
Abstract
The oxidation and hot corrosion behavior of two atmospheric plasma-sprayed NiCoCrAlY–Cr2O3 and CoNiCrAlY–Cr2O3 coatings, which are primarily designed for wear applications at high temperature, were investigated in this study. The two coatings were exposed to air and molten salt (75%Na2SO4–25%NaCl) environment at 800 °C under cyclic conditions. Oxidation and hot corrosion kinetic curves were obtained by thermogravimetric technique. X-ray diffraction analysis and scanning electron microscopy with energy-dispersive x-ray spectrometry were employed to characterize the coatings’ microstructure, surface oxides, and composition. The results showed that both coatings provided the necessary oxidation resistance with oxidation rates of about 1.03 × 10−2 and 1.36 × 10−2 mg/cm2 h, respectively. The excellent oxidation behavior of these two coatings is attributed to formation of protective (Ni,Co)Cr2O4 spinel on the surface, while as-deposited Cr2O3 in the coatings also acted as a barrier to diffusion of oxidative and corrosive substances. The greater presence of Co in the CoNiCrAlY–Cr2O3 coating restrained internal diffusion of sulfur and slowed down the coating’s degradation. Thus, the CoNiCrAlY–Cr2O3 coating was found to be more protective than the NiCoCrAlY–Cr2O3 coating under hot corrosion condition.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
To improve the service life of machine components working at high temperatures, thermally sprayed cermet coatings have been widely used as an effective solution for wear resistance applications (Ref 1, 2). Coatings based on carbides (WC, Cr3C2, etc.) and oxides (Al2O3, Cr2O3, etc.) are frequently used for many applications in gas turbines, internal combustion engines, and power generation apparatus to improve resistance to abrasion. DellaCorte et al. (Ref 3-5) developed PS series plasma-sprayed composite coatings (PS200, PS300, PS400) using Cr2O3 or Cr3C2 as hardening phase with Ni-based alloy as binder; these composite coatings showed excellent tribological properties from room temperature to 800 °C. Murthy et al. (Ref 6) studied the abrasive wear behavior of WC–CoCr and Cr3C2–NiCr composite coatings deposited by high-velocity oxy-fuel (HVOF) spraying, indicating that the WC–CoCr coating had better wear resistance than the Cr3C2–NiCr coating due to the higher hardness of the WC particles and better matrix properties of the CoCr binder material.
However, there are many high-temperature applications where both corrosion and abrasion problems predominate (Ref 7-9). Hot corrosion results from presence of salt contaminants such as Na2SO4 and NaCl in residual fuel oil, which combine to form molten compounds that can destroy the protective oxide layer formed on materials (Ref 10, 11). Although these coatings show excellent wear behavior at high temperature, there is also great demand for coatings for use in applications in hot corrosion environments. However, there is little published literature presenting detailed investigations of the corrosion behavior of these wear resistance coatings. The MCrAlY–Cr2O3 coatings investigated here are primarily chosen for wear applications at high temperatures. In these coatings, Cr2O3 results in wear and oxidation resistance, while MCrAlY (M = Ni, Co, or NiCo) provides excellent mechanical strength and oxidation/corrosion resistance (Ref 12-14). In this work, two kinds of wear-resistant MCrAlY–Cr2O3 coating were prepared by atmospheric plasma spray, and their oxidation and hot corrosion behavior investigated in detail.
Experimental
Development and Characterization of Coatings
GH4169 with nominal composition 52Ni-20Cr-5.1Nb-3Mo-0.9Ti-0.6Co-0.5Al-bal Fe (wt.%) was selected as substrate material, as it possesses excellent high-temperature oxidation and hot corrosion resistance. The specimens were cut to dimensions of 20 mm × 15 mm × 5 mm and blasted with coarse SiC particles before coating deposition using a plasma-spray process. Only one side of the substrate with dimensions of 20 mm × 15 mm was coated.
For fabrication of MCrAlY–Cr2O3 composite powders, two nominal compositions of MCrAlY phase were selected based on two commercially available powders (74Ni-2.5Co-17.5Cr-5.5Al-0.5Y2O3 and 37Co-31.5Ni-25.5Cr-5.5Al-0.5Y2O3) applied in preparation of wear, oxidation, and hot corrosion resistant coatings. Ni, Co, Cr, Al, Y2O3, and Cr2O3 powders, water, and dispersant were blended together mechanically to prepare these composite powders by centrifugal spray granulation. Then, the composite powders were sintered in inert argon atmosphere at 1400 °C for 4 h using solid-state alloying technology. The compositions of the two composite powders are presented in Table 1. These two kinds of powder were deposited by using an APS-2000K plasma spraying system with the spray parameters presented in Table 2. Before MCrAlY–Cr2O3 was coated, commercially available CoNiCrAlY powder with chemical composition of 37Co-31.5Ni-25.5Cr-5.5Al-0.5Y (wt.%) was used to deposit a bonding layer.
The morphology and microstructure of the feedstock powders and as-sprayed coatings were observed using an FEI Quanta 200 FEG scanning electron microscope (SEM, FEI Corp., USA) equipped with an energy-dispersive x-ray (EDX) analysis system. The constituents of the powders and coatings were characterized by x-ray diffraction (XRD) analysis using a Philips X’Pert Pro diffractometer (PANalytical, The Netherlands, Cu Kα, λ = 0.154 nm) over the angular range of 10–90°. The coating porosity was measured using the Archimedes drainage method, with average data obtained from three repeated experiments.
Microhardness and Wear Testing
An HX-1000TM Vickers hardness device with 1.96 N test load and 15 s dwell time was used to measure the samples’ microhardness. In addition, an HT-1000 ball-on-disk high-temperature tribometer (CSM, Switzerland) was used to test the wear resistance of the coatings from room temperature to 800 °C; the wear tests were carried out with load of 9.8 N and sliding distance of 250 m. The volumetric wear rate (R w) was used to evaluate the specimens’ wear rate, calculated as R w = V/F·S, where V is the wear volume (mm3), F is the load (N), and S is the sliding distance (m). The wear volume was calculated as V = 2πr·A, where r = 5 mm is the wear radius and A is the cross-sectional area of the wear scar (mm2). The cross-sectional area was obtained using a 3D white interference surface morphology analyzer (ADE Corp., USA).
Oxidation and Hot Corrosion Testing
Oxidation and hot corrosion studies were performed in air and molten salt (75%Na2SO4–25%NaCl) environment at 800 °C under cyclic conditions. For isothermal oxidation testing, all specimens were placed in a furnace at the same time after the temperature had reached 800 °C. After a chosen time, oxidized specimens were removed and air-cooled to room temperature, then the weight change was measured using an electronic balance with sensitivity of 0.1 mg. For corrosion testing, 75%Na2SO4–25%NaCl, which melts at a relatively low temperature of about 675 °C, is found to be the commonest mixed salt applied in cyclic hot corrosion tests (Ref 15-19). In our experiment, 2.0 ± 0.1 mg/cm2 eutectic salts (75%Na2SO4–25%NaCl) was spread on the coatings and dried at 100 °C in an oven before placing in a furnace at 800 °C. The corroded samples were taken out of the furnace and cooled to room temperature in air after 4 h, then immersed in boiling water for 5 min to wash off residual salts. The washed samples were dried, then weighed using an electronic balance. The hot corrosion study was performed for 35 cycles under cyclic conditions.
XRD and SEM/EDX techniques were used to analysis the oxidation and corrosion products and microstructure of the coatings after oxidation and corrosion.
Results and Discussion
Characterization of Feedstock Powders and As-Sprayed Coatings
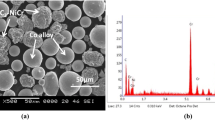
Figure 1 shows the SEM morphology of the NiCoCrAlY–Cr2O3 and CoNiCrAlY–Cr2O3 composite powders. It can be seen that both powders are nearly spherical with relatively uniform size distribution from 45 to 75 μm. Table 3 lists the flowability and apparent density of the NiCoCrAlY–Cr2O3 and CoNiCrAlY–Cr2O3 powders. Both powders showed flowability and apparent density suitable for the plasma spray process. Excellent powder flowability can guarantee stable powder feeding and avoid nozzle clogging. High apparent powder density can ensure that powder will be fed into the flame center and melted well. Figure 2(a) shows the XRD patterns of the NiCoCrAlY–Cr2O3 and CoNiCrAlY–Cr2O3 powders. The main constituent phases of both powders are Cr2O3 [Joint Committee on Powder Diffraction Standards (JCPDS) 84-0312] and Ni- or Co-based solid-solution phase (JCPDS 70-0989, 15-0806); the slight peak shift of the Co-based solid-solution phase appearing for the CoNiCrAlY–Cr2O3 powder is mainly due to the high level of Ni solution. Figure 2(b) shows the XRD patterns of the as-sprayed composite coatings; no new phases are detected in the two composite coatings, indicating that no significant oxidation or decomposition occurred during spraying. The coatings’ XRD patterns show much lower intensity due to presence of amorphous phase that formed during the quick cooling process after spraying.

SEM morphology of (a) NiCoCrAlY–Cr2O3 and (b) CoNiCrAlY–Cr2O3 composite powders

XRD patterns of NiCoCrAlY–Cr2O3 and CoNiCrAlY–Cr2O3 (a) powders and (b) as-sprayed coatings
Figure 3 presents cross-sectional backscatter electron micrographs of the as-sprayed NiCoCrAlY–Cr2O3 and CoNiCrAlY–Cr2O3 coatings, clearly showing the substrate, CoNiCrAlY bonding layer, and composite coating. The bonding layer shows denser structure and has good mechanical bonding with the substrate. It can be seen that both composite coatings show typical flat lamellar structure; the NiCoCrAlY and CoNiCrAlY phases are in white regions, the Cr2O3 phase distributes in gray regions, and the dark regions are pores and cracks. The pores and cracks mostly distribute in the Cr2O3 phase, because Cr2O3 has high melting point (about 2435 °C); it cannot be melted completely during the spraying process, thus reducing the extent of deformation. According to measurements by the Archimedes drainage method, these two coatings showed similar, low porosity of about 3.5 ± 0.3 %, which has a beneficial effect on their cohesive strength.

Cross-sectional backscatter electron micrographs of as-sprayed (a) NiCoCrAlY–Cr2O3 and (b) CoNiCrAlY–Cr2O3 coatings
Microhardness and Wear Testing
The microhardness of the as-sprayed NiCoCrAlY–Cr2O3 and CoNiCrAlY–Cr2O3 coatings was obtained using 1.96 N test load and 15 s dwell time. The results showed that the average hardness of the NiCoCrAlY–Cr2O3 and CoNiCrAlY–Cr2O3 coatings was 645 and 682 HV, respectively, while that of the substrate was about 288 HV. The hardness of the coatings was higher than that of the substrate, which can effectively improve the wear resistance performance. Figure 4 shows the wear rates of the two composite coatings from room temperature to 800 °C in atmosphere. For both coatings, the wear rates are of the order of 10−5 mm3/N m at all testing temperatures, indicating excellent wear resistance. The NiCoCrAlY–Cr2O3 coating had a higher wear rate than the CoNiCrAlY–Cr2O3 coating at temperature below 400 °C, mainly due to more severe brittle fracture caused by the lower microhardness of the NiCoCrAlY–Cr2O3 coating. Due to formation of oxide lubrication film, both coatings showed lower wear rates above 600 °C.

Wear rates of NiCoCrAlY–Cr2O3 and CoNiCrAlY–Cr2O3 coatings from room temperature to 800 °C
Oxidation Kinetics and Scale
According to oxidation testing, the substrate chosen for our experiments showed no obvious mass change at 800 °C, indicating that the substrate did not contribute to mass gain or loss during oxidation and hot corrosion tests of the coatings. The oxidation kinetic curves of the two composite coatings are shown in Fig. 5. The NiCoCrAlY–Cr2O3 coating exhibited better oxidation resistance than the CoNiCrAlY–Cr2O3 coating at 800 °C for 140 h. The oxidation rate of the CoNiCrAlY–Cr2O3 coating was about 1.36 × 10−2 mg/cm2 h, being slightly higher than that of the NiCoCrAlY–Cr2O3 coating (1.03 × 10−2 mg/cm2 h). In the initial stage (0–4 h), the mass gain of both coatings increased sharply. A stable stage occurred from 4 to 30 h, with both coatings following a nearly parabolic oxidation rate law, as seen in Fig. 6, indicating formation of protective oxide scale. In the later stage, the oxidation rates of both coatings became very slow with no obvious weight changes observed. It can be inferred that both coatings formed a diffusion barrier to oxidative substances after rapid oxide formation on the surface and in pores of the coatings. Then, the diffusion of oxidative substances was slowed down and the growth of oxides limited. Furthermore, as-deposited Cr2O3 in the composite coatings also acted as a barrier to diffusion of oxidative substances. The slightly higher weight gains for the CoNiCrAlY–Cr2O3 coating are mainly due to the poorer oxidation behavior of Co compared with Ni. As revealed by the XRD patterns in Fig. 7, the oxidation products of the NiCoCrAlY–Cr2O3 and CoNiCrAlY–Cr2O3 coatings after oxidation for 140 h at 800 °C were similar, being mainly NiO (JCPDS 01-1239), NiCr2O4 (JCPDS 89-6616), and CoCr2O4 (JCPDS 01-1122).

Oxidation kinetic curves of NiCoCrAlY–Cr2O3 and CoNiCrAlY–Cr2O3 coatings at 800 °C

(Weight gain/area)2 vs. time for NiCoCrAlY–Cr2O3 and CoNiCrAlY–Cr2O3 coatings

XRD patterns of oxidation products formed on (a) NiCoCrAlY–Cr2O3 and (b) CoNiCrAlY–Cr2O3 coatings
SEM micrographs showing the surface morphology of the NiCoCrAlY–Cr2O3 and CoNiCrAlY–Cr2O3 coatings before and after oxidation are presented in Fig. 8. Table 4 presents the corresponding EDS element distribution of the oxide scale. Uniform and continuous scale could be found on both rough surfaces in comparison with the as-sprayed coatings. The formed polyhedral oxides mainly consisted of O, Cr, Ni, and Co elements. According to weight percentage analysis, it cold be inferred that the oxides were mainly NiCr2O4 and CoCr2O4, respectively, on the two coatings, consistent with the XRD results. Figure 9 shows cross-sectional back-scattered electron (BSE) images of the two coatings after 140 h of oxidation. The micrographs show that the coatings still maintained their lamellar structure, with no obvious internal oxidation being found for either coating. Compared with the as-sprayed coatings, thin and compact oxide scale formed on both coatings, being linked up with continuous deposited Cr2O3. EDS analysis revealed that the oxide scale was rich in Cr, O, and Ni and Cr, O, and Co, respectively, while a small amount of Al was also present in both scales. Both the deposited and formed oxides provide good barrier protection for the two coatings. Moreover, in areas of pores and cracks, Cr-rich oxides also formed, which could inhibit diffusion of oxide species to the coating interior.

SEM morphology of NiCoCrAlY–Cr2O3 and CoNiCrAlY–Cr2O3 coatings (a1, b1) before and (a2, b2) after oxidation at 800 °C for 140 h

Cross-sectional BSE images of (a) NiCoCrAlY–Cr2O3 and (b) CoNiCrAlY–Cr2O3 coatings after 140 h of oxidation at 800 °C
Hot Corrosion Kinetics and Scale
Weight change plots for the NiCoCrAlY–Cr2O3 and CoNiCrAlY–Cr2O3 coatings subjected to Na2SO4–25%NaCl salt mixture at 800 °C are shown in Fig. 10. The NiCoCrAlY–Cr2O3 coating showed weight gain at all test temperatures, whereas the CoNiCrAlY–Cr2O3 coating showed slight mass loss after the initial stage. In the initial stage (0–4 h), the mass gain of both coatings increased promptly. This is attributed to rapid formation of oxides at the surface and pores of the coatings. Thereafter, the NiCoCrAlY–Cr2O3 coating showed a slow rate of mass gain, and the overall mass gain value reached about 4.5 mg/cm2 after 140 h. For the CoNiCrAlY–Cr2O3 coating, slight mass loss occurred after the initial mass gain stage, and the total mass loss from 8 to 140 h was about 1 mg/cm2. Compared with the air oxidation kinetics (Fig. 5), the NiCoCrAlY–Cr2O3 coating showed much greater weight gain on exposure to the molten salt environment, whereas the CoNiCrAlY–Cr2O3 coating showed stable behavior under both experimental environments.

Hot corrosion kinetic curves of NiCoCrAlY–Cr2O3 and CoNiCrAlY–Cr2O3 coatings
Figure 11 shows the XRD analysis of the NiCoCrAlY–Cr2O3 and CoNiCrAlY–Cr2O3 coatings after hot corrosion for 140 h. In the scale of the NiCoCrAlY–Cr2O3 coating, NiO was observed as the main phase, along with a few indications of NiCr2O4 phase. For the CoNiCrAlY–Cr2O3 coating, CoCr2O4 and Cr2O3 were observed as the main phases.

XRD patterns of (a) NiCoCrAlY–Cr2O3 and (b) CoNiCrAlY–Cr2O3 coatings after hot corrosion at 800 °C for 140 h
The surface morphology of the coatings after hot corrosion is presented in Fig. 12. Uniform and continuous scale formed on the NiCoCrAlY–Cr2O3 coating (Fig. 12a), and EDS analysis (Table 5) revealed the amounts of Ni and O present, suggesting formation of NiO in combination with the XRD analysis. The microcracks occurring in the scale are mainly caused by thermal stress during the cooling and heating process in the cyclic test, and open cracks will accelerate penetration of corrosive species. This in turn will aggravate corrosion of the coating interior. For the CoNiCrAlY–Cr2O3 coating, the corrosion scale was dense and continuous, as shown in Fig. 12(b). The spinel-structure oxide (marked “2”) consisted mainly of Co, Cr, and O, while the schistose oxide (marked “3”) was composed of Cr and O.

SEM morphology of (a) NiCoCrAlY–Cr2O3 and (b) CoNiCrAlY–Cr2O3 coatings after hot corrosion at 800 °C for 140 h
Cross-sectional images of the two coatings after long-term corrosion are shown in Fig. 13. The micrographs show that both coatings retained their as-deposited, lamellar structure. In contrast to the CoNiCrAlY–Cr2O3 coating, the NiCoCrAlY–Cr2O3 coating had thicker oxide scale. Thick scale could aggravate the mismatch between oxide and coating, thus resulting in cracking and spallation, as shown in Fig. 12(a). EDS analysis showed that the upper thin bright part of the scale mainly consisted of NiO whereas the second thick oxide layer was rich in Ni, Cr, and O, being confirmed as NiCr2O4 by quantitative analysis. However, many homogeneous voids were present in this thick oxide layer, and element S could be found around voids. In the inner layer of the coating, the uncorroded metal matrix (bright part) was surrounded by Cr2O3.

Cross-sectional BSE images of (a) NiCoCrAlY–Cr2O3 and (b) CoNiCrAlY–Cr2O3 coatings after 140 h of hot corrosion at 800 °C
At the initial stage of hot corrosion, oxygen ions have low concentration due to coverage by molten salt, thus SO 2−4 will decompose into O2− and SO3, increasing the concentration of oxygen ions. For a Cr-rich coating, Cr ions can react with O2− at the interface of fusant and coating to form oxides:
Molten SO 2−4 would also diffuse into the coating and react with the element Ni to form sulfide:
Ni3S2 will combine with Ni to form the eutectic (Ni·Ni3S2), which has a low melting point of about 635 °C (Ref 15). This eutectic is totally molten at the test temperature of 800 °C. As corrosion proceeds, oxygen will gradually diffuse into the coating, thus the concentration of oxygen is increased. The molten eutectic would oxidize to NiO and SO2, as shown in Eq 3, so that a zone of voids is formed in the oxide scale. The SO2 formed could permeate further into the coating and react with Ni (Eq 4) to accelerate the oxidation process:
After long-time corrosion, the NiO formed would undergo solid reaction with Cr2O3 to form spinel oxide NiCr2O4. NiCr2O4 is a protective oxide that could limit Ni diffusion, thus slowing down the oxidation rate.
The CoNiCrAlY–Cr2O3 coating retained its original structure better than the NiCoCrAlY–Cr2O3 coating. As shown in Fig. 13(b), the dominant phase of the upper thin oxide was CoCr2O4, while the underlying oxide layer was Cr2O3. The compact Cr2O3 formed linked up with the continuous Cr2O3 phase preexisting in the coating. Both CoCr2O4 and Cr2O3 are protective oxides, providing the necessary protection against penetration of corrosive species into the coating. In the initial corrosion period (0–10 h), the rapid increase in weight gain of the CoNiCrAlY–Cr2O3 coating may be due to rapid formation of oxides caused by penetration of oxidizing species. Cr is selected to be oxidized to Cr2O3 first, then the element Co could react with molten SO 2−4 to form a sulfide eutectic Co·Co3S2 with melting point of about 880 °C (Ref 13). Compared with Ni·Ni3S2, Co·Co3S2 possesses a higher melting point and shows more stable behavior at the test temperature. In addition, the diffusion velocity of sulfur in Co is two orders of magnitude lower than that in Ni (Ref 20), so the presence of Co will effectively reduce internal diffusion of sulfur. As Co3S2 is oxidized to CoO, formation of CoCr2O4 spinel occurs via solid-phase reaction between CoO and Cr2O3. Spinel usually has lower diffusion coefficients of cations and anions, which is beneficial for reducing the corrosion rate. At the same time, Cr2O3 will be solved by reaction with NaCl to form the water-soluble salt Na2CrO4 and volatile substance CrO2Cl2:
Na2CrO4 (JCPDS 01-0927) could indeed be detected on the corroded samples before washing, as shown in Fig. 14.

XRD patterns of unwashed CoNiCrAlY–Cr2O3 coating after hot corrosion at 800 °C for 140 h
After the initial stage, slight weight loss of the coating occurred, mainly because the growth of the oxide was slightly slower than dissolution and volatilization. Moreover, as corrosion proceeds, the growth and dissolution of the oxide scale tend to occur at nearly identical rates; this phenomenon can be attributed to formation of protective CoCr2O4 on the upper oxide scale. CoCr2O4 could restrain permeation of oxidative species into the coating with simultaneous outward dissolution of Cr2O3, thus benefiting the coating’s corrosion resistance.
Conclusions
-
1.
NiCoCrAlY–Cr2O3 and CoNiCrAlY–Cr2O3 coatings were successfully prepared by atmospheric plasma spraying. The two coatings showed nearly uniform, dense, and adherent layered microstructure.
-
2.
Both NiCoCrAlY–Cr2O3 and CoNiCrAlY–Cr2O3 coatings showed excellent wear resistance from room temperature to 800 °C.
-
3.
The NiCoCrAlY–Cr2O3 coating showed better oxidation resistance compared with the CoNiCrAlY–Cr2O3 coating. No obvious degradation was observed for either coating because of protection by Cr2O3 deposited in the coatings as well as the spinel oxide NiCr2O4 and CoCr2O4 formed in the upper scales.
-
4.
The NiCoCrAlY–Cr2O3 coating showed poorer resistance to hot corrosion than oxidation, which is attributed to formation of molten Ni·Ni3S2.
-
5.
The melting point as well as stability of Co·Co3S2 are higher than that of Ni·Ni3S2, which means that Co is helpful for enhancement of the corrosion resistance. However, Cr2O3 would be dissolved in the presence of NaCl in the fusant salt before formation of spinel CoCr2O4.
References
C.B. Huang, L.Z. Du, and W.G. Zhang, Effect of Solid Lubricant Content on the Microstructure and Properties of NiCr/Cr2C2-BaF2·CaF2 Composite Coatings, J. Alloys Compd., 2009, 479(1), p 777-784
B.S. Mann and B. Prakash, High Temperature Friction and Wear Characteristics of Various Coating Materials for Steam Valve Spindle Application, Wear, 2000, 240(1), p 223-230
C. DellaCorte and B.J. Edmonds, NASA PS400: A New High Temperature Solid Lubricant Coating for High Temperature Wear Applications, NASA Report, 2009
C. Dellacorte and J.A. Laskowski, Tribological Evaluation of PS300: A New Chrome Oxide-Based Solid Lubricant Coating Sliding Against Al2O3 from 25 °C to 650 °C, Tribol. Trans., 1997, 40(1), p 163-167
H.E. Sliney, C. Dellacorte, and V. Lukaszewicz, The Tribology of PS212 Coatings and PM212 Composites for the Lubrication of Titanium 6A1-4V Components of a Stirling Engine Space Power-System, Tribol. Trans., 1995, 38(3), p 497-506
J.K. Murthy and B. Venkataraman, Abrasive Wear Behaviour of WC-CoCr and Cr3C2-20(NiCr) Deposited by HVOF and Detonation Spray Processes, Surf. Coat. Technol., 2006, 200(8), p 2642-2652
N. Eliaz, G. Shemesh, and R.M. Latanision, Hot Corrosion in Gas Turbine Components, Eng. Fail. Anal., 2002, 9(1), p 31-43
T.S. Sidhu, A. Malik, S. Prakash, and P.D. Agrawal, Oxidation and Hot Corrosion Resistance of HVOF WC-NiCrFeSiB Coating on Ni-and Fe-Based Superalloys at 800 °C, J. Therm. Spray Technol., 2007, 16(5), p 844-849
S.S. Chatha, H.S. Sidhu, and B.S. Sidhu, High Temperature Hot Corrosion Behavior of NiCr and Cr3C2-NiCr Coatings on T91 Boiler Steel in an Aggressive Environment at 750 °C, Surf. Coat. Technol., 2012, 206(19), p 3839-3850
T.S. Sidhu, S. Prakash, and R.D. Agrawal, Hot Corrosion Performance of a NiCr Coated Ni-Based Alloy, Scr. Mater., 2006, 55(2), p 179-182
E. Liu, Z. Zheng, X. Guan, J. Tong, L. Ning, and Y. Yongsi, Influence of Pre-oxidation on the Hot Corrosion of DZ68 Superalloy in the Mixture of Na2SO4-NaCl, J. Mater. Sci. Technol., 2010, 26(10), p 895-899
W. Brandl, G. Marginean, N. Marginean, V. Chirila, and D. Utu, Prevention of Metal Dusting on Ni-Based Alloys by MCrAlY Coatings, Corros. Sci., 2007, 49(10), p 3765-3771
N. Jegadeeswaran, K.U. Bhat, and M.R. Ramesh, Improving Hot Corrosion Resistance of Cobalt Based Superalloy (Superco-605) Using HVOF Sprayed Oxide Alloy Powder, Coating, 2015, 68, p 309-316
K. Yuan, R.L. Peng, X.H. Li, and S. Johansson, Hot Corrosion Behavior of HVOF-Sprayed CoNiCrAlYSi Coatings in a Sulphate Environment, Vacuum, 2015, 122, p 47-53
Q.X. Fan, S.M. Jiang, H.J. Yu, J. Gong, and C. Sun, Microstructure and Hot Corrosion Behaviors of Two Co Modified Aluminide Coatings on a Ni-Based Superalloy at 700 °C, Appl. Surf. Sci., 2014, 311, p 214-223
Q.X. Fan, S.M. Jiang, D.L. Wu, J. Gong, and C. Sun, Preparation and Hot Corrosion Behavior of Two Co Modified NiAl Coatings on a Ni-Based Superalloy, Corros. Sci., 2013, 76, p 373-381
D. Gond, R.S. Lalbondre, D. Puri, and S. Prakash, High Temperature Oxidation and Hot Corrosion Behavior of Yttria-Stabilized Zirconia as Plasma Sprayed Coating in Air and Salt at 900 °C Under Cyclic Condition, J. Miner. Mater. Charact. Eng., 2012, 11, p 285-302
Z.J. Liu and C.G. Zhou, Hot Corrosion Behavior of Si-Y-Co-Modified Aluminide Coating Exposed to NaCl + Na2SO4 Salt at 1173 K, Oxid. Met., 2016, 85, p 205-217
W.Y. Zhao, Y.Z. Liu, S.S. Li, and S.K. Gong, Hot Corrosion Behavior of a Ni3Al-Based IC21 Alloy in a Molten Salt Environment, Oxid. Met., 2014, 81, p 631-644
A.M. Beltran, D.A. Shores, Hot Corrosion, The Superalloys, C.T. Sims and W.C. Hagel, Eds., Wiley, New York, 1972, Chapter 11, p 317-339
Acknowledgment
The authors are grateful for financial support from the National Natural Science Foundation of China (Grant No. 51471159).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhang, T., Huang, C., Lan, H. et al. Oxidation and Hot Corrosion Behavior of Plasma-Sprayed MCrAlY–Cr2O3 Coatings. J Therm Spray Tech 25, 1208–1216 (2016). https://doi.org/10.1007/s11666-016-0433-5
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11666-016-0433-5