
Abstract
This study investigates the optical, structural, and dielectric properties of NiO nanoparticles (NPs) incorporated into a poly(vinyl alcohol)/sodium alginate (PVA/SA) polymer blend. The x-ray diffraction (XRD) pattern confirmed the formation of face-centered cubic NiO NPs with a crystallite size of around 9.9 nm and lattice strain of 5.6 × 10−3, as determined by the Williamson–Hall method. High-resolution transmission electron microscopy (HRTEM) revealed spherical NiO NPs with sizes ranging from 5 nm to 12 nm. The embedding of NiO NPs into the matrix structure of PVA/SA was confirmed by peak shifting and new peaks from the XRD patterns, and the appearance of spherical nanoparticles with size ranging between 5 nm and 12 nm from HRTEM images. Significant shifting was observed in the Fourier transform infrared (FTIR) spectral bands, indicating strong interaction between the blend components and NiO. Optical studies showed that doping of PVA/SA with NiO NPs enhanced the material’s absorption, with a consequent effect on the optical energy band gap, demonstrated by a red shift in the absorption edge with a decrease from 5.0 eV for pristine PVA/SA to 0.09 eV for the 8 wt.% NiO-doped film. In addition, dielectric studies showed an increase in conductivity with the addition of NiO NPs, suggesting their suitability for solid polymer electrolyte applications.
Graphical Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polymer nanocomposites (PNCs) have become a prominent area of study in the field of materials science, presenting remarkable advancements in the physical, chemical, and electrical characteristics of conventional polymers. The composites under consideration comprise a polymer matrix that is combined with nanoparticles or nanofillers. These nanoparticles or nanofillers encompass a variety of elements, including carbon-based materials such as graphene and carbon nanotubes, as well as metal oxides such as titanium dioxide, tungsten trioxide, and zinc oxide. Integrating these nanoscale fillers into polymers results in the improvement of multiple characteristics, such as electrical conductivity, thermal stability, and mechanical strength, rendering PNCs very well suited for electronic applications.1,2,3 The field of polymer nanocomposites for electronic applications is constantly evolving, with continuous progress focused on improving the distribution of nanofillers, strengthening the incorporation between filler and the polymeric matrix to create new nanocomposites with customized characteristics. With the ongoing development of this subject, it is anticipated that PNCs will have a growing significance in the upcoming era of electronic devices, providing resolutions to critical issues in electronics design and manufacturing.4,5,6 Polyvinyl alcohol (PVA) is a synthetic polymer that is soluble in water and is extensively utilized in diverse applications owing to its distinctive characteristics.7
PVA is a semicrystalline polymer that exhibits exceptional characteristics in film formation, emulsification, and adhesion. The suitability of this material for various applications is attributed to its high tensile strength, flexibility, and resistance to oil, grease, and solvents.8 Moreover, PVA exhibits biodegradability, non-toxicity, and biocompatibility, facilitating its extensive application within the medical and pharmaceutical sectors. Sodium alginate (SA), a polymer that is both anionic and soluble in water, is obtained from brown algae through industrial extraction. The material is nontoxic, economical, capable of decomposing naturally, and renewable. SA chains have inherent superhydrophilic and ion-exchange characteristics due to the presence of abundant oxygen-containing groups such as (–OH) and (–COONa). Additionally, these chains are susceptible to chemical modifications.9 SA can be employed as a biodegradable and environmentally friendly material in electrical devices for several applications. The potential of sodium alginate as a dielectric material for capacitors and other electronic components has been investigated owing to its notable characteristics.10 The biodegradability and environmentally favorable characteristics of this material render it a compelling substitute for traditional dielectric materials. Flexible substrates for flexible electronics, such as wearable devices and bendable screens, can be enhanced by using sodium alginate. The mechanical characteristics of the material, such as its flexibility and stretchability, render it well suited for many applications.
Conductive composites can be formed by combining sodium alginate with conductive fillers such as carbon nanotubes or metallic particles. These composite materials have diverse uses in the field of electronics, including but not limited to electromagnetic shielding, antistatic coatings, and conductive inks for printed circuits.11
The potential of sodium alginate as an electrolyte material in electrochemical devices, such as batteries and supercapacitors, has been investigated. The prospective suitability of this material for these applications originates from its ionic conductivity and compatibility with a wide range of electrolyte systems.12 Electrolytes based on PVA and SA have been investigated for use in electrochemical devices, including supercapacitors and batteries. Ionic liquids or other conductive fillers can be added to these materials to increase their ionic conductivity. Nickel oxide (NiO) has exceptional features and qualities, rendering it a highly promising material for a wide range of electrical device applications. NiO is a p-type semiconductor that has a broad energy band gap ranging from 3.6 eV to 4.0 eV, which makes it well suited for use in optoelectronic and electrical devices. The field of electronics has shown considerable interest in the features of this material, including its great chemical stability, low cost, and environmental friendliness. The potential of NiO as a contender for transition metal oxides (TCOs) in a range of optoelectronic devices has been investigated.13,14 The material’s promise for various applications stems from its high transmittance in the visible range and moderate electrical conductivity. NiO has electrochromic characteristics, indicating its ability to undergo reversible alterations in its optical properties when subjected to an electrical potential. The aforementioned attribute has resulted in its application in electrochromic appliances, including intelligent windows, displays, and energy-efficient construction materials.15,16 The high specific capacitance, excellent electrical conductivity, and electrochemical stability of NiO have demonstrated its promise as an electrode material for supercapacitors.17 PVA and SA are typically regarded as compatible and capable of forming a homogeneous mixture at different ratios. The combination of PVA and SA yields materials that possess the advantages of both polymers, including the robust mechanical strength of PVA and the biocompatibility and biodegradability of SA.18,19 The optical characteristics of PVA and SA blends can be adjusted by the inclusion of nanoparticles (NPs), rendering them appropriate for optoelectronic applications. Khalil et al.20 reported that the addition of TiO2 NPs to PVA/SA improved the optical characteristics of the blend, rendering it valuable for implementation in sensors and solar cells, with a direct band gap of 5.5 eV. Ibrahim et al.21 added TiO2 to PVA/SA for use in wound dressing. Eghbalifam et al.18 prepared Ag NPs by gamma irradiation to PVA/SA for antibacterial application. Wang et al.19 added Ag NPs to PVA/SA for antibacterial application. Abdelghany et al.22 added Se NPs to PVA/SA for antibacterial application. NiO nanoparticles are more cost-effective and readily accessible than other metal oxide nanoparticles, making them a cost-efficient option for a wide range of applications.23 NiO nanoparticles are nontoxic and biocompatible, distinguishing them from other metal oxide nanoparticles for use in biomedical applications.24 The reactivity and interaction of NiO nanoparticles with other structural materials are enhanced based on the changes in the volume-to-surface ratio and the embedding of host materials with quantum size effects. This feature is essential for applications in catalysis, sensing, and energy storage.25
Despite the exceptional properties of NiO, to date no studies have explored the effect of NiO on PVA/SA. Thus, the present work deals with the preparation of a blend of PVA/SA (50/50 wt.%) after adding NiO with different weight percentages (2, 4, 6, 8 wt.% NiO). The results confirmed that the addition of NiO NPs led to a greater decrease in the band gap than other materials incorporated with this blend (2.82 eV). This outcome demonstrates that the growth of NiO nanoparticles leads to the formation of conductive routes in the polymer matrix, which in turn promotes electron hopping and improves interactions between carriers. This leads to a displacement of the bands, either valence or conduction, resulting in a decrease in the energy difference between them and an increase in the ability of the PVA/SA blend to conduct electricity, which makes it useful in optoelectronic devices.
Experimental Work
Material and Methods
PVA (Alfa Aesar) and SA (Sigma-Aldrich) were each dissolved in distilled water under continuous stirring. After that, the solutions were combined and maintained under magnetic stirring for about 5 h until a homogeneous solution was formed. NiO (Sigma-Aldrich; particle size < 50 nm) was added to the mixture solution with weight percentage up to 8 wt.%, followed by sonication via a dip sonicator (Hielscher UP100H ultrasonic homogenizer, 100 W, 30 kHz) to ensure that NiO was well dispersed in the PVA/SA solution. The prepared solution was added to a Petri dish and dried at 55°C for 24 h until the polymer nanocomposite film was formed.
Measurement Techniques
The previously prepared samples were subjected to structural analysis using x-ray diffraction (XRD; PANalytical X’Pert PRO, model number PW 3040/60). The vibration investigation was carried out using Fourier transform infrared (FTIR) spectroscopy (Bruker VERTEX 80 spectrometer). The size and shape were confirmed using transmission electron microscopy (HRTEM, JEOL JEM-2100F). Field-emission scanning electron microscopy (FESEM, Quanta 250 FEG) was utilized to analyze the surface morphology. UV–visible (UV–Vis) spectroscopy (Jasco V-630 spectrophotometer) was employed to investigate the optical characteristics. The dielectric characteristics were analyzed using a Novocontrol Alpha high-resolution analyzer (40 type) spanning a frequency range of 10−1 Hz to 107 Hz. The samples were placed between two newly polished brass electrodes, with the top electrode having a 10 mm diameter, forming a parallel-plate capacitor cell. A 1 V sinusoidal voltage was applied in a frequency range covering 10−1 to 107 Hz, yielding complex permittivity, represented by the equation ε = ε′ − iε″. Any of the complex parameters related to one another by the equation below can be used to characterize the investigated composites for complex dielectric spectroscopy.
Results and Discussion
Structural Measurements
A structural characterization with crystallite investigation of NiO NPs was accomplished by employing x-ray diffraction (XRD) over the 2θ range from 10° to 80°, as depicted in Fig. 1a. With the use of XRD patterns, it is possible to observe low and broad diffraction peaks as a result of the small size effect. The XRD patterns clearly show diffraction peaks corresponding to the (111), (200), (220), (311), and (222) planes, indicating a face-centered cubic (fcc)-structured NiO, which matches with card number 00-065-2901.26 It can be seen from the XRD pattern that the NiO NPs appear to be single phase. The Debye–Scherrer formula and the Williamson–Hall (W–H) method were utilized in order to obtain the average crystallite size, denoted by the letter T.27
where k represents a constant value of 0.94 for spherical particles, λ represents the x-ray wavelength (CuKα = 0.1541 nm), β represents the full width at half maximum (FWHM) of the intense and broad peaks, ε represents the lattice strain, and θ represents the Bragg angle or diffraction angle. The average crystallite size calculated using the Scherrer formula (Eq. 1) is 9.9 nm. The lattice strain and average crystallite size of the NiO nanoparticles were determined by analyzing β cos θ against 4sin θ using the W–H method (Eq. 2, Fig. 1b). The value of the component Kλ/T is represented by the intercept of the line, while the slope represents the 4ε value. The average crystallite size was 23.3 nm, while the lattice strain was 5.6 × 10−3. The grain size of the NiO NPs was found to be slightly greater than the findings obtained from Scherrer’s equation. These results were obtained by plotting the W–H relationship for NiO nanoparticles. Consequently, this indicates that Scherrer’s equation was a good fit for the NiO nanoparticles.

(a) XRD of NiO and (b) Williamson–Hall plot of NiO NPs.
Figure 2a and b show the HRTEM and histogram of NiO NPs, respectively. HRTEM is typically used for imaging and analytically characterizing nanoparticles in order to evaluate their form and size. The HRTEM image showed that the NiO NPs had a spherical shape, as displayed in Fig. 2a. A number of aggregated particles are also present. The particle size is 5–12 nm.

(a) HRTEM and (b) histogram of NiO NPs.
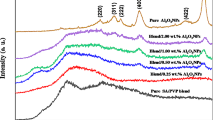
Figure 3 illustrates the XRD of PVA/SA and PVA/SA embedded with different weight percentages of NiO NPs. The figure shows that PVA/SA has a broad diffraction peak at 2θ = 19.6° which is due to the strong interaction because of hydrogen bonding, which is in agreement with previous literature.28,29 After incorporation with NiO NPs, the diffraction peak at 2θ = 19.6° is shifted in the direction of a higher 2θ and its intensity decreased. In addition, there are new peaks appearing at 2θ = 37.4°, 43.5°, 63°, and 75.5° which are related to NiO NPs, confirming the successful incorporation of PVA/SA and NiO NPs.

XRD diffractogram of PVA polymeric matrix and PVA/SA/NiO polymeric nanocomposite.
Figure 4 shows attenuated total reflectance (ATR)-FTIR spectra of the PVA/SA matrix structure before and after embedding with different amounts of NiO NPs. The appearance of transmittance peaks at 3310 cm−1 and 1328 cm−1 in PVA correspond to the functional O–H group with vibrational motion of stretching and bending, respectively. Also, the functional C–H group exhibits vibrational stretching modes at 2938 cm−1 and 2907 cm−1. Additionally, the appearance of a peak at 1725 cm−1 corresponds to the carbonyl group (residue of the acetate group). CH bending and wagging vibrations are identified at 1421 cm−1 and 1373 cm−1. The signal at 1247 cm−1 corresponds to the CH wagging vibration of the remaining acetate group. The 1140 cm−1 shoulder corresponds to C–O stretching vibration in the crystalline component of PVA, while the 1078 cm−1 band is associated with C=O stretching vibration in the amorphous part of PVA. The C–C stretching vibration occurs at 847 cm−1.30,31,32 For SA, the presence of an –OH group is shown at 3291 cm−1. The peak at 2938 cm−1 is associated with the vibration band of –CH alkyl groups. The peak at 1596 cm−1 is characteristic of sodium alginate and represents stretching vibrations of C=O. The 1408 cm−1 asymmetric band is a result of COO– group oscillations of the alginate molecule. The peak at 1066 cm−1 appears from the presence of a C–O functional group, while the band at 1035 cm−1 indicates the presence of a –COC group.33,34 NiO showed a wide band at 2883 cm−1 which is due to the vibrations of the CO2 molecule that are absorbed from the air. These vibrations are symmetric and asymmetric stretching modes combined. There is another band at 1360 cm−1 related to an OH functional group absorbed from the atmosphere. Bands at 560 cm−1, 476 cm−1 , and 418 cm−1 are associated to the bond between Ni and O.35

ATR-FTIR spectra of (a) PVA, SA, NiO, (b) PVA/SA, and the nanocomposite structure of PVA/SA/NiO.
After blending PVA with SA, the appearance of a peak at 1725 cm−1 from the acetate group in PVA is shifted to 1730 cm−1 and the band at 1421 cm−1 from CH bending is shifted to 1428 cm−1. Also, the C=O band is shifted from 1078 cm−1 to 1088 cm−1. A new vibrational peak appears at 1655 cm−1 , and the peak at 1408 cm−1 from COO disappears. All of these findings indicate that PVA and SA work well together. After the incorporation of NiO into the PVA/SA blend, new bands appear at 1572 cm−1 and 587 cm−1, indicating the successful interaction between PVA/SA and NiO.
Figure 5 presents FESEM images of PVA/SA and PVA/SA doped with 2 wt.% and 8 wt.% NiO. As seen in Fig. 5a, PVA/SA clearly shows a smooth and homogeneous surface and Fig. 5b shows the 3D images of PVA/SA with root-mean-square roughness (Rq) of 64.2 nm and mean roughness (Ra) of 53.6. With the addition of NiO, there are white spots distributed on the surface of PVA/SA which are attributed to NiO NPs. The Rq for 2 wt.% NiO and 8 wt.% NiO are 66.2 nm and 70.8 nm, respectively. Also, Ra for 2 wt.% NiO and 8 wt.% NiO are 54.7 nm and 53.1 nm, respectively, which confirmed that the roughness is increased with insertion of NiO NPs into the matrix.

FESEM images and 3D images of (a, b) PVA/SA, with (c, d) 2 wt.% NiO and (e, f) 8 wt.% NiO.
Optical Measurements
Studying optical absorption is crucial for understanding a material’s energy gap. Figure 6 shows absorption spectra of NiO NPs with different amounts in the PVA/SA blend. Increasing the concentration of the doped NiO NPs clearly enhanced the absorption of the PVA/SA blend. The development of the NiO NPs in the host mixture is believed to be responsible for this behavior. Two peaks appeared at 216 nm and 277 nm related to n–π* and π–π*, respectively, which are attributed to the chromphoric group of PVA and SA polymer.20 Addition of NiO causes a change in the absorption edge with shifting the absorption peak at 216 nm. At a higher concentration of NiO (8 wt.% NiO), two peaks appeared at 354 nm and 381 nm which are attributed to the absorption peaks of NiO.36,37

UV–Vis absorbance curve of PVA/SA and PVA/SA/NiO.
Discrepancies in absorbed radiation levels can be used to differentiate between different types of electronic transitions. The sudden shift in the basic absorption is known as the "absorption edge." The absorption coefficient (α) signifies the speed at which light intensity diminishes and it is calculated from the following equation.38
The absorption edge value is determined by extrapolating the linear component of Fig. 7 to a point where α (cm−1) = 0. The absorption edge of PVA/SA is 5.0 eV, decreasing to 0.09 eV in 8 wt.% doped NiO film, showing a red shift caused by doping of NiO NPs. Table I displays the absorption edge values for both PVA/SA and PVA/SA/NiO films.

Absorption coefficient versus photon energy curve of PVA/SA and PVA/SA/NiO.
Also, the optical transitions are studied by the following formula.38
where α represents the absorption coefficient, hυ denotes the photon energy, B is a constant, Eg is the optical energy band gap, and L represents the transition types (1/2, 3/2, 2, and 3 for transitions labeled as direct allowed/forbidden, indirect allowed/forbidden, respectively).39 To obtain the optical energy band gap value Eg, we plot (αhυ)1/L versus hυ. Figure 8 represents the plot of (αhʋ)2 and (αhʋ)1/2 versus hυ. It is possible to obtain the optical energy band gaps by extrapolating the linear section of the plots to the abscissa. A modest decrease in Eg was observed when the content of NiO increased. The variance in the computed values of the optical energy gap likely reflects the role that NiO plays in modifying the electronic structure of the PVA/SA matrix. This modification arises as a result of the emergence of different polaronic and defect levels. The decrease in the optical energy band gap with doping is likely attributed to the formation of charge transfer complexes, resulting from the integration of small amounts of dopant into the host matrix.40 Because these charge transfer complexes provide extra charges, they are able to boost the electrical conductivity of the material, which in turn leads to a reduction in the optical energy gap. Increasing the concentration of the dopant causes the molecules of the dopant to begin bridging the gap that separates the two localized states and lowering the potential barrier that exists between them. This, in turn, makes it easier for charge carriers to move between the two localized states.

Relation between (αhʋ)2 and (αhʋ)1/2 versus hʋ of PVA/SA and PVA/SA/NiO.
An intermediate band gap state can be estimated via the width of the defect bands in PVA/SA/NiO nanocomposites. This state has a band tail that stretches from the bottom of the CB to the depth of the band gap. Similarly, defect states near the valence band also undergo deformation at the edge of the valence band, which is deep inside the gap. The energy that is associated with the damaged tail is referred to as the Urbach tail, and its abbreviation is Eu.41
As shown in Fig. 9, the Urbach energy can be calculated by graphing lnα versus hυ. For the linear component below the optical band gap, one can find the value of Eu by calculating the reciprocal of the slopes. The calculated values of the Urbach energy increased as the content of NiO NPs increased, as shown in Table I. The defect energy increases as the band gap decreases. Our contention that sub-band states can be formed between the conduction and valence bands and hence lower energy band gaps is strongly supported by this.

Relation between lnα and hʋ of PVA/SA and PVA/SA/NiO.
Dielectric Studies
The conductivity of PVA/SA and PVA/SA/NiO was determined using a specific relation.
ρ indicates electrical resistance while σ represents conductivity. Figure 10 shows the graph of the conductivity log(σ) versus log(f) for all samples taken at room temperature. At low frequencies, the conductivity is low because of interfacial impedance or space charge polarization. This shows that the thin films being studied do not have Debye qualities. As the frequency increases, the conductivity also increases. The conductivity of PVA/SA/NiO was higher than that of the PVA/SA. This suggests that adding NiO NPs to the PVA/SA blend can enhance the charge conduction mechanism to a moderate extent. Also, this improvement could mean that there is more disorder, which makes it harder for charge carriers to move. This can cause a linked percolating chain to form, which is good for charge transfer processes.42 The increase in conductivity of the PVA/SA/NiO nanocomposite indicates its suitability for creating solid polymer electrolytes in nanocomposites.

Relation between log(σ) and log(f) of PVA/SA and PVA/SA/NiO.
The following equation can be used to calculate the dielectric constant (ε′) and dielectric loss (ε″).43
where C represents capacitance, L stands for the thickness of samples, εo denotes the permittivity in free space, A represents the area of the electrode, and D represents the dissipation factor. Figure 11a illustrates the variation of the dielectric constant with frequency for the films produced at room temperature. As frequency increases, the dielectric constant decreases, leading to diminished rotational capacity of the dipole and reduced oscillation in the field.44 Figure 11b shows the frequency dependence of dielectric loss (ε″) for all PVA/SA/NiO films prepared at room temperature. In Fig. 11b, the decrease in dielectric loss as frequency rises is due to ion motion, which is the main cause of nanocomposite dielectric loss at lower frequencies. Ion hopping and ion motion conduction slows while ion polarization decreases at lower frequencies, causing more dielectric loss.45 High-frequency ion vibrations are the primary cause of dielectric loss and result in a reduction in dielectric loss. The dielectric constant (ε′) and dielectric loss (ε″) values of the doped films in Fig. 11a, b are higher than those of the PVA/SA polymer matrix because NiO NPs have a higher dielectric constant. The increase in value indicates that the electrostatic interactions between the functional groups of PVA/SA and NiO NPs result in a more pronounced alignment of dipoles in parallel, hence enhancing dielectric polarization in the doped samples. The increased ε″ values of the doped films indicate a larger energy dissipation every cycle. The enhanced dielectric loss in these nanocomposite films is caused by the limited dipolar realignment and the influence of space charges on the internal AC conduction loss.

Relation between (a) log (ε′) and (b) log (ε″) against log (f) of PVA/SA and PVA/SA/NiO.
The dielectric relaxation mechanism was analyzed using the complex electric modulus (M*), defined by the following equation.46
where M′ and M′′ are the real and imaginary components of the complex electric modulus, respectively. The electric modulus is used to analyze conductivity relaxation by mitigating the impact of electrode polarization. M′ and M″ were determined based on the dielectric constant and dielectric loss values utilizing specific relationships.46
Figure 12a shows how M′ changes with the frequency of the electric field for PVA/SA and PVA/SA/NiO. The figure shows that M′ is very low, close to zero. This means that electrode polarization has little effect on the materials.47 Additionally, M′ grows at an exponential rate as the frequency rises. Adding NiO NPs to films lowers the value of M′ because segmental chains move and free volume increases due to temperature expansion. This makes polymer segments more mobile, which improves free carriers and conductivity.

Relation between (a) M′ and (b) M″ against log (f) of PVA/SA and PVA/SA/NiO
Figure 12b shows the change in M″ in relation to the frequency of PVA/SA and PVA/SA/NiO films. The peak displayed in M″ signifies the relaxation of mobile ion conductivity and is associated with the kinetics of ion translation. As the quantity of NiO NPs increases, the peak moves towards a higher frequency and its intensity diminishes. These results indicate that the relaxation process shows polarity. The presence of dopant nanoparticles weakens the connection at the interface of the polymeric matrix and promotes the movement of segments inside the matrix in nanocomposite films, as indicated by the shift in the modulus relaxation peak. The results above validate the actual alterations in the structural characteristics observed in previous investigations such as FTIR and x-ray.
Conclusions
This study successfully demonstrated the incorporation of NiO nanoparticles into a PVA/SA polymer blend, followed by optoelectronic investigation. The NiO nanoparticles exhibit an fcc structure, with a crystallite size that varies depending on the measurement technique, either Scherrer’s formula or the Williamson–Hall method. The HRTEM images confirm the spherical shape of the NiO nanoparticles and their aggregation behavior. The embedding of NiO NPs into the polymeric matrix of PVA/SA was evidenced by shifts and new peaks in the XRD patterns and FTIR spectra, indicating a complexation affecting both the structural and chemical properties of the blend. Optical measurements revealed an enhancement in absorption and a red shift in the absorption edge with increasing NiO content, which was related to the charge transfer complexes and polaronic states. The optical band gap decreased from 5.0 eV for pristine PVA/SA to 0.09 eV for the 8 wt.% NiO-doped film. The Urbach energy increased with increasing NiO content, supporting the formation of sub-band states between the conduction and valence bands, leading to a decrease in the optical band gap. The decrease in the optical band gap as the concentration of NiO increased is ascribed to the creation of additional energy levels inside the band structure of the polymer matrix. These states enable more efficient electronic transitions, hence reducing the energy needed to accomplish these transitions. The dielectric studies showed an increase in conductivity and dielectric constant with the addition of NiO NPs, suggesting their suitability for solid polymer electrolyte applications. The electric modulus analysis revealed a weakening of the interface between the polymeric matrix and NiO NPs, promoting segmental mobility and charge carrier movement within the nanocomposite films. The embedding of NiO NPs into the PVA/SA polymer blend resulted in significant changes in the physicochemical properties. This result shows that the increase in NiO nanoparticles enables the creation of conductive pathways within the polymer matrix, facilitating electron hopping and enhancing carrier–carrier interactions. This results in a shift of the valence band and conduction band, thereby reducing the band gap and increasing the conductivity of the PVA/SA blend, opening up potential applications in fields such as energy storage, optoelectronics, and sensing.
Data availability
All data generated or analyzed during this study are included in this published article.
References
A.D. de Oliveira and C.A.G. Beatrice, Polymer nanocomposites with different types of nanofiller, in Nanocomposites - Recent Evolutions (2018), p. 230.
J. Huang, J. Zhou, and M. Liu, Interphase in polymer nanocomposites. JACS Au 2(2), 280 (2022).
J.H. Koo, Polymer Nanocomposites (New York: McGraw-Hill Professional Publisher, 2006).
B. Nivedha, H. Mohit, M.R. Sanjay, N.S. Suresh, S. Siengchin, and P. Ramesh, Electrical properties of polymer nanocomposites, Advanced Polymer Nanocomposites. (Woodhead Publishing, 2022), pp. 73–90.
K. Müller, E. Bugnicourt, M. Latorre, M. Jorda, Y. Echegoyen Sanz, J.M. Lagaron, O. Miesbauer, A. Bianchin, S. Hankin, U. Bölz, G. Pérez, and M. Schmid, Review on the processing and properties of polymer nanocomposites and nanocoatings and their applications in the packaging, automotive and solar energy fields. Nanomaterials 7(4), 74 (2017).
M.M.Y. Zaghloul, M.M.Y. Zaghloul, and M. Fuseini, Recent progress in epoxy nanocomposites: corrosion, structural, flame retardancy and applications—a comprehensive review. Polym. Adv. Technol. 34(11), 3438 (2023).
J. Wang and L. Ye, Structure and properties of polyvinyl alcohol/polyurethane blends. Compos. B Eng. 69, 389 (2015).
C.C. DeMerlis and D.R. Schoneker, Review of the oral toxicity of polyvinyl alcohol (PVA). Food Chem. Toxicol. 41(3), 319 (2003).
Y. Wang and Y. Lu, Sodium alginate-based functional materials toward sustainable applications: water treatment and energy storage. Ind. Eng. Chem. Res. 62(29), 11279 (2023).
B. Wang, Y. Wan, Y. Zheng, X. Lee, T. Liu, Z. Yu, J. Huang, Y.S. Ok, J. Chen, and B. Gao, Alginate-based composites for environmental applications: a critical review. Crit. Rev. Environ. Sci. Technol. 49(4), 318 (2019).
G.T. Padma, T.S. Rao, and K.C.B. Naidu, Preparation, characterization and dielectric properties of sodium alginate/titanium dioxide composite membranes. SN Appl. Sci 1(1), 75 (2019).
C. Li, K. Zhang, X. Cheng, J. Li, Y. Jiang, P. Li, B. Wang, and H. Peng, Polymers for flexible energy storage devices. Progress Polym. Sci. 143, 101714 (2023).
C.G. Granqvist, Transparent conductors as solar energy materials: A panoramic review. Sol. Energy Mater. Sol. Cells 91(17), 1529 (2007).
H. Sato, T. Minami, S. Takata, and T. Yamada, Transparent conducting p-type NiO thin films prepared by magnetron sputtering. Thin Solid Films 236(1–2), 27 (1993).
G.A. Niklasson and C.G. Granqvist, Electrochromics for smart windows: thin films of tungsten oxide and nickel oxide, and devices based on these. J. Mater. Chem. 17(2), 127–156 (2007).
A. Agrawal, S.H. Cho, O. Zandi, S. Ghosh, R.W. Johns, and D.J. Milliron, Localized surface plasmon resonance in semiconductor nanocrystals. Chem. Rev. 118(6), 3121–3207 (2018).
U.K. Chime, A.C. Nkele, S. Ezugwu, A.C. Nwanya, N.M. Shinde, M. Kebede, M. Maaza, and F.I. Ezema, Recent progress in nickel oxide-based electrodes for high-performance supercapacitors. Curr. Opin. Electrochem. 21, 175 (2020).
N. Eghbalifam, M. Frounchi, and S. Dadbin, Antibacterial silver nanoparticles in polyvinyl alcohol/sodium alginate blend produced by gamma irradiation. Int. J. Biol. Macromol. 80, 170 (2015).
T. Wang, F. Zhang, R. Zhao, C. Wang, K. Hu, Y. Sun, C. Politis, A. Shavandi, and L. Nie, Polyvinyl alcohol/sodium alginate hydrogels incorporated with silver nanoclusters via green tea extract for antibacterial applications. Des. Monomers Polym. 23(1), 118 (2020).
R. Khalil, N.A. Kelany, M.A. Ibrahim, G.M. Al-Senani, and A.M. Mostafa, Linear and nonlinear optical properties of PVA: SA blend reinforced by TiO2 nanoparticles prepared by flower extract of aloe vera for optoelectronic applications. Coatings 13(4), 699 (2023).
M.A. Ibrahim, G.M. Nasr, R.M. Ahmed, and N.A. Kelany, Physical characterization, biocompatibility, and antimicrobial activity of polyvinyl alcohol/sodium alginate blend doped with TiO2 nanoparticles for wound dressing applications. Sci. Rep. 14(1), 5391 (2024).
A.M. Abdelghany, D.M. Ayaad, and S.M. Mahmoud, Antibacterial and energy gap correlation of PVA/SA biofilms doped with selenium nanoparticles. Biointerface Res. Appl. Chem 10, 6280 (2020).
M.G. Berhe and Y.T. Gebreslassie, Biomedical applications of biosynthesized nickel oxide nanoparticles. Int. J. Nanomed. 18, 4229 (2023).
Y.C. Lv, E.E. Shahri, A. Mahmoudi, R.K. Naughabi, S. Abbaspour, and R. Tayebee, Bioinspired nickel oxide nanoparticle as an efficient nanocarrier in the delivery of doxorubicin as an anti-bladder cancer drug. Inorg. Chem. Commun. 152, 110650 (2023).
S.J. Hong, H.J. Mun, B.J. Kim, and Y.S. Kim, Characterization of nickel oxide nanoparticles synthesized under low temperature. Micromachines 12(10), 1168 (2021).
S. Sagadevan and J. Podder, Investigations on structural, optical, morphological and electrical properties of nickel oxide nanoparticles. Int. J. Nanopart. 8(3–4), 289 (2015).
S. Mustapha, J.O. Tijani, M.M. Ndamitso, A.S. Abdulkareem, D.T. Shuaib, A.T. Amigun, and H.L. Abubakar, Facile synthesis and characterization of TiO2 nanoparticles: X-ray peak profile analysis using Williamson–Hall and Debye–Scherrer methods. Int. Nano Lett. 11(3), 241 (2021).
U. Duru Kamaci and A. Peksel, Enhanced catalytic activity of immobilized phytase into polyvinyl alcohol-sodium alginate based electrospun nanofibers. Catal. Lett. 151, 821 (2021).
M.S. Islam and M.R. Karim, Fabrication and characterization of poly (vinyl alcohol)/alginate blend nanofibers by electrospinning method. Colloids Surf. A 366(1–3), 135 (2010).
E. Olewnik-Kruszkowska, M. Gierszewska, E. Jakubowska, I. Tarach, V. Sedlarik, and M. Pummerova, Antibacterial films based on PVA and PVA–chitosan modified with poly (hexamethylene guanidine). Polymers 11(12), 2093 (2019).
A.M. Ismail and R.A. Nasr, Structural characterization of PVDF/PVA polymer blend film doped with different concentration of NiO NPs for photocatalytic degradation of malachite green dye under visible light. J. Appl. Polym. Sci. 139(13), 51847 (2022).
A. Kharazmi, N. Faraji, R.M. Hussin, E. Saion, W.M.M. Yunus, and K. Behzad, Structural, optical, opto-thermal and thermal properties of ZnS–PVA nanofluids synthesized through a radiolytic approach. Beilstein J. Nanotechnol. 6(1), 529 (2015).
M. Kuczajowska-Zadrożna, U. Filipkowska, and T. Jóźwiak, Adsorption of Cu (II) and Cd (II) from aqueous solutions by chitosan immobilized in alginate beads. J. Environ. Chem. Eng. 8(4), 103878 (2020).
O.S. Reddy, M.C.S. Subha, T. Jithendra, C. Madhavi, K.C. Rao, and B. Mallikarjuna, Sodium alginate/gelatin microbeads-intercalated with kaolin nanoclay for emerging drug delivery in Wilson’s disease. Int. J. Appl. Pharm. 15, 71 (2019).
S.D. Khairnar and V.S. Shrivastava, Facile synthesis of nickel oxide nanoparticles for the degradation of Methylene blue and Rhodamine B dye: a comparative study. J. Taibah Univ. Sci. 13(1), 1108 (2019).
M.W. Alam, A. BaQais, T.A. Mir, I. Nahvi, N. Zaidi, and A. Yasin, Effect of Mo doping in NiO nanoparticles for structural modification and its efficiency for antioxidant, antibacterial applications. Sci. Rep. 13(1), 1328 (2023).
Y. Qi, H. Qi, J. Li, and C. Lu, Synthesis, microstructures and UV–vis absorption properties of β-Ni (OH) 2 nanoplates and NiO nanostructures. J. Cryst. Growth 310(18), 4221 (2008).
J. Tauc, A. Menth, and D.L. Wood, Optical and magnetic investigations of the localized states in semiconducting glasses. Phys. Rev. Lett. 25(11), 749 (1970).
O.G. Abdullah and B.K. Aziz, Effect of kaolin light concentration on optical characteristic of PVA films. Asian Trans. Sci. Technol. 1(4), 12 (2011).
V. Raja, A.K. Sarma, and V.N. Rao, Optical properties of pure and doped PMMA-CO-P4VPNO polymer films. Mater. Lett. 57(30), 4678 (2003).
B. Choudhury, M. Dey, and A. Choudhury, Defect generation, d-d transition, and band gap reduction in Cu-doped TiO2 nanoparticles. Int. Nano Lett. 3, 1–8 (2013).
P. Stoller, V. Jacobsen, and V. Sandoghdar, Measurement of the complex dielectric constant of a single gold nanoparticle. Opt. Lett. 31(16), 2474 (2006).
M.A. Morsi, A. Rajeh, and A.A. Al-Muntaser, Reinforcement of the optical, thermal and electrical properties of PEO based on MWCNTs/Au hybrid fillers: nanodielectric materials for organoelectronic devices. Compos. B Eng. 173, 106957 (2019).
M.R. Anantharaman, S. Sindhu, S. Jagatheesan, K.A. Malini, and P. Kurian, Dielectric properties of rubber ferrite composites containing mixed ferrites. J. Phys. D Appl. Phys. 32(15), 1801 (1999).
S.A. Mansour, I.S. Yahia, and G.B. Sakr, Electrical conductivity and dielectric relaxation behavior of fluorescein sodium salt (FSS). Solid State Commun. 150(29–30), 1386 (2010).
M.M. Abdelhamied, A.M. Abdelreheem, and A. Atta, Influence of ion beam and silver nanoparticles on dielectric properties of flexible PVA/PANI polymer composite films. Plast. Rubber Compos. 51(1), 1–12 (2022).
N.S. Alghunaim, Effect of CuO nanofiller on the spectroscopic properties, dielectric permittivity and dielectric modulus of CMC/PVP nanocomposites. J. Market. Res. 8(4), 3596 (2019).
Acknowledgments
The authors extend their sincere appreciation to Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2024R38), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. Also, this study is supported via funding from Prince Sattam bin abdulaziz University project number (PSAU/2024/1445). Furthermore, the authors are thankful to the Deanship of Graduate Studies and Scientific Research at University of Bisha for supporting this work through the Fast-Track Research Support Program.
Funding
This research was funded by Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2024R38), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Author information
Authors and Affiliations
Contributions
All authors have the same contribution.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Alkallas, F.H., Salem, A., Al-Ahmadi, A.N. et al. Structural, Optical, and Dielectric Characterization of NiO Nanoparticles and Their Integration into PVA/SA Blends for Enhanced Nanocomposite Properties. J. Electron. Mater. 53, 6209–6220 (2024). https://doi.org/10.1007/s11664-024-11283-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11664-024-11283-7