not yet well established as the characteristics of the matching properties of the coating-substrate pair materials also play a very important role (Ref 4,5,6). It is known that when a particle is sprayed onto the substrate, mutual plastic deformation of particle and substrate is desirable for proper bonding to occur, depending on the relative mechanical properties of both substrate and spraying material. Oxides from the substrate are removed upon impact, which provides an ideal surface for effective adhesion of the particle: either metallurgical bonds and/or mechanical anchoring have been reported (Ref 1,2,3, 6). The effective contact between the surface and particle and the high pressures during spraying, causing compression of the particles on the substrate and on the previous deposited layer, are ideal conditions for adhesion of the particle/substrate and particle/deposited material (Ref 6). Other phenomena that are often responsible for the adhesion of materials such as interfacial melting and diffusion of materials between phases were investigated (Ref 3, 6). However, the results show that these factors do not have a large influence in the case of CGS. This is because the average of the kinetic energy of the particles during the spraying process and the thermal energy of particles during spraying is much smaller than the energy necessary to produce melting and diffusion; therefore, the formation of the coatings is a solid-state process (Ref 6). Furthermore, plastic deformation is generally considered an energy dissipation mechanism (Ref 7). Thus, even the increase in temperature at the interfacial region due to the impact of the particles is not significant to promote diffusion processes between the materials (Ref 3, 7).
Several works have revealed that for effective bonding and coating formation, the sprayed material needs to achieve a critical velocity during the spray process (Ref 8,9,10,11,12). This is basically the speed above which the particle will be able to adhere on the surface upon impact. For this purpose, it is important to understand all the variables affecting the process, i.e., the chemical, physical, and mechanical properties of powder and substrate, as well as the spraying parameters. The spraying parameters, such as gas pressure, gas temperature, spraying distance, feeding rate, use of different guns, nozzle, carrier gas, and surface preparation or even substrate heating are conditions that influence the properties of the final product and should be considered in the coating preparation (Ref 1,2,3). The influence of these variables on the coating preparation has been reported in some studies (Ref 9,10,11).
Interestingly, several works have reported the influence of oxidation of the starting material in the coating formation by CGS (Ref 9,10,11,12,13,14). Previous studies (Ref 10, 11) have shown that the presence of an oxide film on the surface of the particles had a significant influence on the deposition efficiency, microstructure, and properties of cold-sprayed coatings. The oxide inclusions at the particle interface could inhibit bonding and decrease the bonding strength between the coating and substrate. Some studies using experimental and computational simulation (Ref 9, 10, 14) have shown that the original existing oxide film on the particle surface can be disrupted during the impact with the surface and pushed out from the interface by the outward metal jet. However, it has been observed that the cracked oxides cannot be entirely cleaned up; part of them will be left at the interface after particle deposition and these remaining oxides significantly influence the metallic bonding strength and the final coating quality (Ref 10, 15, 16). Wei et al. studied copper feedstock powders fabricated through electrolysis (EP), gas-assisted water atomization (WA), inert gas atomization (GA) and annealed before depositing by cold spray. The strain release and low GB angle play a role in increasing the bonding strength of cold-sprayed coatings. The asymmetric dendritic morphology is detrimental to intensive plastic deformation, resulting in the lowest bonding strength of cold-sprayed coatings. The recrystallized twin grains are beneficial for plasticity which may increase the bonding strength (Ref 16).
Few researchers have reported the corrosion resistance of copper coatings deposited by CGS (Ref 17,18,19,20,21). In our previous study (Ref 17), copper coatings showed a dense microstructure, with thickness above 500 µm, and a porosity of 0.5%. The coating was able to protect the steel substrate against corrosion for ~ 1100 h of immersion in 3.5 wt.% NaCl solution. Winnicki et al. (Ref 20) produced copper coatings with 1% porosity and high corrosion resistance. According to the Tafel curves, the coatings showed passivation due to the formation of a passive oxide layer covering the copper coating surface, leading to a decrease in the corrosion rate. However, none of these reports investigated the corrosion resistance of copper coatings obtained from raw materials with different degrees of oxidation, which is presented in this work. For that purpose, coatings were prepared using copper powders with different oxidation degrees and mainly their electrochemical behavior was studied.
The purpose of this work is to evaluate the deposition of different copper feedstock powders subjected or not to different oxidation treatments and investigate the electrochemical behavior and some mechanical properties of the prepared coatings. Oxidized copper powders at room temperature (≈25 °C, for 5 months), at 100 °C for 1 h, and at 200 °C for 1 h, and a gas-atomized powders were sprayed. The influence of these different feedstock materials in the coating formation, microstructure, and wear and corrosion resistance was investigated. Different from reports by other researchers, the corrosion resistance of coatings was evaluated using open-circuit potential (EOCP) during a relatively long immersion time, electrochemical impedance spectroscopy (EIS) and salt fog spraying, and corrosion mechanisms are proposed.
Experimental Design
Feedstock Materials
To evaluate the influence of powder oxidation degree on the coating formation, five copper powders were used as feedstock as described in Table 1.
Cu-Safina powder was supplied by Safina A.S (Vestec, Prague, Czech Republic-Cu ≥ 99.9%) and Cu-FST powder supplied by Flame Spray Technologies - FST (Duiven, Netherlands-Cu ≥ 99%). Three oxidation treatments were applied to the Cu-FST powder to obtain the other three oxidized feedstock powders.
Substrate and Reactants
A low carbon steel alloy with the composition Al (0.46 wt.%), Ti (0.22 wt.%), Mn (0.18 wt.%), C (0.15 wt.%), Cr (0.091 wt.%), Zn (0.080 wt.%), and Fe balance, determined using an EDX-720/800HS Energy-Dispersive x-ray Spectrometer (Shimadzu, Barueri-SP, Brazil), was used as the substrate. Rectangular and flat (50 × 20 × 5 mm) and cylindrical (∅ = 25.4 mm and h = 25.4 mm) substrate samples were degreased with acetone and grit blasted with alumina (MPA, Spain) 220-grade corundum using a low-pressure cold gas spray (LPCGS) system. The resultant roughness was Ra = (4.7 ± 0.4) µm as an average value obtained from at least ten roughness measures on each sample surface. The measures were performed using a Mitutoyo Surftest 301 Precision (Mitutoyo, Japan) reference specimen calibrated with a calibrator 2.97 µm rough. NaCl (Sigma-Aldrich) 99.8% and Milli-Q water (18.2 MΩ cm) were used for preparing the electrolyte for electrochemical experiments.
Coating Preparation
Copper coatings were produced at the Thermal Spray Center, Barcelona University. Cold spray equipment Kinetics 4000/17 kW (Cold Gas Technology, Ampfing, Germany) was used with a WC nozzle (type D24) and nitrogen as the carrier gas. The copper coatings were obtained under the conditions: traverse speed of 500 mm/s, standoff distance of 40 mm, nitrogen gas temperature of 400 °C and pressure of 3 MPa. (Ref 17).
Structural, Morphological, and Chemical Characterization
The particle size distribution of powders was analyzed by laser scattering (LS) using a Beckman Coulter LS 13320 instrument. Feedstock materials and coatings were structurally characterized by scanning electron microscopy (SEM) using a JEOL JSM-5310 scanning electron microscope coupled to an energy-dispersive x-ray spectroscopy (EDS). SEM images were obtained at different energy values as indicated in the corresponding figures. For coatings prepared with oxidized powders and subjected to long immersion times, SEM images were only obtained after drying for around 24 h at 100 °C in a vacuum chamber.
The powders and coatings were analyzed by x-ray diffraction (XRD) using an x-ray diffractometer (SIEMENS model D5000). Structural characterization of the Cu-Safina, Cu-treat, and Cu-treat2 powders was performed with x-ray photoelectron spectroscopy (XPS), using a UNI-SPECS UHV instrument. The powder was fixed onto a small flat disk on an XYZ manipulator and placed in the analysis chamber. The residual pressure in this chamber was maintained below 10−8 Torr, while data were being obtained. The Mg Kα line was used (hν = 1253.6 eV) and the analyzer pass energy set to 10 eV. XPS was used to investigate the core level of C 1s, O 1s, Si 2p. The intensities were estimated by calculating the area under each peak after smoothing and subtraction of the shaped background and fitting the experimental curve to a combination of Lorentzian and Gaussian lines of variable proportions.
The coating thickness was determined from the transverse section of SEM images using Image J software making 10 measures for each sample. For that, the samples were cut, embedded in resin, and polished using grit sandpaper of 320, 600, 1200, and 4000 grit, and polished with diamond suspension of 6 and 1 µm. The porosity of coatings was also estimated by analyzing the transverse section using optical microscopic (OM) images using Image J software, according to the ASTM E2109-01 standard practice.
Physical and Mechanical Characterization
For coatings prepared with oxidized powders, mechanical properties were not measured due to the great material loss in tests made using different loads (5-20 N). This is probably related to the low cohesion between oxidized copper particles (Ref 11, 12). The tribological properties of coatings were characterized by two standardized wear tests: sliding and abrasive. The sliding wear test was carried out using a CM4 ball-on-disk instrument to evaluate the friction coefficient (ASTM G99-17). A stainless steel counterpart ball, with a sample relative velocity of 131 rpm, a total sliding distance testing of 1000 m, and a normal force load of 5 N for a radius of 14 mm, were the selected parameters for test purposes. Humidity and temperature were maintained below 20% and 25 °C, respectively. The wear tracks produced on the coatings were studied using SEM, and a Leica confocal equipment was used to measure the sliding wear rate of the wear tracks and recreate the wear path images.
Abrasive tests were carried out using a CM4 Rubber Wheel (ASTM G65-16) machine with a rotation rate of 139 rpm, and a load of 50 N with a flow of silica (∅ = 0.4-0.8 mm) between 250 and 310 g min−1. Material loss was measured by weighing the samples every 1 min during the first 5 min and then every 5 min until the end of the test (total duration: 30 min). To express the wear rate in terms of the removed material (mm3 N−1 m−1), the density of the coatings was used to transform mass into volume.
The adhesion of the coatings was performed according to the ASTM C633-13 (Standard Test Method for Adhesion or Cohesion Strength of Thermal Spray Coatings), using a cylindrical substrate (∅ = 25.4 mm and height = 25.4 mm). The test consists of coating one face of a substrate fixture, bonding this coating to the face of a loading fixture, and subjecting this assembly of coating and fixtures to a tensile load normal to the plane of the coating. The tests were carried out on CSM-Revetest equipment with a constant speed of 10 mm/min with a progressive increase in the force traction until the adhesive failure of the coating. The measurements were performed in triplicate.
The electrical conductivity and resistance of the coatings were measured by eddy current measurements with the use of FOERSTER SIGMATEST 2.069 equipment. The measurements were conducted using a probe, with a current frequency of 960 kHz and 22.5 °C.
Corrosion Studies
All experiments were carried out using a conventional Tait type cell (Ref 22) with 80 mL of 3.5 wt.% NaCl solution and three-electrodes setup. The reference electrode was an Ag|AgCl|KCl3mol L−1 coupled to a Luggin capillary, with a Pt-network as the auxiliary, and the substrates (ground with P240 SiC paper) or as-prepared coated substrates were used as working electrodes. The specimen area exposed to the electrolyte solution was 1 cm2. The corrosion resistance of the samples was evaluated through open-circuit potential (EOCP) measurements, small amplitude linear polarization (SALP), potentiodynamic polarization curves (PPC), and (EIS).
Using SALP, Rp was determined from I–E curves by applying ± 10 mV/EOCP and recorded at 0.166 mV s−1 after 18 h of immersion. Potentiodynamic polarization experiments were performed after 18 h of EOCP measuring, using a scan rate of 0.166 mV s−1 at the potential interval from − 0.050 V/EOCP to a potential where the anodic current is near 10-3 A cm-2. EIS and EOCP were monitored for relatively long immersion times: Cu-Safina 1100 h; Cu-FST 600 h, Cu-treat1 360 h, Cu-treat 380 h, and Cu-treat2 for 340 h of immersion. For the coatings obtained with the oxidized powders, this time corresponds to the change in EOCP values, indicating that the electrolyte may have reached the substrate. EIS measurements for coated samples were performed at 1 h and every 24 h of immersion, in the frequency range from 100 kHz to 5 mHz, by applying a sinusoidal potential perturbation of 10 mV rms on EOCP and recording 10 points/frequency decade. All experimental data were tested for consistency with the Kramers–Kronig transform (KKT) available in GAMRY system software. The equivalent electrical circuit (EEC) theory using the Z-view® software was employed for quantitative analysis of the EIS responses. All measurements were performed using GAMRY Reference 600 systems, in aerated and unstirred 3.5 wt.% NaCl solution at (25 ± 1) °C. Salt spray tests of as-deposited Cu-Safina and Cu-FST coatings were performed in a neutral solution of 5 wt.% NaCl at ≈35 °C for 1000 h, in accordance with ASTM B117-11. As no signals of corrosion were observed up to 1000 h, a time established by the Standard Practice, the salt spray test was continued for a total of 2000 h. For instance, considering that electrochemical techniques (EIS and EOCP) clearly indicated the deterioration of the coatings prepared with oxidized powders around 350 h compared to Cu-Safina (1000 h) and Cu-FST (600 h), the salt spray test for oxidized coatings was considered of less importance.
Results and Discussion
Structural, Morphological, and Chemical Characterization
LS analyses (Fig. S1, SD-supplementary data) showed that the particle size of all powders was micrometric with approximate particle size distribution in the range of 18-45 µm (Cu-Safina), 15-56 µm (Cu-FST), 19-54 µm (Cu-treat1; 100 °C, 1 h), 19-55 µm (Cu-treat; air for 5 months at ~ 25 °C), and 19-50 µm (Cu-treat2; 200 °C, 1 h). SEM images of the powder surface morphology (Fig. S2, SD) show that the powder had a similar spherical shape. The powders present a micrometer particle size with submicron particles (called satellites) formed during solidification in the gas atomization process (Ref 23). The LS and powder morphology analysis show that the oxidation treatments did not appreciably change the average particle size. This avoids any influence that the particle size may induce during the spraying of the coating. These characterizations show that the feedstock material has an ideal size and morphology for spraying by CGS, mainly because smaller and regular shaped particles can achieve higher speeds during spraying, reaching the substrate with higher kinetic energy, allowing greater plastic deformation and formation of more dense and compact coatings (Ref 24).
XRD patterns of the powders (Fig. 1) and coatings (Fig. S3, SD) apparently revealed only peaks attributed to the Cu phase. Peaks observed at 2θ: 43.3°; 50.4°; 74.1°; 89.9°; and 95.1° are attributed to metallic copper. It seems that no change in the chemical composition of the starting material occurs and the amount of surface oxides, generated during the treatment of the powders, was not enough to be detected by the XRD analysis performed. Since CGS is a solid-state process, the chemical composition of the feedstock powder is maintained in the deposited coating. The reason for the lack of change to the powders during the coating buildup is that CGS uses kinetic energy instead of thermal energy and relatively low temperatures, leading to oxide-free coatings, free of unwanted chemical reactions (Ref 2, 25). However, a closer look at the feedstock copper powders indicated the presence of copper oxides, mainly for FST and the oxidized powders (Fig. 1). Peaks of low intensity were observed from 2θ: 32° to 40° and from 2θ:57° to 65° for FST and oxidized FST powders, respectively. The peak at 2θ = 36.45°, with very low intensity for FST, is associated with the presence of Cu2O (2θ = 36.441°) (Ref 26, 27) and increases in the order Cu-FST < < Cu-treat1 (100 °C, 1 h) < Cu-treat2 (200 °C, 1 h) powders. Another peak related to the presence of Cu2O is observed at 2θ = 61.41° (Fig. 1) with intensity following the order: Cu-treat ≅ Cu-treat1 < Cu-treat2 powders. These results indicate that the thermal treatment favored the Cu2O formation, which is grown by diffusion of copper ion vacancies, resulting in a gross defect structure of Cu2O, in confront to CuO (Ref 26,27,28,29,30,31). The oxidation of Cu-FST powder at room temperature (~25 °C) and air atmosphere (5 months) formed both Cu2O and CuO oxides (Ref 26, 27, 31). Peaks corresponding to Cu2O and CuO (2θ = 35.33 ° and 38.78°) were observed for Cu-treat powder. To confirm the nature of the oxides on the surface of the copper powders, Cu-Safina, Cu-treat, and Cu-treat2 powders were analyzed by XPS.

X-ray diffractograms of Cu powders
Figure 2 shows the XPS spectra for the O1s and Cu2p for Cu-Safina, Cu-treat, and Cu-treat2 powders. Table S1(SD) shows the binding energy values, area, and full width at half-maximum (FWHM). It is important to remember that the XPS technique, under the conditions used, allows analysis to a depth of ~ 5 nm from the sample surface. The presence of metallic copper in the XPS spectrum means that a very thin oxide layer covers the Cu particles. Previous works showed that the thickness of the native oxide layer can range from 2.5 to 5.2 nm for copper exposed during 112 days at atmosphere condition (Ref 26). XPS data show that all powders have a thin copper oxide layer mainly composed of Cu2O and CuO. At the surface of the three powders analyzed, the oxygen content followed the order: Cu-treat > Cu-treat2 > Cu-Safina (Table S1, SD). This result demonstrates that the oxidation treatments allowed copper oxides to grow on the surface, and despite this, SEM images of the cross section and surface of the powders (Fig. S4, SD) did not clearly show morphological differences between the powders, probably because a very thin oxide layer was formed. With the aim of supporting the XRD and XPS data were performed a detailed morphological analysis of copper powder splats. SEM images of powder splats (Fig. S5, SD) showed differences in their morphology. The Cu-Safina splat showed a smooth morphology, and no oxide layer was observed. On the other hand, the Cu-treat2 splat showed a rougher appearance due to the plastic deformation of copper particle covered with an oxide layer. These oxide films could obstruct the particle deformation, and crushing the oxide films could dissipate part of the kinetic energy (Ref 11, 12). Therefore, more kinetic energy should be provided to obtain a proper particle deformation (Ref 11, 12). So, this splat morphology can inhibit the bonding between the deposited particles, which can decrease the deposition efficiency and coating thickness. The oxides above the particles started to grow as islands which can contribute to the formation of a coating with more cracks, defects, inclusion of oxides, and low deposition efficiency. The effect of the presence of this thin oxide layer was also observed in the electric conductivity and resistivity experiments of coating. The presence of oxides significantly reduces the coating conductivity and increases the coating resistivity (Table S2, SD).

XPS fitting spectra for O1s and Cu2p. (a) Cu-Safina, (b) Cu-treat, and (c) Cu-treat2 powders
The XPS can detect oxygen bonded to different chemical species present in the XPS chamber even considering the very low pressure (~10−7 Pa). These species may be organic molecules, carbon dioxide, and others containing O–C=O and O–C groups that appear in the XPS spectra but cannot be detected by XRD due to the chemical nature and the quantity below the technique detection limit. The metallic hydroxides detected by XPS could not be detected by XRD considering the small quantity adsorbed on the coating surface. It means that XPS is much more sensitive than XRD.
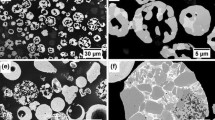
Figure 3 shows cross-sectional SEM images of copper coatings. The particles appear to be severely deformed, and the coatings are highly dense. High particle velocities in the order of 500-1200 m/s during the spraying process cause severe plastic deformation of the material on impact with the substrate to produce good bonding between particle/substrate and between particles, allowing thick and dense coatings to be formed (Ref 32, 33). The top layer of the coatings was less compact than the bottom layer (Fig. 3), agreeing with the literature (Ref 32,33,34) that describes CGS coatings as mainly consisting of a top layer with some porosity and a dense bottom layer. However, for the coatings prepared with oxidized powders, some cracks were observed along the coating cross section (Fig. S7 and S8, SD). On the other hand, for the gas-atomized powders (Fig. 3 and S8, SD), no cracks were observed at the metal/coating interface or along the coating. The porosity measured at the bulk of the sample (medium of the coating cross section) was around 0.6% for all coatings, the average value being the same, within the experimental error, for all coatings (Table 3S, SD); therefore, the coatings can be considered compact.

Cross-sectional SEM images of as-prepared coatings: (a) Cu-Safina, (b) Cu-FST, (c) Cu-treat1, (d) Cu-treat, (e) Cu-treat2
The dark particles at the coating/substrate interface (Fig. 3) are alumina as showed by EDS analysis (Fig. S9, SD). These particles adhere to the surface during the grit blasting process for surface preparation. The images did not show any defect or delamination at the coating/substrate interface. The adherence of the coatings obtained with optimized parameters was found to be in the range from 20 to 25 MPa. The coatings failed at the substrate/coating interface, meaning that the interlayer bonding was stronger than that of the substrate-coating interface (Ref 17). However, the cross section of the Cu-treat2 sample shows a great number of small particles that contrast with the metallic areas, which are probably covered by oxides, while the cross section of the entire Cu-treat shows a porous layer at the surface with cracks and Cu-treat bottom (Fig. S6 and S7, SD) with cracks and porosity.
The thickness of the studied copper coatings follows the order: Cu-Safina > Cu-FST > Cu-treat1 > Cu-treat > Cu-treat2 (Table 2). The coatings sprayed with the oxidized powder at 200 ºC or stored in an air atmosphere (5 months) presented smaller thicknesses. This demonstrates that the oxide on the surface of the particles and, possibly, its chemical nature, even as a thin layer, have a strong influence on the deposition efficiency (Fig. 4) and, consequently, on the thickness of coatings.

Deposition efficiency of different copper powders
The deposition efficiency for all coatings was also calculated as the previously described procedure (Ref 13) using Eq 1:
Kindly note that the equations are renumbered to ensure sequential ordering. Accordingly citations were also changed. Kindly check and confirm.It is ok.
The sprayed mass was obtained from the cold spraying parameters. The deposited mass was determined by measuring the mass of the sample before and after spraying and subtracting the mass of the substrate. Figure 4 indicates low deposition efficiencies when oxidized feedstock materials are used.
The differences in deposition efficiency and thickness are consequence of the presence of oxides on the particle surface that leads to a decrease in the cohesion between particles and particle/substrate during the coating formation (Ref 11, 12). The occurrence of oxides on the particle surface makes the interaction between the surfaces less effective, leading to worse performance in the deposition and coating building, which has also been observed earlier (Ref 11, 12, 16). The difficulty in spraying oxidized particles and achieving good deposition efficiency can be explained by the previously described mechanism (Ref 12, 16).
Another study suggests that the oxide films could impede particle deformation and crushing the oxide films would dissipate part of the kinetic energy (Ref 11). Therefore, more kinetic energy should be provided to obtain the critical velocity and particle deformation, as well as which, the gas condition for deposition of the oxidized powder should be adjusted. However, when changing the spraying parameters, the oxidized Cu still demonstrates a thinner coating compared with the coating deposited with the gas-atomized powder. This reveals that the presence of oxides on the feedstock powder plays an important role in the coating formation.
Wear Behavior
Sliding Wear Resistance
Tests performed with oxidized coatings showed high wear damage at the first 30-60 s of the experiment, making it difficult to quantify the results of wear tests. Therefore, the wear tests were performed only for Cu-Safina and Cu-FST coatings. After a sliding distance of 1000 m, both samples demonstrated almost similar values of friction coefficient and Sliding wear rate (Table 3).
Figure 6 shows the wear track of the coatings after the sliding test. The smeared appearance of the surface is typical of adhesive wear (Ref 34, 35), and the wear track profiles show evidence of plowing and extrusion of the worn material outside the wear track. The profiles of wear paths obtained by confocal microscopy also evidence the adhesive wear mechanism. The images show that the material is plastically deformed during the test and part was taken to the wear track edges. The loose wear debris had the same color and appearance as the coating material, again suggesting an adhesive wear mode (Ref 34,35). It was also observed Cu adhered to the steel counter ball surface, indicating an adhesive wear mechanism (Ref 34, 35). On the other hand, the ball appears to be virtually unworn. This is a case of adhered material being transferred from the softer coating to the harder counter ball material. All these facts clearly support an adhesive wear mechanism.

Morphological features of the wear tracks of the cold spray copper coatings: (a) Safina coating and (b) FST coating

Open-circuit potential vs. time for all studied samples measured in aerated and unstirred 3.5 wt.% NaCl solution for 18 h at 25 °C
Abrasion Tests
Abrasive wear tests were performed only for Cu-Safina and Cu-FST coatings due to the same reason already explained before. For the test, as-prepared specimens without grinding or polishing the surface were used. Abrasion tests showed that the abrasive wear rate slowly decreased for a short time (2-3 min) and then remained constant until the end of the test. The abrasive wear rate was almost the same with values of (3.8 ± 0.3) × 10-4 mm3 N−1 m−1 (Cu-Safina) and (3.2 ± 0.2) × 10-4 mm3 N−1 m−1 (Cu-FST). The SEM images of the wear tracks (Fig. S10, SD) show a plowing aspect and areas of different shades of a gray color. EDS analysis (Fig. S11, SD) before and after abrasive tests demonstrated a drastic increase in oxide content on the coating surfaces after abrasion.
Electrochemical Corrosion Studies
Open-Circuit Potential and Polarization Studies
Firstly, the electrochemical results obtained at short immersion times (~18 h) will be discussed. Figure 6 shows the EOCP curves in 3.5 wt.% NaCl solution during 18 h of immersion. For the carbon steel substrate, EOCP values decreased during the first hours of immersion and potential oscillations were observed with the increase in immersion time. The oscillations can be related to the attack of chloride ions to the oxide thin layer present at the surface followed by precipitation/dissolution of corrosion products. It is well known that the native iron oxide grows on the steel surface for thermodynamic reasons and common steel is not passivated in chloride solution, mainly in neutral highly concentrated chloride, acid, or extremely alkaline solutions (Ref 34). The EOCP of the substrate tends to stabilize at ~-0.74 (V/Ag|AgCl|KCl3 mol L−1).
For all copper coatings and until 1 h of immersion, the EOCP values remain between − 0.14 V and − 0.24 (V/Ag|AgCl|KCl3 mol L−1), close to that obtained for the bare massive copper in chloride medium (Ref 37), reflecting the copper behavior for Cu-Safina and Cu-FST. Cu-Safina, Cu-FST, and Cu-treat1 coatings show EOCP values of − 0.18 (V/Ag|AgCl|KCl3 mol L−1) after 18 h of immersion. Cu-treat1 sample presents some amount of Cu2O and keeps the EOCP value almost constant during the time course of the experiment, suggesting the small influence of this film on the open-circuit potential. For the Cu-treat2 coating, the EOCP decreases after ~ 1 h and stabilizes at − 0.53 (V/Ag|AgCl|KCl3mol L−1), reflecting a mixed potential between steel and copper, a consequence of the defected-oxide grown on the powder surface, allowing the electrolyte to achieve the coating/substrate interface in a few hours. For Cu-treat coating, the potential decreases to − 0.26 (V/Ag|AgCl|KCl3 mol L−1) after ~ 3 h, remains constant at ~ − 0.28 (V/Ag|AgCl|KCl3 mol L−1) until 12 h, and then continuously diminishes to reach − 0.40 (V/Ag|AgCl|KCl3 mol L−1) at the end of the test. This result may attribute to the same reasons given above, and the difference in the potentials is related to the presence of a mixture of Cu2O and CuO on the powder surface. If EOCP value of the coating is closer to the substrate, which reflects the existence of pores, it means an open way to expose the substrate to the used solution (Ref 21).
After 18 h of immersion, the anodic and cathodic Tafel slopes were obtained from polarization curves, Rp values from SALP measurements and icorr estimated using the Stern–Geary equation (Ref 38,39,40). All parameters are shown in Table 4. The Rp values have followed the order: Cu-Safina > Cu-FST > Cu-treat1 > Cu-treat > Cu-treat2. The Cu-Safina and Cu-FST coatings presented higher Rp values than the others due to the lower porosity (0.55% Cu-Safina, 0.58% Cu-FST), higher thickness (Table 1), and a dense microstructure (Fig. 3a, b and Fig. S8), thus acting like a barrier, making slower the diffusion of the electrolyte through the coating. On the other hand, the oxidized powder coatings showed low Rp values.
The low Rp values can be attributed to the diffusion of electrolyte through the pores, cracks, and defects (Fig S6 and S7, SD), of the oxidized coatings that allows the corrosion of coating/substrate interface. This is also supported by EOCP measurements. At 18 h of immersion the Cu-treat2 and Cu-treat (Fig. 6), and after 48 h for the Cu-treat1 (Fig. 8), the coatings showed negative EOCP values almost close to the substrate, which clearly indicates an active state due to the electrolyte that reaches the coating/substrate interface.
It is important to notice that the potentials at i → 0 are like those obtained from the open-circuit measurements, indicating that the SALP experiments do not move the system from an apparent steady state, i.e., it demonstrates that the systems returned to the initial state after each linear polarization.
Figure 7 shows the potentiodynamic polarization curves for the substrate and coatings. The current density increases for the coatings due to the dissolution of copper to cuprous and/or cupric ions (Ref 37) or iron oxidation (Ref 34). The substrate and Cu-treat2 demonstrate similar behavior with a current increase after the corrosion potential and going in an anodic direction. These results suggest that similar processes are dominating the i–E response, which seems to be the iron dissolution, since the potential for the dissolution of copper has not yet been attained. This would be possible only if iron takes part in the electrode reactions, which suggests that the solution reached the substrate during the first hours of immersion. This hypothesis is also supported by E−t measurements (Fig. 6). For Cu-Safina, Cu-FST, and Cu-treat1, a fast increase in the current density occurs for potentials between − 0.10 and 0.0 (V/Ag|AgCl|KCl3 mol L−1) due to the copper oxidation, mainly to cuprous ions (Ref 41). Copper dissolution mainly begins in zones of material with stress and defects, as well as at oxide defects and the regions surrounding them to continue all over the copper surface. Regarding the oxidized coatings like Cu-treat and Cu-treat2, the current density is slightly lower (Table 3 and Fig. 7), due to the decrease in the active area and/or precipitation of corrosion products. Cross-sectional SEM images, after polarization (Fig. S12, SD), indicated that a brittle zone is formed inside the coatings, a region up to ≈50 µm from the top of the coating, in which cracks are easily developed during the cross section preparation. The applied potential also favors the increase in stress at the top region due to the acceleration of the growth of copper oxides, the dissolution of cuprous chloride, and electrolyte penetration. The electrochemical results obtained so far indicate, as expected, that the potential region where the main electrochemical processes occur depends on the chemical nature of the surface of the feedstock powders and the coating structure, even when considering that all coatings are copper.

Linear polarization of steel substrate and coated samples recorded in non-deaerated and quiescent 3.5 wt.% NaCl solution at 0.166 mV/s and after ~ 18 h of immersion
The cathodic Tafel slopes obtained from Fig. S13 (SD) at potentials less negative than those of the current plateau of oxygen reduction reaction (ORR) were around − 170 mV/dec, like the values obtained by other researchers (Ref 42, 43) using the same conditions. The main reactions involved in the coating degradation in the aggressive medium can be suggested based on those proposed for iron and copper dissolution in a chloride medium. For iron dissolution in halide-containing acid solutions, the mechanism is similar to that proposed in the acid medium by Bockris, Drazic, Despic (BDD) (Ref 44), and modified by Kelly (Ref 45). It is also believed that (FeOH)ad species can partially cover the electrode surface, dissolve, or grow to form a passive layer, depending on the applied potential and pH of the solution at the electrode surface. In our copper coating system, halides may inhibit the iron dissolution when reaching the substrate due to the strong adsorption or accelerate the dissolution due to the decrease in the local pH by the iron ion hydrolysis, according to previous studies performed for iron in chloride-containing acid solution (Ref 46,47,48,49). Summarizing, in chloride medium the following equations can be considered (Ref 47):
For [Cl−] < 1.9 mol L−1, Chin and Nobe (Ref 48) have proposed the formation of (FeClOH)− adsorbed species that can be oxidized and dissolved to form Fe2+ and Cl− ions, as recently described (Ref 24). The (FeCl)ad intermediate was proposed by Kuo and Nobe (Ref 49) for [Cl−] > 1.9 mol L−1. However, the effect of chloride ions is a function of the current density at certain pH, and of [Cl−], and can diminish the iron dissolution to a level at which this process becomes negligible.
In chloride-containing neutral aqueous solution and at low anodic potentials, Eq 2-5 are accepted to be involved in the iron dissolution (Ref 50, 51) and, depending on the chloride concentration (≤ 0.01 mol L−1), the repassivation of iron is possible. However, at high chloride concentrations, the effect of chloride on the dissolution of the passive film is dominant and accelerates the iron dissolution. Burstein and Davis (Ref 50) considered that (FeCl)ad species are formed parallel to Fe(OH−)ad and FeOHad species, and therefore, a mechanism like that proposed by Kuo and Nobe (Ref 49) can be considered.
In neutral and oxygenated NaCl solution, the ORR produces predominantly hydroxide groups on both iron and copper (Ref 52):
This ORR reaction on copper in air dissolved in neutral NaCl aqueous solution was demonstrated to occur on a modified surface where both CuClad and CuOHad compete for the surface (Ref 42, 43). These species may form a Cu2O (very low corrosion product) layer both through hydrolysis of CuCl2- (CuCl + Cl- → CuCl2-; CuCl2- + H2O → Cu2O + 2HCl) and 2CuOHad → Cu2O + H2O. Deslouis et al. observed that the diffusion-limited current (iL) of ORR is the same in the absence or presence of Cu2O; the reduction of this oxide originates a current peak at around − 0.9 V/SCE, located at the potential region of ORR (Ref 45). It is possible that the ORR occurs on a copper-modified surface (CuClad, CuOHad or Cu2O sub-monolayer). At less negative potentials, the cathodic Tafel slope could be assumed to be a purely kinetic-controlled reaction (Ref 49, 50). However, EIS results suggested that the effect of a CuCl layer should also be considered to explain the behavior of ORR at less negative potentials than those where the current plateau for ORR is observed (Ref 53).
In our work, the anodic Tafel slope of copper dissolution was around 60 mV/dec, like those described in the literature (Ref 42, 54, 55). The anodic process of copper in NaCl solution is much more elaborated. Initially, chloride ion is adsorbed on copper according to the reaction (Ref 56):
This adsorbed species has low solubility and may accumulate to form CuClfilm (Ref 57,58,59) and/or react with chloride ions to form a soluble species (Ref 60, 61)
The CuClfilm at the film/solution interface is partially dissolved to form CuCl2-(Ref 62, 63), and at [Cl−] > 1 mol L−1 NaCl CuCl32-, CuCl43- complexes, depending on the chloride ion concentration (Ref 64). For instance, the following reaction could occur:
On the other hand, in the presence of O2 and excess of CuCl2- the following reaction may be occurring at the electrode surface (Ref 65):
which is responsible for the powdery patina Cu2Cl(OH)3 on archeological bronzes. The sum of reactions 7 and 8 may also consider giving the reaction 11 (Ref 61):
and in an excess of CuCl2- at the electrode surface and in the absence of O2, the following reaction has been suggested (Ref 65):
Therefore, it is possible that in the presence of O2, chloride-containing species can be found at the electrode surface, while in the holes or sublayers of copper, where oxygen is consumed fast, it is more probable that an equilibrium between a cuprous chloride porous film and soluble copper-chloride complexes species (CuClfilm/CuClx-(x−1)) will be stablished.
Electrochemical Experiments for Long Immersion Times
To collect more information about the coating behavior, EOCP measurements (Fig. 8) were performed for relatively long immersion times and the EOCP of the substrate was also measured for comparison: Cu-Safina (~1100 h), Cu-FST (~600 h), Cu-treat (~384 h), Cu-treat1 (~360 h), Cu-treat2 (~330 h), and carbon steel substrate (160 h). The testing for different times was because the EOCP response indicated deterioration of some coatings earlier than others, and there was no sense in continuing the measurement after receiving a clear indication that the electrolyte had reached the coating/substrate interface.

EOCP vs. time measured in aerated and unstirred 3.5 wt.% NaCl solution for the different coated samples for relatively long immersion times
For all coatings, at the initial immersion times, the EOCP was around − 0.20 (V/Ag|AgCl|KCl3 mol L−1), near the values expected for massive copper in chloride solution (Ref 36). For Cu-treat, Cu-treat1, and Cu-treat2 coatings after ~ 100 h the EOCP decreased from − 0.20 (V/Ag|AgCl|KCl3 mol L−1) to − 0.58 (V/Ag|AgCl|KCl3 mol L−1) (Fig. 8), which is very close to that obtained for the substrate, suggesting that the electrolyte may have reached the coating/substrate interface (Ref 21). During the immersion tests of these coatings, the dissolution of the oxides around the copper particles and formation of pores may occur. Therefore, the remaining oxides between the particles allow the formation of interconnected porosity, defects, and paths which facilitate the transport of the electrolyte throughout the coating up to the coating/substrate interface. Cross-sectional SEM images for these coatings show that the top and some inner parts of some coatings are severely damaged (Fig. 9 and S14, SD). The coatings were damaged to a great extension, with pathways, which allow the electrolyte to reach the substrate at some specific points, but not all over the coating and coating/substrate interface. EDS analysis (Fig. S15, SD) of the cross section show the presence of copper, oxygen, and iron for Cu-treat, Cu-treat1, and Cu-treat2. The amount of oxygen is greater at the point near the surface, iron was found close to the top of the coating and in a great extension of the cross section. This indicates that the electrolyte has reached the coating/substrate interface.

SEM images of the (a) Cu-Safina/steel, (b) Cu-FST/steel, (c) Cu-treat1/steel, (d) Cu-treat/steel, and (e) Cu-treat2/steel coatings after immersion in chloride solution for relatively long immersion times/h: (a) 1000 h; (b) 600 h; (c) 384 h; (d) 360 h, and (e) 330 h
For the Cu-Safina and Cu-FST coatings, EOCP values slightly decreased in the first hours of immersion (Fig. 8), probably due to the dissolution of oxides on the surface. After 48 h the potential was around − 0.20 (V/Ag|AgCl|KCl3 mol L−1) and remained constant until the last hour of immersion (Fig. 8). This suggests that the electrolyte does not reach the coating/substrate interface after relatively long immersion times: Cu-Safina (~1100 h) and Cu-FST (~600 h). This hypothesis is supported by the observation of the cross-sectional SEM images (Fig. 9 and S14, SD) and EDS analyses (Fig. S15, SD) performed after long immersion times. Corrosion of the substrate is not observed since these coatings are dense and compact. Only oxides formation can be seen at the region close to the top surface. EDS analysis at the cross section revealed only copper and oxygen as the main constituents, indicating that the electrolyte did not reach the substrate. The detection of copper and oxygen in the EDS spectra suggests that Eq 6, 7, 8, and 12 could be involved in the copper dissolution to form cuprous oxide and soluble copper species. Cracks observed on the cross-sectional images of Cu-treat, Cu-treat1, and Cu-treat2 samples were not seen at the cross section analysis before immersion, meaning that the formation of these defects was at least facilitated by the immersion in sodium chloride solution for longer times.
Electrochemical Impedance Spectroscopy (EIS)
Electrochemical impedance spectroscopy measurements were performed for all samples every 24 h for relatively long immersion times: steel substrate (~120 h), Cu-Safina (~1100 h), Cu-FST (~600 h), Cu-treat (~384 h), Cu-treat1 (~360 h), and Cu-treat2 (~330 h).
Figure 10 shows EIS diagrams that illustrate the impedance behavior of the substrate in aerated and neutral sodium chloride solution with the increase in immersion time. The symbols correspond to experimental data and solid lines correspond to the fitting of the impedance data.

(a) Complex plane and (b) Bode phase plots obtained for the carbon steel during different immersion times in 3.5 wt.% NaCl solution at 25 °C
EIS measurements performed for the steel substrate show one semicircle in the complex plane plot (Fig. 10a) at 24 h of immersion and the Bode phase angle diagram (Fig. 10b) depicts one asymmetric time constant at the medium frequency range (MF). The shape of this plot suggests the presence of more than one time constant, which was verified when treating the experimental data using equivalent electrical circuits (EEC). The semicircle of the complex plane increases until 96 h and then the real impedance decreases at low frequencies (LF) until 120 h of immersion, suggesting ion desorption and/or dissolution of a non-protective film. Accordingly, the impedance modulus values also decrease at LF range, which is assigned to the iron oxidation and iron oxide porous film dissolution. The cathodic reaction is mainly represented by Eq 6 as previously described for iron in the same electrolyte and under similar conditions (Ref 47).
EIS diagrams were also obtained for all studied coatings during the time course of immersion at intervals of 24 h, and some of these were chosen as representative of the impedance response evolution with time (Fig. 11, 12, 13, 14 and 15, and Fig. S16, SD). At 1 h of immersion, the Nyquist plot (Fig. S14, SD) demonstrates one asymmetric semicircle and a second time constant seems to be appearing at low frequency. At this immersion time (24 h), the amplitude of the semicircles followed the order: Cu-Safina > Cu-FST > Cu-treat1 > Cu-treat > Cu-treat2. Accordingly, the Cu-Safina coating showed the highest modulus of impedance (Fig. S16b, SD), suggesting that powders prepared under controlled-atmosphere produce coatings with fewer defects and porosity, and with higher corrosion resistance at short immersion times.

(a) Complex plane and (b) Bode phase plots obtained for the Cu-Safina coating during different immersion times in 3.5 wt.% NaCl solution at 25 °C

(a) Complex plane and (b) Bode phase plots obtained for the Cu-FST coating during different immersion times in 3.5 wt.% NaCl solution at 25 °C

(a) Complex plane and (b) Bode phase plots obtained for the Cu-treat1 coating during different immersion times in 3.5 wt.% NaCl solution at 25 °C

(a) Complex plane and (b) Bode phase plots obtained for the Cu-treat coating during different immersion times in 3.5 wt.% NaCl solution at 25 °C

(a) Complex plane and (b) Bode phase plots obtained for the Cu-treat2 coating during different immersion times in 3.5 wt.% NaCl solution at 25 °C
For Cu-Safina and Cu-FST coatings at 24 h, the complex plane diagrams (Fig. 11a and 12a) demonstrate one asymmetric and incomplete semicircle, but a second time constant seems to be appearing at low frequency. The Bode phase angle diagrams (Fig. 11b and 12b) show one asymmetric time constant with a phase angle of − 60° (~200 Hz). The shape of this plot suggests the presence of more than one time constant, which was verified when treating the experimental data using EECs. So, for all samples the asymmetric Bode diagrams suggest the presence of at least two time constants. Figure 11, 12, 13, 14 and 15 illustrates the EIS diagrams for some immersion times, and the changes evolution are mainly observed in the Bode phase angle diagrams indicated with the letter b and in the fitting of experimental data using equivalent electric circuits (EECs). Depending on the sample, the shape of the Bode diagrams becomes more complex, which requires a third time constant for fitting the experimental data. Thus, two time constants are enough to adjust the impedance diagrams for Cu-Safina for immersion times equal or higher than 432 h; for Cu-FST this time decreases to 360 h and for Cu-treat to 24 h. The third time constant is required to adjust data for t ≥ 520 h (Cu-Safina), t ≥ 408 h (Cu-FST) and t ≥ 96 h for Cu-treat, while for Cu-treat1 and Cu-treat2 two time constants were enough for adjusting the EIS diagrams for all the studied immersion time (360 h for Cu-treat1 and 330 h for Cu-treat2).
The impedance diagrams for the coatings prepared with oxidized powders change continuously with immersion time. For the Cu-treat1 at 24 h, Zreal is 15 kΩ cm2 (5 mHz) and an abrupt decrease occurs after 120 h, when the Zreal is 2 kΩ cm2 (Fig. 13a). The EIS results for the Cu-treat coating demonstrate a gradual decrease in Zreal values with increasing immersion time (Fig. 14a). At 24 h, Zreal = 5 kΩ cm2 (5 mHz), and then decreases to ≈1 kΩ cm2 at ~ 170 h. An asymmetric time constant in the phase diagrams (Fig. 14b) is observed, suggesting the presence of two partially overlapped time constants in the medium/high frequency (MF-HF) range. With the increase in immersion times, the time constant shifts to low frequencies and the phase angle decreases from − 55° at 24 h to − 44° after ~ 170 h. This can be attributed to the decrease in the capacitive behavior and corrosion resistance and also to some contribution of the substrate response. Asymmetric Bode phase angle diagrams are also observed for sample Cu-treat2 (Fig. 15b) and after 48 h the phase angle decreases and the time constant shifts toward low frequency, and the impedance response is dominated by the response of the substrate due to the many pores and defects present in the coating.
The modulus of impedance at low frequency (Fig. 11b, 12, 13, 14 and 15b and Fig. 15S, SD) shows different behavior for all coatings. For Cu-Safina and Cu-FST coatings, no changes are observed in the impedance module throughout the immersion time and these coatings have a higher impedance module than the others. For Cu-treat the impedance response is stable up to 24 h, then slightly decreases and at ~ 170 h abruptly decreases and remains constant until the end of the test, with a value of |Z| = 2.9 kΩ m2 when compared to the initial one |Z| = 3.7 kΩ cm2). For Cu-treat1 the impedance response is stable up to 48 h, then abruptly decreases at 120 h and remains constant until the end of the experiment. For Cu-treat2, the impedance value decreases abruptly at 24 h and remains at the same value during the time course of the experiment. In summary, the performance of the copper coatings at relatively long immersion times has the following order: Cu-Safina ≈ Cu-FST > Cu-treat1 > Cu-treat > Cu-treat2.

Electrical equivalent circuits used to fit the EIS data: (a) steel for t ≤ 24 h of immersion, (b) steel for t ≥ 96 h of immersion
The EIS results for the substrate were also quantitatively analyzed using EECs (Fig. 16) like the previously suggested EEC (Ref 17), which were chosen considering the structure of the samples, the best fitting of experimental data, and the lower residual error of each parameter of the equivalent circuit. The values of the elements of the EECs are shown in Table S4 (SD). For the substrate, the EEC of Fig. 16(a) fits the EIS diagrams for the first 24 h of immersion and the EEC of Fig. 16(b) fits the EIS diagrams obtained for 96 and 120 h. The first time constant CPEdl//Rct at medium frequency (MF) is attributed to iron oxidation and oxygen reduction, CPEdl being the admittance related to the capacitance of the electrical double layer, ndl is the exponent, and Rct the charge transfer resistance. At low frequency (LF), the CPEfilm//Rfilm subcircuit is attributed to iron ions desorption and non-protective film formation/dissolution, where CPEfilm is composed by the CPE-Tfilm, the admittance, which is related to the capacitance of the film (adsorbed species and iron oxides-hydroxides) and the CPE-Pfilm(nfilm) the exponent, and Rfilm is the resistance of the film and solution inside the pores and defects of the film (Ref 17, 29).
Figure 17 presents the scheme used to represent the coatings and the corresponding EECs used to fit the experimental impedance data obtained for different immersion times. To facilitate the discussion, the studied coatings are divided into two groups: the first formed by the coatings obtained with non-treated copper powders (Cu-Safina and Cu-FST) and the second in which the coatings were prepared using treated copper powders (Cu-treat, Cu-treat1, and Cu-treat2). Firstly, the scheme shows the as-prepared coating (Fig 17a). The coating is formed by a top layer with some pores and small defects and a compact layer at the bottom as shown in Fig. 17(a) and SEM images (Fig. 3). For the Cu-Safina and Cu-FST coatings, the copper oxidation occurs at short immersion times in chloride solution and the first time constant CPEdl//Rct at HF-MF frequencies is attributed to this oxidation reaction (Fig. 17b). The localized dissolution of copper at some areas of the coating allows the formation of pores which explain the second time constant (CPEpo//Rpo) that can be assigned to the pore’s response (Fig. 17b), CPE-Tpo being the admittance of the pores, which is related to the capacitance, npo the exponent, and Rpo the resistance of the solution inside the pores (Ref 17, 41, 66,67,68,69,70). The pores are formed due to the dissolution of copper at specific sites (Ref 17, 34, 71,72,73,74,75,76,77,78,79), and at some coatings defects(Fig. 3 and S8, SD). As previously described, the EOCP remained constant ≈-0.20 (V/Ag|AgCl|KCl3 mol L−1) for the Cu-Safina and Cu-FST until the last hour of immersion (Fig. 6). Cross-sectional SEM images (Fig. 9, S14a and b, SD) and EDS analyses (Fig. S15, SD), performed after long immersion times, show no corrosion at the coating/substrate interface. These results indicate that the electrolyte does not reach the coating/substrate interface.

Scheme representing the coatings before and after different times of immersion and electrical equivalent circuits used to fit the EIS data: (a) Scheme of coatings before immersion in 3.5 wt.% NaCl solution at 25 °C; (b) EECs for Cu-Safina (t ≤ 432 h), Cu-FST (t ≤ 360 h) and Cu-treat (t ≤ 24 h); (c) EECs for Cu-Safina (s ≥ 520 h), Cu-FST (t ≥ 408 h); (d) EECs for Cu-treat (t ≤ 24 h) (e) EECs for Cu-treat (t ≥ 96 h), Cu-treat1, and Cu-treat2
However, after 520 h (Cu-Safina) and 408 h (Cu-FST) of immersion, the pores become deeper, but not sufficiently deep to reach the coating/substrate interface. At this immersion time, a third time constant (CPEbott//Rbott) is required to fit the EIS diagrams (Fig. 17c) and was assigned to the capacitance and charge transference resistance due to the copper oxidation reaction at the bottom of pores, where some corrosion products may accumulate.
For the coatings prepared using treated copper powders (Cu-treat, Cu-treat1 and Cu-treat2), the scheme representing the coatings before and after different immersion times and the EECs used to fit the EIS data are show in Fig. 17. In this case, Cu-treat presents a more complex behavior than Cu-treat1 and Cu-treat2 samples, since two time constants were required to adjust the experimental data of Cu-treat up to 24 h (Fig. 17d) and three time constants were required to adjust the EEC for times ≥ 96 h (Fig. 17e). The first time constant, CPEdl//Rct (Fig. 17d and e), at HF-MF frequency for the coatings of treated powders has the same attribution given for Cu-Safina and Cu-FST. The Rct values (Table S4, SD) are slightly different depending on the nature and amount of copper oxide around the particles, remembering that this amount is always very small. The remaining oxides between the particles allow the formation of interconnected porosity, defects, and paths. The second time constant, represented by the CPEpo//Rpo (Fig. 17e), subcircuit for Cu-treat, was attributed to the response of the pores and defects developed in the coating. It seems that all the coating is wetted by the electrolyte. This was suggested considering the difficulty in maintaining a vacuum during the SEM analysis, which needed drying the sample for at least 1 day to remove the electrolyte contained in pores and defects of the coating. This effect seems less pronounced, mainly for Cu-treat2 samples, for which the electrolyte would have reached the substrate almost immediately after immersion (see Fig. 9). The time constant represented by the CPE//R (Fig. 17d and e) subcircuit is related to the response of the substrate, i.e., iron oxidation. For the Cu-treat1 sample, however, the response corresponding to the second time constant is dominated by the substrate oxidation only after 48 h of immersion and for shorter times there are contributions of pores and defects of the coating without reaching the substrate. For longer times, the response is dominated by the corrosion of the substrate. Cross-sectional SEM images for these coatings show that the coating/substrate interface is severely damaged after the EIS studies (Fig. 9 and Fig. S14, SD).
Table S4 and Fig. 18 show the evolution of Rct and Rpo values with immersion time. For Cu-Safina and Cu-FST, the Rct values are almost constant during all immersion times and relatively high for the Cu-treat coating for times up to 100 h and then similar to the other coatings prepared with oxidized powders (Fig. 18). Thus, comparing the Rct values along the immersion time, the following order is obtained: Cu-treat > Cu-treat2 > Cu-treat1 > Cu-Safina ≈ Cu-FST.

(a) Rct and (b) Rpo values vs. time for copper coatings in 3.5 wt.% NaCl solution at 25 °C
Figure 18b shows the evolution of Rpo values: For Cu-Safina, Rpo values increase from 11 kΩ cm2 at initial immersion times to 17 kΩ cm2 at 96 h, slightly decrease to 14 kΩ cm2, and then increase to 17 kΩ cm2 at 432 h; for the Cu-FST, Rpo increases from 10.8 to 14.8 kΩ cm2 in the first 96 h, remains around this value until 240 h, and then slightly decreases to 11.3 kΩ cm2 at 360 h. The variation in Rct values can be associated with the number and size of pore depths until achieving certain values. The npo values during this time interval are around 0.5 which can be assigned to the diffusion of oxygen, chloride ions, or products of copper corrosion through the pores. After 520 h (Cu-Safina) and 408 h (Cu-FST), when the third time constant is necessary to fit the experimental data, Rbott was around 10 kΩ cm2 for Cu-Safina and increased to 13 kΩ cm2 at 1100 h and for Cu-FST was 10.7 kΩ cm2 at 408 h and increased to 15.3 kΩ cm2 at 600 h. The increase in the Rbott value can be assigned to the deposition of the corrosion products at the bottom of the pores.
For coatings prepared with treated copper powders, the R values (Table S4, SD) were related to the iron dissolution at the coating/substrate interface. These values are relatively low due to the hydrolysis of iron ions which diminishes the local pH and allows the iron dissolution to continue. For these coatings, mainly for Cu-treat and Cu-treat2, the electrolyte has attained the coating/substrate interface at a significant area of the interface, as can be seen from the SEM images (Fig. 9, Fig. S14, SD).
In summary, EIS results for long immersion times show that Cu-Safina and Cu-FST coatings protect the substrate due to their dense and thick characteristics, allowing them to act as a barrier that impedes the electrolyte to reach the substrate. On the other hand, the coatings prepared from the oxidized powders presented higher porosity and defects, which agrees with (Ref 15). Pores and defects between the particles allow electrolyte diffusion through the coating, causing its deterioration and the corrosion of the substrate, implying in significant changes in the impedance response (Ref 80).
The characteristics of Cu-Safina and Cu-FST coatings led these coatings to be also evaluated by the salt fog spray test (5 wt.% NaCl) at 35 °C for 2000 h. Figure 19 shows the samples before and during different times of the test. After 500 h for the Cu-Safina and 600 h for the Cu-FST coatings, copper dissolution can be observed and some areas are covered by corrosion products (oxides/hydroxides), but even after 2000 h there are no signs of substrate corrosion. To confirm this hypothesis, EDS surface analyses (Fig. S17 and S18, SD) were performed at different points on the surface of the coatings after 2000 h of testing. Only oxygen, sodium, chloride, and copper, but no iron were detected for the coatings, confirming that these two coatings act as a barrier and protect the substrate even after 2000 h of exposure in 5 wt.% NaCl.

Surface images of the copper coatings after salt spray tests (5 wt.% NaCl) at different immersion times: (a) Cu-Safina coating, (b) Cu-FST coating
Kindly note that the citation of Figure 20 present in the text but the appropriate Figure is not provided, so we changed Figure 20 as Figure 19, amend if necessary. Kindly check and confirm.Thank you, now it is ok.
Conclusions
The study shows that the characteristics of the feedstock material have significant influence on the coating deposition and properties. Coatings prepared from the Cu-treat and Cu-treat2 powders present low deposition efficiencies and lower thicknesses; on the other hand, Cu-Safina and Cu-FST coatings prepared from the gas-atomized feedstock powder showed higher thickness values up to ~ 500 µm. The mechanical evaluation performed with Cu-Safina and Cu-FST coatings demonstrated that these coatings are dense and compact and have good mechanical resistance to sliding and to abrasion compared to the coatings prepared from oxidized powders. Furthermore, the electrochemical results and SEM images of Cu-Safina and Cu-FST coatings showed no corrosion at the coating/substrate interface even after ~ 1000 and ~ 600 h of immersion in 3.5 wt.% NaCl solution, respectively, due to the dense coating microstructure obtained by CGS. This dense microstructure also explains the very high performance of Cu-Safina and Cu-FST coatings against corrosion in the salt fog tests (~2000 h).Reference [35] was provided in the reference list; however, this was not mentioned or cited in the manuscript. As a rule, if a citation is present in the text, then it should be present in the list. Please provide the location of where to insert the reference citation in the main body text. Kindly ensure that all references are cited in ascending numerical order.Thank you for the corrections. The reference 35 was added in the Sliding wear resistance section.
Data Availability
The raw/processed data required to reproduce these findings cannot be shared at this time as the data also form part of ongoing study research and submitted at any time.
References
R.N. Raoelison, Y. Xie, T. Sapanathan, M.P. Planche, R. Kromer, S. Costil, and C. Langlade, Cold Gas Dynamic Spray Technology: A Comprehensive Review of Processing Conditions for Various Technological Developments till to Date, Addit. Manuf., 2018, 19, p 134-159.
S. Yin, P. Cavaliere, B. Aldwell, R. Jenkins, H. Liao, W. Li, and R. Lupoi, Cold Spray Additive Manufacturing and Repair: Fundamentals and Applications, Addit. Manuf., 2018, 21, p 628-650.
R.N. Raoelison, Ch. Verdy, and H. Liao, Cold Gas Dynamic Spray Additive Manufacturing Today: Deposit Possibilities Technological Solutions and Viable Applications, Mater. Des., 2017, 133, p 266-287.
Q.J. Zhu, K. Wang, X.H. Wang, and B.R. Hou, Electrochemical Impedance Spectroscopy Analysis of Cold Sprayed and Arc Sprayed Aluminium Coatings Serviced in Marine Environment, Surf. Eng., 2012, 28, p 300-305.
A. Bruera, P. Puddu, S. Theimer, M. Villa-Vidaller, A. List, G. Bolelli, F. Gärtner, T. Klassen, and L. Lusvarghi, Adhesion of Cold Sprayed Soft Coatings: Effect of Substrate Roughness and Hardness, Surf. Coat. Technol., 2023, 466, p 129651.
V.K. Champagne, The Cold Spray Materials Deposition Process Fundamentals and Applications, Woodhead Publishing, 2007.
R. Drehmann, T. Grund, T. Lampke, B. Wielage, K. Manygoats, T. Schucknecht, and D. Rafaja, Splat Formation and Adhesion Mechanisms of Cold Gas-Sprayed Al Coatings on Al2O3 Substrates, J. Therm. Spray Technol., 2014, 23, p 68-75.
H. Singh, T.S. Sidhu, S.B.S. Kalsi, and J. Karthikeyan, Development of Cold Spray from Innovation to Emerging Future Coating Technology, J. Braz. Soc. Mech. Sci. Eng., 2013, 35, p 231-245.
M. Yu, W. Li, X. Guo, and H. Liao, Impacting Behavior of Large Oxidized Copper Particles in Cold Spraying, J. Therm. Spray Technol., 2013, 22, p 433-440.
S. Yin, X. Wang, W. Li, H. Liao, and H. Jie, Deformation Behavior of the Oxide Film on the Surface of Cold Sprayed Powder Particle, Appl. Surf. Sci., 2012, 259, p 294-300.
W.Y. Li, C.J. Li, and H. Liao, Significant Influence of Particle Surface Oxidation on Deposition Efficiency, Interface Microstructure and Adhesive Strength of Cold-Sprayed Copper Coatings, Appl. Surf. Sci., 2010, 256, p 4953-4958.
Y. Ichikawa and K. Ogawa, Effect of Substrate Surface Oxide Film Thickness on Deposition Behavior and Deposition Efficiency in the Cold Spray Process, J. Therm. Spray Technol., 2015, 24, p 1269-1276.
C.-J. Li, W.-Y. Li, and H. Liao, Examination of the Critical Velocity for Deposition of Particles in Cold Spraying, J. Therm. Spray Technol., 2006, 15, p 212-222.
W.Y. Li, H. Liao, C.J. Li, H.S. Bang, and C. Coddet, Numerical Simulation of Deformation Behavior of Al Particles Impacting on Al Substrate and Effect of Surface Oxide Films on Interfacial Bonding in Cold Spraying, Appl. Surf. Sci., 2007, 253, p 5084-5091.
A.A. Hemeda, C. Zhang, X.Y. Hu, D. Fukuda, D. Cote, I.M. Nault, A. Nardi, V.K. Champagne, Y. Ma, and J.W. Palko, Particle-Based Simulation of Cold Spray: Influence of Oxide Layer on Impact Process, Addit. Manuf., 2021, 37, p 101517.
F.-J. Wei, B.-Y. Chou, K.-Z. Fung, S.-Y. Tsai, and C.-W. Yang, Influence of Powder Plasticity on Bonding Strength of Cold-Sprayed Copper Coating, Coatings, 2022, 12, p 1197.
F.S. da Silva, N. Cinca, S. Dosta, I.G. Cano, J.M. Guilemany, C.S.A. Caires, A.R. Lima, C.M. Silva, S.L. Oliveira, A.R.L. Caires, and A.V. Benedetti, Corrosion Resistance and Antibacterial Properties of Copper Coating Deposited by Cold Gas Spray, Surf. Coat. Technol., 2019, 361, p 292-301.
R. Partovi-Nia, S. Ramamurthy, D. Zagidulin, J. Chen, R. Jacklin, P. Keech, and D.W. Shoesmith, Corrosion of Cold Spray Deposited Copper Coating on Steel Substrates, Corrosion, 2015, 71, p 1237-1247.
D. Rui, L. Xiangbo, W. Jia, and X. Likun, Electrochemical Corrosion and Mathematical Model of Cold Spray Cu-Cu2O Coating in NaCl Solution-Part I: Tafel Polarization Region Model, Int. J. Electrochem. Sci., 2013, 8(4), p 5902-5924.
M. Winnicki, A. Małachowska, A. Baszczuk, M. Rutkowska-Gorczyca, D. Kukla, M. Lachowicz, and A. Ambroziak, Corrosion Protection and Electrical Conductivity of Copper Coatings Deposited by Low-Pressure Cold Spraying, Surf. Coat. Technol., 2017, 318, p 90-98.
H. Koivuluoto, J. Lagerbom, M. Kylmälahti, and P. Vuoristo, Microstructure and Mechanical Properties of Low-Pressure Cold-Sprayed (LPCS) Coatings, J. Therm. Spray Technol., 2008, 17, p 721-727.
W.S. Tait, An Introduction to Electrochemical Corrosion Testing for Practicing Engineers and Scientists, Wisconsin Pair Odocs, 1994.
S. Lagutkin, L. Achelis, S. Sheikhaliev, V. Uhlenwinkel, and V. Srivastava, Atomization Process for Metal Powder, Mater. Sci. Eng. A, 2004, 383, p 1-6.
L. Pawlowski, The Science and Engineering of Thermal Spray Coatings, Wiley, 2008.
R.B. Heimann, J.I. Kleiman, S. Marx, R. Ng, S. Petrov, M. Shagalov, R.N.S. Sodhi, and A. Tang, High-Pressure Cold Gas Dynamic (CGD) -Sprayed Alumina-Reinforced Aluminum Coatings for Potential Application as Space Construction Material, Surf. Coat. Technol., 2014, 252, p 113-119. https://doi.org/10.1016/j.surfcoat.2014.04.053
I. Platzman, R. Brener, H. Haick, and R. Tannenbaum, Oxidation of Polycrystalline Copper Thin Films at Ambient Conditions, J. Phys. Chem. C, 2008, 112, p 1101-1108.
H. Wieder and A.W. Czanderna, The Oxidation of Copper Films To CuO.067, J. Phys. Chem., 1962, 66(5), p 816-821.
J. Aromaa, M. Kekkonen, M. Mousapour, A. Jokilaakso, and M. Lundström, The Oxidation of Copper in Air at Temperatures up to 100 °C, Corros. Mater. Degrad., 2021, 2, p 625-640.
S.-K. Leea, H.-C. Hsua, and W.-H. Tuan, Oxidation Behavior of Copper at a Temperature below 300 °C and the Methodology for Passivation, Mater. Res., 2016, 19(1), p 51-56.
V.-H. Castrejón-Sánchez, A.C. Solís, R. López, C. Encarnación-Gomez, F.M. Morales, O.S. Vargas, J. Mastache-Mastache, and G.V. Sánchez, Thermal Oxidation of Copper Over a Broad Temperature Range: Towards the Formation of Cupric Oxide (CuO), Mater. Res. Expr., 2019, 6, p 075909.
J.-P. Wang and W.D. Cho, Oxidation Behavior of Pure Copper in Oxygen and/or Water Vapor at Intermediate Temperature, ISIJ Int., 2009, 49(12), p 1926-1931. https://doi.org/10.2355/isijinternational.49.1926
R.M. Souto, M.M. Laz, and R.L. Reis, Degradation Characteristics of Hydroxyapatite Coatings on Orthopaedic TiAlV in Simulated Physiological Media Investigated by Electrochemical Impedance Spectroscopy, Biomaterials, 2003, 24, p 4213-4221.
X. Zhou and P. Mohanty, Electrochemical Behavior of Cold Sprayed Hydroxyapatite/Titanium Composite in Hanks’ Solution, Electrochim. Acta, 2012, 65, p 134-140.
F.S. da Silva, J. Bedoya, S. Dosta, N. Cinca, I.G. Cano, J.M. Guilemany, and A.V. Benedetti, Corrosion Characteristics of Cold Gas Spray Coatings of Reinforced Aluminum Deposited onto Carbon Steel, Corros. Sci., 2017, 114, p 57-71.
S. Dosta, M. Couto, and J.M. Guilemany, Cold Spray Deposition of a WC-25Co Cermet Onto Al7075-T6 and Carbon Steel Substrates, Acta Mater., 2013, 61, p 643-652.
M. Couto, S. Dosta, M. Torrell, J. Fernández, and J.M. Guilemany, Cold Spray Deposition of WC-17 and 12Co Cermets onto Aluminum, Surf. Coat. Technol., 2013, 235, p 54-61.
K. Rahmouni, M. Keddam, A. Srhiri, and H. Takenouti, Corrosion of Copper in 3% NaCl Solution Polluted by Sulphide Ions, Corros. Sci., 2005, 47, p 3249-3266.
M. Stern and A.L. Geary, Electrochemical Polarization: I. A Theoretical Analysis of the Shape of Polarization Curves, J. Electrochem. Soc., 1957, 104(56), p 63.
E. McCafferty, Validation of Corrosion Rates Measured by the Tafel Extrapolation Method, Corros. Sci., 2005, 47, p 3202-3215.
W.J. Lorenz and F. Mansfeld, Determination of Corrosion Rates by Electrochemical DC and AC Methods, Corros. Sci., 1981, 21, p 647-672.
W.A. Badawy, K.M. Ismail, and A.M. Fathi, Effect of Ni Content on the Corrosion Behavior of Cu-Ni Alloys in Neutral Chloride Solutions, Electrochim. Acta, 2005, 50, p 3603-3608.
C. Deslouis, B. Tribollet, G. Mengoli, and M.M. Musiani, Electrochemical Behaviour of Copper in Neutral Aerated Chloride Solution. I. Steady-State Investigation, J. Appl. Electrochem., 1988, 18, p 374-383.
F. King, M. Quinn, and C. Litke, Oxygen Reduction on Copper in Neutral NaCl solution, J. Electroanal. Chem., 1995, 385, p 45-55. https://doi.org/10.1016/0022-0728(94)03705-8
J.O.’M. Bockris, D.R. Drazic, and A. Despic, The Electrode Kinetics of the Deposition and Dissolution of Iron Electrochim, Acta, 1961, 4(325), p 361. https://doi.org/10.1016/0013-4686(61)80026-1
E.J. Kelly, The active iron electrode I. Iron Dissolution and Hydrogen Evolution Reactions in Acidic Sulfate Solutions, J. Electrochem. Soc., 1965, 112(124), p 131. https://doi.org/10.1149/1.2423480
W.J. Lorenz, Der Einfluss Von Halogenidionen Auf Die Anodische Aufl Dsung Des Eisens, Corros. Sci., 1965, 5, p 121-131. https://doi.org/10.1016/S0010-938X(65)90478-6
E. Mccafferty and N. Hackerman, Concentrated Acidic Chloride Solutions, J. Electrochem. Soc., 1972, 119, p 999-1009. https://doi.org/10.1149/1.2404426
R.J. Chin and K. Nobe, Electrodissolution Kinetics of Iron in Chloride Solutions, J. Electrochem. Soc., 1972, 119, p 1457-1461. https://doi.org/10.1149/1.2404023
H.C. Kuo and K. Nobe, Electrodissolution Kinetics of Iron in Chloride Solutions, J. Electrochem. Soc., 1978, 125, p 853-860. https://doi.org/10.1149/1.2131567
G.T. Burstein and D.H. Davies, The Effects of Anions on the Behaviour Scratched Iron Electrodes in Aqueous Solutions, Corros. Sci., 1980, 20, p 1143-1155. https://doi.org/10.1016/0010-938X(80)90145-6
F. Hilbert, Y. Miyoshi, G. Eichkorn, and W.J. Lorenz, Correlations Between the Kinetics of Electrolytic Dissolution and Deposition of Iron: I. The Anodic Dissolution of Iron Correlations between the Kinetics of Electrolytic Dissolution and Deposition of Iron, J. Electrochem. Soc., 1971, 118, p 1919-1926. https://doi.org/10.1149/1.2407868
D. Wang, B. Xiang, Y. Liang, S. Song, and C. Liu, Corrosion control of copper in 3.s5 wt.% NaCl Solution by Domperidone : Experimental and Theoretical Study, Corros. Sci., 2014, 85, p 77-86. https://doi.org/10.1016/j.corsci.2014.04.002
C. Deslouis, B. Tribollet, G. Mengoli, and M.M. Musiani, Electrochemical Behaviour of Copper in Neutral Aerated Chloride Solution. II. Impedance Investigation, J. Appl. Electrochem., 1988, 18, p 384-393. https://doi.org/10.1007/BF01093752
C.H. Bonfiglio, H.C. Albaya, and O.A. Cobo, The Kinetics of the Anodic Dissolution of Copper in Acid Chloride Solutions, Corros. Sci., 1973, 13, p 717-724. https://doi.org/10.1016/S0010-938X(73)80010-1
M. Braun and K. Nobe, Electrodissolution Kinetics of Copper in Acidic Chloride Solutions, J. Electrochem. Soc., 1979, 126, p 1666-1671. https://doi.org/10.1149/1.2128773
J.-P. Diard, J.-M. Le Canut, B. Le Gorrec, and C. Montella, Copper Electrodissolution in 1M HCl at low Current Densities. I. General Steady-State Study, Electrochim. Acta, 1998, 43, p 2469-2483. https://doi.org/10.1016/S0013-4686(97)10155-4
H.P. Lee, K. Nobe, and A.J. Pearlstein, Film Formation and Current Oscillations in the Electrodissolution of Cu in Acidic Chloride Media I. Experimental Studies, J. Electrochem. Soc., 1985, 132(1031), p 1037. https://doi.org/10.1149/1.2114010
C.I. Elsner, R.C. Salvarezza, and A.J. Arvia, The Influence of Halide Ions at Submonolayer Levels on the Formation of Oxide Layer and Electrodissolution of Copper in Neutral Solutions, Electrochim. Acta, 1988, 33, p 1735-1741. https://doi.org/10.1016/0013-4686(88)85008-4
F.K. Crundwell, The Anodic Dissolution of 90% Copper-10% Nickel Alloy in Hydrochloric Acid Solutions, Electrochim. Acta, 1991, 36, p 2135-2141. https://doi.org/10.1016/0013-4686(91)85221-R
A.V. Benedetti, P.T.A. Sumodjo, K. Nobe, P.L. Cabot, and W.G. Proud, Electrochemical Studies of copper, Copper-Aluminium and Copper-Aluminium-Silver Alloys: Impedance Results in 0.5M NaCl, Electrochim. Acta, 1995, 40, p 2657-2668. https://doi.org/10.1016/0013-4686(95)00108-Q
G. Kear, B.D. Barker, K. Stokes, and F.C. Walsh, Electrochemical Corrosion Behaviour of 90–10 Cu–Ni Alloy in Chloride-Based Electrolytes, J. Appl. Electrochem., 2004, 34, p 659-669. https://doi.org/10.1023/B:JACH.0000031164.32520.58
A.K. Hauser and J. Newman, Singular Perturbation Analysis of the Faradaic Impedance of Copper Dissolution Accounting for the Effects of Finite Rates of a Homogeneous Reaction, J. Electrochem. Soc., 1989, 136, p 2820-2831. https://doi.org/10.1149/1.2096293
O.E. Barcia, O.R. Mattos, N. Pebere, and B. Tribollet, Mass-Transport Study for the Electrodissolution of Copper in 1M Hydrochloric Acid Solution by Impedance, J. Electrochem. Soc., 1993, 140, p 2825-2832. https://doi.org/10.1149/1.2220917
K. Nobe and G.L. Bauerle, Technical Note: Effects of Chloride Ions on the Anodic Dissolution of 90/10 and 70/30 Cu-Ni Alloys in H2SO4, Corrosion, 1981, 37, p 426-427. https://doi.org/10.5006/1.3578205
J. Wang, C. Xu, and G. Lv, Formation Processes of CuCl and Regenerated Cu Crystals on Bronze Surfaces in Neutral and Acidic Media, Appl. Surf. Sci., 2006, 252, p 6294-6303. https://doi.org/10.1016/j.apsusc.2005.08.041
A.M. Fenelon and C.B. Breslin, The Electrochemical Synthesis of Polypyrrole at a Copper Electrode: Corrosion Protection Properties, Electrochim. Acta, 2002, 247, p 4467-4476. https://doi.org/10.1016/S0013-4686(02)00518-2
T. Tüken, B. Yazici, and M. Erbil, The Use of Polyindole for Prevention of Copper Corrosion, Surf. Coatings Technol., 2006, 200, p 4802-4809. https://doi.org/10.1016/j.surfcoat.2005.04.023
E.M. Sherif and S.-M. Park, Inhibition of Copper Corrosion in 3.0% NaCl Solution by N-Phenyl-1,4-phenylenediamine, J. Electrochem. Soc., 2005, 152, p B428-B433. https://doi.org/10.1149/1.2018254
J.-P. Diard, J.-M. Le Canut, B. Le Gorrec, and C. Montella, Copper Electrodissolution in 1M HCl at Low Current Densities. II. Electrochemical Impedance Spectroscopy Study, Electrochim. Acta, 1998, 43, p 2485-2501. https://doi.org/10.1016/S0013-4686(97)10156-6
K. Rahmouni, M. Keddam, A. Srhiri, and H. Takenouti, Corrosion of Copper in 3% NaCl Solution Polluted by Sulphide Ions, Corros. Sci., 2005, 47, p 3249-3266. https://doi.org/10.1016/j.corsci.2005.06.017
Y. Bai, Z.H. Wang, X.B. Li, G.S. Huang, C.X. Li, and Y. Li, Corrosion Behavior of Low Pressure Cold Sprayed Zn-Ni Composite Coatings, J. Alloys Compd., 2017, 719, p 194-202. https://doi.org/10.1016/j.jallcom.2017.05.134
Y. Van Ingelgem, A. Hubin, and J. Vereecken, Investigation of the First Stages of the Localized Corrosion of Pure Copper Combining EIS, FE-SEM and FE-AES, Electrochim. Acta, 2007, 52, p 7642-7650. https://doi.org/10.1016/j.electacta.2006.12.039
R. Babić, M. Metikoš-Hukovi, and A. Jukić, A Study of Copper Passivity by Electrochemical Impedance Spectroscopy, J. Electrochem. Soc., 2001, 148, p B146-B151. https://doi.org/10.1149/1.1354608
B. Rosborg and J. Pan, An Electrochemical Impedance Spectroscopy Study of Copper in a Bentonite/Saline Groundwater Environment, Electrochim. Acta, 2008, 53, p 7556-7564. https://doi.org/10.1016/j.electacta.2008.04.021
Y. Feng, K.S. Siow, W.K. Teo, K.L. Tan, and A.K. Hsieh, Corrosion Mechanisms and Products of Copper in Aqueous Solutions at Various pH Values, Corrosion, 1997, 53, p 389-398. https://doi.org/10.5006/1.3280482
I. Betova, B. Beverskog, M. Bojinov, P. Kinnunen, K. Mäkelä, S.-O. Pettersson, and T. Saario, Corrosion of Copper in Simulated Nuclear Waste Repository Conditions, Electrochem. Solid-State Lett., 2003, 6, p B19-B22. https://doi.org/10.1149/1.1557033
X. Zhang, W. He, I. Odnevall Wallinder, J. Pan, and C. Leygraf, Determination of Instantaneous Corrosion Rates and Runoff Rates of copper From Naturally Patinated Copper During Continuous Rain Events, Corros. Sci., 2002, 44, p 2131-2151. https://doi.org/10.1016/S0010-938X(02)00015-X
R.D.P.B. Hernández, I.V. Aoki, B. Tribollet, and H.G. De Melo, Electrochemical Impedance Spectroscopy Investigation of the Electrochemical Behaviour of Copper Coated with Artificial Patina Layers and Submitted to Wet and Dry Cycles, Electrochim. Acta, 2011, 56, p 2801-2814. https://doi.org/10.1016/j.electacta.2010.12.059
A. Srivastava and R. Balasubramaniam, Electrochemical Impedance Spectroscopy Study of Surface Films Formed on Copper in Aqueous Environments, Mater. Corros., 2005, 56, p 611-618. https://doi.org/10.1002/maco.200503866
J.M. Guilemany, N. Espallargas, P.H. Suegama, and A.V. Benedetti, Comparative Study of Cr3C2–NiCr Coatings Obtained by HVOF and Hard Chromium Coatings, Corros. Sci., 2006, 48, p 2998-3013. https://doi.org/10.1016/j.corsci.2005.10.016
Acknowledgments
The authors would like to thank the Prof. Irene García Cano and Prof. Dr. Josep Maria Guilemany from Thermal Spray Center–Barcelona University Spain. Ministerio de Ciencia y Tecnología (Proyecto MAT2009-10827), the Generalitat de Catalunya (Proyecto 2009SGR390) for the financial support, CNPq–Conselho Nacional de Pesquisa (Procs. 153177/2014-4 and 201325/2014-4), CAPES PNPD (Coordenação de Aperfeiçoamento do Pessoal de Ensino Superior) for the financial support and scholarships, and CEMPEQC (Centro de Monitoramento e Pesquisa da Qualidade de Combustíveis, Biocombustíveis, Petróleo e Derivados) for the analysis of low carbon steel.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
da Silva, F.S., Montoya, K.Z., Dosta, S. et al. Influence of Different Copper Feedstock Powders on the Microstructure and Corrosion Resistance of Coatings Prepared by Cold Gas Spray. J Therm Spray Tech 33, 1365–1388 (2024). https://doi.org/10.1007/s11666-024-01783-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11666-024-01783-7