
Abstract
We report the origin of capacity fading in Li2FeSiO4 cathode material, while addressing the challenges associated with the synthesis of phase-pure Li2FeSiO4 positive electrode materials in bulk and nanostructured geometries. The process has been optimized to remove iron oxide (Fe2O3) impurities and for effective carbon coating on a Li2FeSiO4 nanostructured core using a carbon source precursor during the synthesis process. X-ray diffraction measurements confirm the phase purity and nanocrystalline nature of synthesized materials. UV–visible spectroscopic measurements are carried out to understand the electronic/optical characteristics, and their correlation with materials’ electrochemical performance is investigated. The impact of iron charge state in an FeO4 tetrahedra configuration is correlated to the electrochemical performance together with the lithium-ion transporting states. The Jahn–Teller active FeO4 tetrahedra may show structural distortion in the sub-lattice of Li2FeSiO4 with different charge states and may be the main source of capacity fading in Li2FeSiO4 cathode material from the first cycle onwards.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lithium-ion batteries with high capacity and high energy density are attracting increasing attention for their probable applications in power electronics, hybrid electrical vehicles (HEVs) and electrical energy storage for power plants. These applications demand specific cathode materials to be explored with more than one lithium atom in one formula unit which can be extracted/inserted during charge/discharge cycles to get high energy density. The interest in silicate-based polyanionic materials, i.e. Li2TMSiO4 (TM = Fe, Mn, Co, and Ni), has grown recently because of their high theoretical capacity of about 333 mAh g−1. LiFePO4is considered as the first polyanionic material having P–O bond which makes it more stable. However, the specific capacity of this material is limited to 170 mAh g−1, and the electrical conductivity of this material is very poor, posing challenges for electron transport across the current collectors. There are efforts to find materials with higher storage capacity and better electrical conductivity of the electrode materials.1
Silicate-based polyanionic Li2TMSiO4 materials are promising, and synthesis of Li2TMSiO4 by Nyten et al.2 containing Fe showed promise as a high-energy-capacity material. Theoretically, two Li ions can be extracted, and the maximum specific capacity of ~ 333 mAh g−1 can be achieved, which is twice than that of LiFePO4. Further, Si–O bonds should make this structure highly stable.1,2,–3 Thus, Li2TMSiO4 (TM = transition metal = Fe, Mn, and Co) cathode materials seem to be very promising for high-energy and high-capacity rechargeable lithium-ion batteries. However, the phase-pure synthesis of these materials is very challenging. The commonly used synthesis processes are prone to impurities, especially transition metal oxides. These impurities are responsible for the poor electrochemical performance of these materials. In addition, the high bandgap of Li2TMSiO4 makes them insulating, and thus poor electrical conductivity through the electrode, hampering efficient electrical response. In spite of these factors, various approaches, such as nanostructuring, composites with conductive matrix such as graphene, and carbon nanotubes, are used to overcome some of the issues. However, the capacity fading is still a challenge and is commonly observed in such cathode materials.
Li2FeSiO4 falls into the family of tetrahedral structures, and several structures have been proposed in literature.4,5,6,–7 The first structure, Pmn21, was proposed by Tarte and Cahay,6 which is the most common polymorph because of its higher stability.1,3 In 1970, Li2MnSiO4 isostructural to β-Li3PO4 (synthesized by solid-state route) where LiO4, MO4 and SiO4 tetrahedra share corners only and run along the same direction was produced. Later in 1979, Setoguchi et al. reported a new orthorhombic structure with parameters a and b nearly twice in comparison to that reported by Tarte et al.7 In both structures, there were some unidentified diffraction peaks, which were indexed as impurities. Nishimura et al. reported a new monoclinic P21 (synthesized by ceramics route at 800°C) structure with larger cell parameters and reported it as γS. This structure was further corrected by Nyten et al.2 to P21/n. In this structure, FeO4 shares one edge with a LiO4 tetrahedron. In 2010, Sirisopanaporn et al.7 reported a Pmnb (γI) structure (synthesized at 900°C using ceramics route). Here, FeO4 shares two edges with LiO4 tetrahedra in this structure. Because of all these different structures and orientation of tetrahedra, there is an effect on bond lengths and its intercalation properties.3 In all these structures, SiO4 tetrahedra never share their edges because of the small Si–O bond lengths.3 Thus, Li2FeSiO4 has been synthesized by using different routes such as solid-state (ceramics route),2,3,4,5,6,–7 sol–gel,3,4,–5 hydrothermal,3,4,5,–6,8 microwave-solvothermal,3 and supercritical fluid synthesis.3. Sometimes, the reducing gaseous atmosphere of Ar with H2 or CO/CO2 is also used to suppress the oxidation of Fe2+ to Fe3+. Carbonic precursors have been used during synthesis which suppresses the crystal growth, leading to smaller-diameter nanoparticle formation,9 where Li+ ion diffusion may become easier because of smaller particle size. Further, the appropriate amount of carbonic precursor will also assist in preventing the oxidation of Fe2+ and in reduction of Fe3+.8 This carbon coating also improves the electronic conductivity of a material. The presence of impurities such as Li2SiO3 and FeOx after synthesis is also reported, which are also responsible for capacity degradation during the charge/discharge process.2,10,11,–12 The Pmn21structure is the most stable structure for Li2FeSiO4, but after cycling (delithiation of one or more Li+ ions), it changes to a more stable structure, Pmn21-mod.3 The Li+ diffusion pathway will also change during charge/discharge cycles because of such structural changes and needs to be considered for their performance.2,13,14,–15 Deng et al. also reported the presence of lithium carbonate (Li2CO3) using Fourier transform infrared (FTIR) analysis and attributed it as the impurity phase formation because of Li2FeSiO4(Fe2+) oxidation in contact with air and thus changes in LiFeSiO4(Fe3+) + Li2CO3.3
There is a very small formation energy difference between orthorhombic (Pmn21) and monoclinic (P21) structures, so both structures can be realized simultaneously during synthesis.16 In an orthorhombic structure, FeO4-SiO4 tetrahedra are oriented in the same direction, while in the monoclinic case, these take opposite direction after every unit. A common synthesis procedure is followed for both these structures, and longer heating duration is required for tetrahedra to take only one of these structures.3 An additional interesting issue is the iron tetrahedra in this system, which is Jahn–Teller-active, and the change in iron valence states while taking lithium out during charging and putting it back during discharging may cause local structural changes, and thus cause structural disturbance, hampering effective lithium insertion in the structure. This will also hamper the overall electrochemical performance. In the present work, we will emphasize some of the synthesis challenges, its impact on physical properties and the consequences of Jahn–Teller distortion on electrochemical performance of a Li2FeSiO4 system. In this work, we investigated the process of synthesizing phase-pure materials in different crystallographic geometries and evaluated their electrochemical performance. The optical studies are correlated to understand the structural correlations, such as Jahn–Teller distortion, on the electrochemical performance.
Experiment
The bulk Li2FeSiO4 was synthesized by solid-state method using lithium acetate (CH3COOLi.2H2O), iron chloride (FeCl2.4H2O), silicon acetate [Si(CH3COO)4] and silicon dioxide (SiO2) precursors. These precursors were mixed in stoichiometric ratio where only one type of Si precursor was used for each synthesis, using a ball mill at 100 rpm in ethanol medium. This mixture was dried to evaporate ethanol before annealing. First, it was heated at 300°C for 3 h in an Ar + H2 (10%) mixture. This dried mixture was ground using a mortar and pestle and further heated at 700°C and 750°C for 8 h under continuous flow of Ar + H2 (10%) gas. Here, H2 was used as a reducing agent to prevent the iron converting into Fe3+ .
Additionally, two variants of sol–gel methods were used to obtain orthorhombic (Pmn21) and monoclinic (P21) crystallographic phases of Li2FeSiO4. Lithium acetate, iron nitrate [Fe(NO3)3·9H2O] and tetra ethyl orthosilicate [Si(OC2H5)4] were used as precursors for synthesizing Pmn21crystallographic phase together with citric acid as the precursor for carbon shell coating. All precursors with a molar ratio of 2:1:1 were dissolved in a mixture of deionized (DI) water and ethanol at a ratio of 4:1 and stirred. Four to five milliliters of aqueous ammonia was added to hydrolyze the silicate precursor in the above solution and left under stirring for 1 h. Citric acid (0.02 mol) was added to the solution and stirred continuously. This solution was dried at 80°C in an oven to obtain the powder, which was ground with a mortar and pestle. The ground powder was heated at 300°C for 3 h and followed by 650°C for 8 h with intermediate grinding under continuous flow of Ar + H2 (10%).
For the synthesis of P21 crystallographic phase, lithium acetate (0.02 molar), iron nitrate (0.01 molar) and silicon acetate (0.01 molar) were dissolved in ethanol separately. These different solutions were mixed and stirred for 1 h. Lauric acid was dissolved in ethanol and used as the carbon coating agent with different molarities by mixing in the abovementioned solution and stirred for 3 h. This final solution was dried at 80°C to obtain powder, which was ground using a mortar and pestle for heating. The two-step annealing was followed by one annealing at 300°C for 3 h and another annealing at 750°C for 8 h under continuous flow of Ar + H2 (10%) gas mixture. Further, the weight ratio of 77:15:8 was used for synthesized Li2FeSiO4, with carbon super-P as a conducting agent and polyvinylidene fluoride (PVDF) as a binder. These were mixed in a mortar and pestle, and N-methyl-2-pyrrolidone (NMP) solution was used to make a slurry which was coated via the drag (dr) blade technique on aluminum foil. After coating, it was dried in an oven at 90°C for 6 h under atmospheric conditions and punched into circular shapes. Lithium as the reference anode material and LiPF6 in EC:DEC:DMC (1:1:1) solvent as electrolyte were used for fabricating coin cells. We used double-collared separators to ensure that both electrodes were not be in contact with each other.
The structural information for synthesized powders was investigated using an X-ray diffractometer (D8 Advance Bruker diffractometer) with Cu Kαincident radiation. A Bruker vertex 70 was used for Fourier transform infrared (FTIR) analysis to understand the bonding between different atoms and to ensure the presence of carbon, as used in synthesizing the carbon shell on nano-Li2FeSiO4 materials. UV–visible (UV–Vis) spectroscopy using a Carry 4000 UV–Vis spectrometer was performed on powder samples to correlate the bandgap with the electrochemical performance. The cyclic voltammetry and charge/discharge characteristics were analyzed by using an AUTOLAB Pgstat302N and a Neware battery testing system.
Results and Discussion
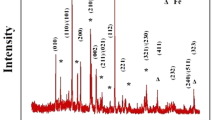
Low reactivity and chemical stability of silicon-based compounds make it difficult to synthesize phase-pure Li2FeSiO4. Figure 1 shows XRD patterns of Li2FeSiO4 synthesized using solid-state reaction route in different conditions. The material annealed at 750°C with SiO2 as Si precursor, Fig. 1a, shows a monoclinic P21 phase, where all the peaks are indexed with reference (ICDD #01-016-1988), together with minor impurity peaks. Further, pure orthorhombic Pmn21 phase (indexed with ICDD # 01-080-6279) using Si(CH3COO)4 as Si precursor and synthesized at 750°C is shown in Fig. 1c with Fe2SiO4, LiFeO2 and Li2Si2O5 impurity peaks.2,10,11,–12 Therefore, the final structure also depends on the precursors used during synthesis. The material synthesized using a similar procedure but annealed at lower temperature (700°C) showed the mixed phase, i.e. presence of both Pmn21 and P21 crystallographic phases (Fig. 1b). It was clear that the thermal treatment of the material synthesized via the solid-state route affected the crystallographic structure of the Li2FeSiO4 system.

XRD of Li2FeSiO4synthesized using a solid-state route: (a) P21 phase, (b) P21 with phase impurity and (c) Pmn21 phase with impurities.
The phase-pure nanostructured P21crystallographic phase was synthesized using sol–gel method, and the recorded XRD pattern is shown in Fig. 2a. The second sol–gel method (as discussed in the experimental sub-section) led to the formation of orthorhombic Pmn21 phase (Fig. 2b). Further, the annealing at 700°C of sol–gel-derived material led to the mixed crystallographic (Pmn21 + P21) phases (Fig. 2c). The phase-pure Li2FeSiO4 material was synthesized using the sol–gel route only, whereas solid-state reaction has always ended up with mixed phases. The crystallite size was estimated from XRD data using Scherrer's formula: particle size d = (0.9λ)/[FWHM × cos(θ)], where λ is x-ray wavelength (1.541 Å), θ is the Bragg angle and FWHM is full width at half maximum in radian for the corresponding diffraction angle. The particle crystallite sizes for sol–gel-synthesized nanostructured Li2FeSiO4 are 31 nm, 13 nm and 24 nm for orthorhombic, monoclinic and mixed-phase systems, respectively. Thus, the final structure and the purity of the Li2FeSiO4 depends on the precursor, synthesis route and annealing temperature. All these parameters need to be considered carefully to obtain a specific phase-pure material.

XRD of Li2FeSiO4 synthesized via sol–gel route: (a) P21 phase, (b) Pmn21 phase and (c) mixed phase.
We further investigated these materials using FTIR spectroscopic measurements by collecting data in transmission mode (shown in Fig. 3). The peaks in the 890–940-cm−1 range are attributed to the stretching vibration of Si–O bonds in SiO4 tetrahedra, while the peaks between 508 cm−1 and 590 cm−1 correspond to the bending vibration of Si–O bonds of SiO4 tetrahedra. The peaks in the 443–460-cm−1 range show the vibration of Li–O bonds in LiO4 tetrahedra.4,11,17,18,–19 The bandgap of these materials was investigated using UV–Vis spectroscopic measurements by collecting the diffuse reflectance measurements on powder samples. The Kubelka–Munk function \( F\left( R \right) = \frac{{\left( {1 - R} \right)^{2} }}{2R} \), where R is the reflectance of the sample, was used to calculate the absorbance. The calculated (α.hν)2 versus energy E (= hν) data were plotted to estimate the optical bandgap of these materials, as these materials are direct-bandgap systems. The measured bandgap value was 2.18 eV for Pmn21, Fig. 4b, which was lower than that of P21, which was 3.56 eV (Fig. 4d). The bandgap for mixed-phase samples was around 3.3 eV (Fig. 4f). However, the low bandgap effect also seems effective, as high absorption can be observed above 2.18 eV or near that region as well. The bandgap of the materials may affect the operating voltage conditions and thus affect the electrochemical properties.

FTIR analysis of (a) P21 phase, (b) P21 with impurity, (c) mix of P21 and Pmn21, (d) Pmn21 phase and (e) Pmn21 phase with impurities.

Cyclic voltammetry with a scan rate of 0.5 mVs−1and UV analysis for the bandgap of (a) and (b) Pmn21 phase, (c) and (d) P21 phase and (e) and (f) mix phase.
Cyclic voltammetry measurements were carried out to understand the electrochemical performance, and the data is shown in Fig. 4. Li2FeSiO4 material with Pmn21 phase in the first cycle during charging showed two oxidation peaks at 2.95 V and 3.42 V, corresponding to Fe2+ to Fe3+ and Fe3+ to Fe4+transitions, respectively. The measurements were repeated for the second cycle, whereas in subsequent cycles, one peak started fading and only one oxidation peak was noticed at lower voltages. This supports that both Li+ ions in Li2FeSiO4 were delithiated in the first two initial cycles at respective voltages, and later only one Li+ ion delithiation could be realized. There was only one peak at ~ 2.6 V in the reduction cycle, corresponding to the insertion of one Li+ ion back into Li2-xFeSiO4cathode material. Thus, we observed that two Li+ ions were delithiated in the first two cycles and further reduced to only one Li+, as evident from only one oxidation peak after two cycles (Fig. 4a). Further, Li2FeSiO4 cathode material with a P21 space group and monoclinic crystallographic phases showed only one oxidation peak in the first cycle at 3.68 V, which corresponded to the transition of Fe2+ into Fe3+ , whereas after the first cycle, the two oxidation peaks around 3.31 V and 3.68 V were observed, corresponding to Fe2+ to Fe4+ transitions, successively (Fig. 4.2b). However, only one peak around 3.15 V was noticed in reduction during discharging and can be attributed to one Li+ insertion back into the Li2FeSiO4 cathode material. In the mixed orthorhombic and monoclinic crystallographic phases, Li2FeSiO4 cathode material initially showed an oxidation peak at 3.94 V during charging, which disappeared after one cycle. This can be attributed to the phase change or segregation of Li2FeSiO4 after one cycle. However, in contrast, one oxidation peak was observed at 3.23 V, and two reduction peaks at 2.45 V and 1.84 V were observed in successive cycles (Fig. 4e). These results were correlated to the electronic bandgap of the materials as oxidation peaks were observed at higher voltage for the monoclinic P21 phase, whose bandgap is higher than that of orthorhombic Pmn21 phase. The response of the mixed phase was somewhat intermediate, and higher oxidation voltage was observed in the first cycle and attributed to the presence of a higher-bandgap monoclinic phase, which shifted to the lower oxidation/reduction voltages, suggesting the participation of the orthorhombic phase.
The charge/discharge characteristics for orthorhombic Pmn21 Li2FeSiO4 phase are shown in Fig. 5a, and the specific charge and discharge capacities were 130 mAh g−1 and 98 mAh g−1 for first charge/discharge cycle, respectively. This cathode material was relatively stable under charge/discharge cycling, as shown in Fig. 5b for 10 cycles. Further, the charge/discharge specific capacities were 8.79 mAh g−1 and 5.68 mAh g−1in first charge/discharge cycle for P21 Li2FeSiO4phase cathode material shown in Fig. 5c, which is much lower than orthorhombic Pmn21 Li2FeSiO4 cathode material. However, these values are very low with respect to theoretical capacity for Li2FeSiO4 cathode material, which is about 333 mAh g−1. The noticed lower specific capacity suggests that complete delithiation and lithiation were not achieved and about 0.6 Li for Pmn21is taking place in place of two Li+ ions, which is extremely low or negligible for P21 phase and remains very low even after various charge/discharge cycles (Fig. 5d). This suggests that the monoclinic phase is not transformed into orthorhombic phase after different charge/discharge cycling where better performance is expected.19,20 We further explored the electrochemical performance of mixed-phase Li2FeSiO4 material and observed 82 mAh g−1 and 64 mAh g−1 specific capacities in the first charging and discharging cycle (Fig. 5e). The cyclic stability for mixed phase is shown in Fig. 5f, suggesting the stability during charge/discharge cyclings. These specific capacity values lie between orthorhombic and monoclinic Li2FeSiO4 cathode materials, suggesting that only contribution is coming from orthorhombic Li2FeSiO4 phase and that the monoclinic phase is not contributing in electrochemical performance. The reduction in the capacity may be equivalent to respective fraction of the monoclinic phase. Thus, the present studies suggest that a moderate, however much lower than the theoretically predicted, electrochemical performance can be achieved, and the poor performance is attributed to the structural instability in these systems (Fig. 5).

Galvanostatic charge/discharge performance and cyclability for Pmn21 phase in (a) and (b), P21 phase in (c) and (d), and mixed phase in (e) and (f) of nanostructured Li2FeSiO4 phases.
The observed much lower specific capacities for Li2FeSiO4 cathode material is a subject of intensive exploration, as either smaller capacities (extraction/insertion of only 50% or lesser lithium atoms) or large capacity fading is observed not only in this system but also in similar systems such as Li2MnSiO4 systems.15,21 Thus, it seems that bulk and localized crystal structures are responsible for such capacity fading. Here, iron forms tetrahedra with oxygen, i.e. FeO4, which shares an edge or corner with SiO4 tetrahedra, yet the complete crystallographic structure is not very clear.15,19,20,22,23 The 2+ oxidation state of iron leads to the 3d6 electronic configuration, and as per Hund’s rule, the initial configuration will prefer to be in the high-spin state, thus making FeO4 Jahn–Teller (J–T)-active and likely exhibiting corresponding transitions.
We also plotted differential capacity (dQ/dV) versus voltage for cycles 1, 5, 9 and 10 in the entire potential window (Fig. 6), similar to Loveridge et al.’s analysis carried out for anode material.24 The clear peaks at two different voltages are observed in the first half cycles with a broad peak at ~ 3.0 V together with a peak at ~ 4.0 V while charging the cathode material. The first peak at 3.0 V suggests 50% of delithiation where Fe changes from the +2 to +3 state, whereas the peak at 4.0 V signifies the removal of more than 50% lithium where Fe starts changing from the +3 state to the +4 charge state to ensure the electrical neutrality of the cathode material. The process at +4 V has reduced significantly or nearly disappeared in later cycles, suggesting the difficulty in moving lithium again inside the matrix of cathode material. This is attributed to the probable J–T distortion, causing localized changes in the lattice structure and not allowing the additional lithium inside the cathode material. In the remaining lithiation/delithiation cycles, the peak is nearly not showing much change in position in both charging/discharging cycles, and the process limits lithium up to 50% in the cathode material. This is consistent with the observed nearly constant capacity in later cycles. The presence of a broad peak near 3.0 V may indicate simultaneous delithiation, contributing to different valence states of Fe. Thus, there is a clear evidence of absence of the +4 state of Fe during the delithiation process after the first cycle, suggesting J–T distortion causes change in the structure locally and thus hinders in achieving the maximum electrochemical capacity of the investigated cathode material.

Differential capacity versus voltage for different cycles for Pmn21 phase, derived from Fig. 5a; vertical lines are marked for peak identification.
Further, we also plotted absorption versus energy data in Fig. 7 together with two probable transitions at 2.04 eV (E1) and 3.03 eV (E2).25 The transition states are clearly visible for P21, and relatively less obvious for Pmn21 and mixed-phase materials, suggesting the strong J–T effect in P21 phase. Further, during charge/discharge cycles, the charge state of iron changes from 2+ to 3+ for the first half-lithium extraction from the cathode, leading to the localized deformation in the system. The situation will become more complicated while extracting additional, i.e. > 50%, lithium from the cathode material, where iron will start changing its charge state from 3+ to 4+, causing additional localized distortion. The active Jahn–Teller distortion in these systems may lead to partially irreversible lithium insertion in the cathode material and thus hamper the electrochemical performance. The observed high degree of J–T signature in P21 phase seems to be the main source of poor electrochemical performance. Further, the system may show stable charge/discharge cyclability for less than 50% lithium extraction, and may lead to large capacity fading for higher lithium extraction/insertion.

Optical absorption versus energy for Pmn21, P21 and the mixed phase together with two dashed lines, indicating the probable iron transitions in the different valence states.
Conclusion
Li2FeSiO4 material is synthesized in two crystallographic, i.e. orthorhombic and monoclinic, phases using solid-state and sol–gel techniques. The solid-state method resulted in secondary phases and thus was unable to synthesize the phase-pure materials. However, the sol–gel method resulted in phase-pure orthorhombic and monoclinic phases by varying the post-synthesis thermal treatment conditions. We also synthesized mixed-phase materials as well. The cathode material in the orthorhombic phase showed relatively higher specific charge capacity of ~ 130 mAh g−1, which is equivalent to 0.6 Li+ extraction/insertion material but much lower than that of the theoretical specific capacity values. The very poor electrochemical performance is observed in case of the monoclinic phase, whereas the mixed phase also showed relatively better (~ 60 mAh g−1) specific capacity. The disappearance of the 4.0-V differential capacity peak also suggests localized structural changes in Li2FeSiO4 cathode material, which is associated to J–T distortion in such multivalent transition metal-based materials. Thus, the probable reason for the lower capacity or the capacity fading after the first cycle is attributed to the localized structural distortion because of Jahn–Teller distortion in FeO4 tetrahedra in the Li2FeSiO4 system, suggesting it may be an intrinsic characteristic in such cathode materials.
References
A.K. Padhi, K.S. Nanjundaswamy, C. Masquelier, S. Okada, and J.B. Goodenough, J. Electrochem. Soc. 144, 1609 (1997).
A. Nytén, A. Abouimrane, M. Armand, T. Gustafsson, and J.O. Thomas, Electrochem. Commun. 7, 156 (2005).
G. Mali, C. Sirisopanaporn, C. Masquelier, D. Hanzel, and R. Dominko, Chem. Mater. 23, 2735 (2011).
C. Sirisopanaporn, R. Dominko, C. Masquelier, A.R. Armstrong, G. Mali, and P.G. Bruce, J. Mater. Chem. 21, 17823 (2011).
M. Setoguchi, S. Kobayashi, H. Yamaguchi, C. Sakamoto, and K. Akatsuka, Mem. Osaka Kyioku Univ. Ser. III 28, 27 (1979).
P. Tarte and R. Cahay, C. R. Acad. Sci. Paris C 271, 777 (1970).
C. Sirisopanaporn, A. Boulineau, D. Hanzel, R. Dominko, B. Budic, R. Armstrong, P.G. Bruce, and C. Masquelier, Inorg. Chem. 49, 7446 (2010).
D. Lv, J. Bai, P. Zhang, S. Wu, Y. Li, W. Wen, Z. Jiang, J. Mi, Z. Zhu, and Y. Yang, Chem. Mater. 25, 2014 (2013).
N. Yabuuchi, Y. Yamakawa, K. Yoshii, and S. Komaba, Dalton Trans. 40, 1846 (2011).
G. Haegawa, M. Sannohe, Y. Ishihara, K. Kanamori, K. Nakanishi, and T. Abe, Phys. Chem. Chem. Phys. 15, 8736 (2013).
H. Hao, J. Zhang, X. Liu, T. Huang, and A. Yu, Int. J. Electrochem. Sci. 8, 10976 (2013).
S. Sun and Z. Wu, J. Chem. Pharm. Res. 6, 221 (2014).
X. Lu, H. Wei, H.C. Chiu, R. Gauvin, P. Hovington, A. Guerfi, K. Zaghib, and G.P. Demopoulos, Sci. Rep. 5, 8599 (2015).
P. Vjeeston and H. Fjellvag, RSC Adv. 7, 16843 (2017).
H.N. Girish and G.Q. Shao, RSC Adv. 5, 98666 (2015).
Y. Hong, Z. Ying, and C. Xuan, J. Electrochem. 19, 565 (2013).
D. Xiao, L. Shao, R. Ma, M. Shui, J. Gao, F. Huang, K. Wu, S. Qian, D. Wang, N. Long, Y. Ren, and J. Shu, Int. J. Electrochem. Sci. 8, 7581 (2013).
A. Saracibar, A. Van der Ven, and M.E. Arroyo-de Dompablo, Chem. Mater. 24, 495 (2012).
C. Sirisopanaporn, C. Masquelier, P.G. Bruce, R.A. Armstrong, and R. Dominko, J. Am. Chem. Soc. 133, 1263 (2011).
R.A. Armstrong, N. Kuganathan, M.S. Islam, and P.G. Bruce, J. Am. Chem. Soc. 133, 13031 (2011).
P. Babbar, B. Tiwari, B. Purohit, A. Invanishchev, A. Churikov, and A. Dixit, RSC Adv. 7, 22990 (2017).
A. Boulineau, C. Sirisopanaporn, R. Dominko, A.R. Armstrong, P.G. Bruce, and C. Masquelier, Dalton Trans. 39, 6310 (2010).
Y. Wang, D. Su, and G. Wang, Acta Phys. Pol. A 123, 279 (2013).
M.J. Loveridge, M.J. Lain, I.D. Johnson, A. Roberts, S.D. Beattie, R. Dashwood, J.A. Darr, and R. Bhagat, Sci. Rep. 6, 37787 (2016).
S.L. Reddy, T. Endo, and G.S. Reddy, Electronic (Absorption) Spectra of 3d Transition Metal Complexes, Chapter 1 in Advanced Aspects of Spectroscopy, Ed. M. A. Farrukh (InTech Open 2012) https://www.intechopen.com/books/advanced-aspects-of-spectroscopy.
Acknowledgments
Author Ambesh Dixit acknowledges the Department of Science and Technology (DST) through project #INT/RUS/RFBR/320, and author Alexander Ivanishchev acknowledges the Russian Foundation for Basic Research (Projects # 18-53-45004 and # 16-33-00328) for financial assistance to carry out this research work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Babbar, P., Tiwari, B., Ivanishchev, A.V. et al. Capacity Fading in Li2FeSiO4 Cathode Material: Intrinsic or Extrinsic. J. Electron. Mater. 50, 1059–1066 (2021). https://doi.org/10.1007/s11664-020-08620-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11664-020-08620-x