
Abstract
In this work, the kinetics of decarburization and demanganization of Fe–15Mn–1C alloy by bubbling mixtures of Ar–O2 into the melt at 1823 K was studied. Experiments were conducted at total gas flow rates of 200 and 300 Nml/min and gas mixtures of Ar containing 6.7 to 20 pct O2. Increasing the gas flow rate and oxygen in the gas mixture resulted in higher overall rates of decarburization and demanganization. However, the experiments with the lowest oxygen concentration were the most efficient in terms of oxygen utilization for decarburization. The ratio of manganese loss to decarburization was found to be controlled by the relative mass transport of manganese and carbon in the metal. Based on the estimated mass transfer coefficient for either carbon or manganese, the reaction time for each bubble was estimated to be 0.001 seconds which is about 1 pct of the residence time of the bubble in the liquid. Although the initial competition for oxygen between manganese and carbon was controlled by relative mass transport rates, this work found no evidence that manganese and carbon repartitioned towards the equilibrium over the remaining lifetime of the bubble.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Advanced high-strength steels (AHSS) rely heavily on the manganese content as an important alloying element. High manganese twinning-induced plasticity (TWIP) steels with 15 to 30 pct Mn[1,2] attracted the automotive industry’s interest due to their outstanding combination of strength and elongation. Since the 1980s, the properties of this grade of steels have been researched extensively; a review of these studies is summarized by Bouaziz et al.[3] However, less attention was paid to the processing of these steels. Currently to the knowledge of the authors only POSCO[4,5] and Thyssen Krupp[6] have commercialized the production of high manganese steels containing up to 26 pct Mn and 0.2 to 0.6 pct C. The important challenge for steelmakers is to reduce carbon content while minimizing the manganese losses during the oxygen refining process because high temperature and the presence of oxygen can lead to excessive manganese losses.[7,8] The yield of Mn in the Argon Oxygen Decarburization (AOD) processing of high carbon ferromanganese is reported to be 92 pct and higher.[9] Hence, AOD can be an appropriate route to produce high manganese steels. AOD is broadly researched experimentally and mathematically for the behavior of chromium in stainless steels.[10,11,12,13,14,15,16,17,18,19,20] For stainless steel, there is a critical carbon content Ccrit above which no chromium is oxidized and the rate of decarburization is controlled by the supply of oxygen. Below Ccrit, chromium oxidizes, and mass transfer of carbon in the liquid controls the decarburization rate.[21] For AOD processing of stainless steel, Krivsky[22] proposed that argon must be injected deep in the melt to be dispersed thoroughly to be efficient in gas dilution. This finding was later confirmed by Saccomano et al.[23] for plant data and by Fruehan[24] in laboratory experiments. In Fruehan’s study,[24] a mixture of Ar–O2 was injected into a shallow bath of 7.5 cm of stainless steel containing 18 pct Cr-8 pct Ni-0.5 pct C. He reported that the ratio of the rates of Cr to C removal was nearly identical to their concentrations in the liquid despite the fact that thermodynamics heavily favored CO formation. Fruehan interpreted these results to suggest that chromium oxidized more rapidly than carbon because of faster mass transport. Due to the short residence time of the bubble in the bath, there was insufficient time for reduction of chromium oxide (Cr2O3) by C to bring the Cr2O3/CO back towards the equilibrium. These findings were the basis of a model for chromium recovery in AOD[11] wherein it was assumed that in the vicinity of the tuyeres, chromium is mainly oxidized and as the bubbles ascend Cr2O3 at the surface of the bubbles is reduced by dissolved carbon. The current work aims to determine whether during AOD refining, the competition for oxygen between manganese and carbon follows the same mechanism as that of chromium and carbon.
Compared to stainless steel, there are only a few published studies on the oxygen refining of high manganese alloys.[25,26,27,28,29,30] Yamamto et al.[25] studied the simultaneous decarburization and manganese loss in Ar–O2 refining of Fe–76Mn–6.8C– melts in a pilot converter with top lance O2 blowing and bottom plug Ar or Ar-O2 injection. Carbon and manganese concentrations changed in three stages with respect to time. First, there was manganese loss, followed by a reversion stage and then a final rapid manganese loss. Initially, the rate of carbon removal was slow, proceeding to a higher rate and finally slowing to an almost negligible rate. Manganese loss was reported to be due to oxidation and evaporation, however, the kinetics of decarburization and manganese losses were not addressed. Lee et al.[26] analyzed Yamamoto’s data and proposed that the rate-controlling steps for decarburization were chemical reaction at the gas-liquid interface in stage 1, mass transfer of oxygen in the gas phase during stage 2, and the mass transfer of carbon in the liquid phase in stage 3. These researchers reported the excess oxygen enhanced manganese evaporation to form MnO fume according to the mechanism of Turkdogan et al.[31] However, no mention was made of metal phase mass transfer control in reference to manganese which makes sense at very high manganese concentrations.
You[27] studied decarburization of Fe–75Mn–7C–0.3Si by combined top and bottom Ar-O2 blowing in an 87 kg AOD converter. Decarburization proceeded quickly initially until reaching approximately 2 pct at which point demanganization started and decarburization ceased. Later, You et al.[28,29] studied the decarburization of Fe–75Mn–6C using Ar–O2 injection in a laboratory setup reporting that the utilization of oxygen for decarburization increased remarkably with increasing temperature and decreasing fraction of oxygen in the gas mixture. This is in accordance with thermodynamic predictions that at higher temperatures, CO formation is favored over MnO. In a further study, these workers[30] conducted oxygen refining of high carbon ferromanganese in a 2-ton AOD converter with combined blowing. The reported total manganese loss by oxidation and evaporation was between 2 and 25 pct. In contradiction to the previous work of this group,[28,29] they recommended lower temperatures to avoid evaporative loss of manganese.
Liu et al.[32] examined CO2-O2 injection into a Fe–16Mn–3C alloy showing that the dilution of O2 with CO2 enhances the rate of decarburization while retaining manganese in the bath. These workers did not offer a detailed analysis, proposing that manganese loss was due to evaporation and oxidation.
Despite several studies of argon-oxygen refining of high carbon ferromanganese, no full picture of the behavior of manganese in AOD is presented in the published literature. It might be expected that manganese and chromium behave similarly in AOD refining but researchers have highlighted the role of MnO fume formation which might lead to some differences. As proposed by Fruehan for AOD refining of stainless steel the Cr2O3 layer formed at the bubble-melt interface is reduced by carbon during the bubble rise through the melt. In the case of manganese, if the oxide is formed as a fume inside the bubble it may not be as accessible for reduction by carbon. Furthermore, there is little discussion in the Mn literature regarding the concept of a critical carbon concentration.
Previous work by the authors[33] focused on the kinetics of manganese loss and decarburization in Ar–O2 bubbling into Fe–Mn–C alloys containing 0.05 to 0.42 pct C and 5 to 25 pct Mn at 1823 K. The rate of total manganese loss and the competition between carbon and manganese for oxygen appeared to be at least partly controlled by the relative rates of manganese and carbon transport in the liquid metal. This observation agrees with the findings of Fruehan for Cr–C alloys. The current work will explore further, the nature of the competition between manganese and carbon for oxygen. Furthermore, the authors’ previous work[33] for alloys with between 0.05 and 0.42 pct C, showed that the total manganese loss could only be explained by invoking an evaporation-condensation mechanism. The current work will determine if this mechanism operates when the carbon concentration is increased to 1 pct.
In summary, the current work will address gaps in the published literature regarding the behavior of Mn in AOD refining. Specifically, this work will investigate the competition between Mn and C for O2 including, the role of relative mass transport rates in the metal, the possibility of oxide fume formation via Mn vapor, and the existence or otherwise of critical carbon content. The authors will also examine whether their previously proposed mechanism for Mn evaporation–condensation continues to operate at higher C contents.
Experimental Method
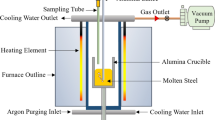
The experimental method identical to the previous work used by authors,[33] is summarized here for the convenience of the readers. The steel composed of 15 pct Mn and 1 pct C was prepared by mixing electrolytic iron powder (99.977 pct), manganese flakes (99.990 pct), and graphite (99.9999 pct). To remove the oxide layer from manganese flakes, a 5 pct HCl solution was used for acid pickling before the mixing. 330 g of the steel mixture was placed in an alumina crucible with the outer and inner diameter of 4 and 3.8 cm and a height of 9 cm and positioned in the hot zone of the furnace. A vertical resistance furnace with an alumina tube of an inner diameter of 7.9 cm and a height of 76.2 cm was used as shown in Figure 1. The furnace was sealed using O-rings and water-cooled stainless steel caps at both ends. The metal was melted under an argon atmosphere that before entering the furnace was passed through a column of anhydrous CaSO4. To ensure that the furnace was fully sealed before each experiment it was evacuated using a vacuum pump and backfilled with argon. The crucible was then heated in the furnace to the target temperature of 1823 K (1550 °C) and held for one hour to homogenize the melt. The temperature was controlled within ± 8 K using a B-type (Pt30Rh–Pt6Rh) thermocouple. At this temperature, the system was evacuated and backfilled with argon again. Then the entrance and exit gas were opened and, the nozzle was lowered into the melt where the height of the bath above the tip of the nozzle was 3 cm. Then, a mixture of Ar–O2 was injected into the melt through a single bore alumina tube with the outer and inner diameters of 0.48 cm and 0.16 cm, respectively. The total flow rates were 200 and 300 Nml/min and the gas mixture varied from 6.7 to 20 pct. It should be noted that for each experiment, the gas flow rate and composition were constant. The metal samples were taken frequently and were analyzed by ICP-OES for manganese content and by LECO for carbon content.

Schematic diagram of the experimental setup used in this work-not to scale
Results
Effect of Gas Composition and Flow rate on Decarburization and Manganese Loss
To investigate the effect of gas composition on the rate of decarburization and demanganization, experiments were conducted at 1823 K using Ar–O2 mixtures containing 6.7 to 20 pct O2 at total flow rates of 200 and 300 Nml/min. The results are shown in Figures 2 and 3. For both gas flow rates, the decarburization rate increased with increasing oxygen in the gas mixture. This is in agreement with studies by Fruehan[11] for AOD processing of Fe–11Cr–1.2C alloys but contrary to the finding of You et al.[28,29] for decarburization of Fe–75Mn–6C alloys.

Variation of carbon concentration of Fe–15Mn–1C alloy with time at 1823 K for the flow rates of (a) 200 Nml/min, (b) 300 Nml/min, variable gas composition

Variation of manganese concentration of Fe–15Mn–1C alloy with time at 1823 K for the flow rates of (a) 200 Nml/min, (b) 300 Nml/min, variable gas composition
Figure 3 shows data for the effect of gas composition on the rate of demanganization of Fe–15Mn–1C alloy. As found in previous work by the authors[33] demanganization proceeds in three stages. For lower flow rates and lower oxygen in the gas mixture, manganese loss in stage 1 is discernible but low, making it difficult to distinguish between stage 1 and stage 2. With increasing oxygen in the gas mixture, the rate of demanganization increased in stages 1 and 3. This finding is consistent with the reported data by You et al.[29] for Fe–75Mn–6C and Liu et al.[32] for Fe–16Mn–3C alloys.
From the comparison of the slopes of the rate plots in Figures 2(a) and (b), it is seen that the rate of decarburization increases with the flow rate. The corresponding curves for manganese presented in Figure 3 show that the demanganization rate also increases with flow rate. Quantitative comparison of the rates (Table I) shows that for the same gas composition the rate increases in direct proportion to the total flow rate. This is in agreement with the work of Liu et al.[32]
Discussion
Thermodynamic Assessment
To compare the experimental data with thermodynamic predictions, the equilibrium composition of liquid metal and gaseous and oxide products was calculated using FactSage 8.0, with FactPS, FToxid, and FSstel databases in the Equilib module. For these calculations, the input amount and composition of the metal, gas flow rate, and gas composition, were chosen to be the same as used in the experiments presented above. It was assumed that the temperature remained constant at 1823 K during the processing time.
Figure 4 shows the calculated changes in the carbon and manganese concentration in the bath at different oxygen levels in the gas mixture for a flow rate of 200 Nml/min. The prediction is entirely consistent with the concept of critical carbon content showing a two-stage trend. Above the critical carbon content, no manganese is predicted to be oxidized, and all oxygen is consumed for decarburization. Below the critical carbon concentration, most of the oxygen is predicted to be consumed by the oxidation of manganese and decarburization is much slower.

FactSage prediction for (a) carbon concentration vs. time, and (b) manganese concentration vs. time at a total gas flow rate of 200 Nml/min, variable: oxygen concentration in the gas mixture
A critical finding of the current work is that, although Figures 4(a) and (b) are essentially thermodynamic predictions of the data presented in Figures 2(a) and 3(a), respectively, there is no agreement between the prediction and the experimental observations. This is definitive proof that although the reaction between injected bubbles and the melt is expected to be very fast, the injected bubbles do not achieve equilibrium with the bulk melt.
Rate of Decarburization and Demanganization
Figure 5 shows that the rates of decarburization normalized for oxygen flow rate versus the oxygen percentage in the gas mixture are independent of the total gas flow rate. With increasing oxygen concentration in the gas mixture, the overall rate of decarburization is increased, however, the efficiency of oxygen for decarburization decreases. In the work of You et al.[28] for Fe–75Mn–6C alloys, the rate of decarburization decreased with increasing oxygen concentration in the Ar–O2 mixture. These workers stated that by lowering the O2/(Ar + O2) ratio, the equilibrium carbon concentration at the liquid-bubble interface is decreased. The difference in Mn and C concentration compared to the current work could be one reason for this apparent contradiction.

The rate of decarburization of Fe–15Mn–1C alloys versus percent oxygen in the gas mixture normalized for oxygen flow rate
In Figure 6, the rate of manganese loss is normalized for oxygen flow rate (dMn/dO2) and plotted versus oxygen concentration in the gas mixture. In stage 1, dMn/dO2 increased linearly with oxygen concentration in the gas mixture, but it is hardly affected in stage 3. It is interesting to note that while the oxygen that goes to carbon is nearly constant with time, in terms of oxygen that goes to manganese there is a significant difference between stages 1 and 3. It seems that carbon oxidizes at an almost constant rate and the competition for what is left is between Fe and Mn. In stage 2, it would appear that either all the oxygen went to Fe or that Fe and Mn oxidized in proportion to their concentration in the alloy, giving the appearance of no manganese loss. With increasing gas flow rate, the increased oxygen supply resulted in the observed higher rate of manganese loss (Table I). However, in both stages, the normalized rates of demanganization were independent of flow rate (Figure 6). For all oxygen levels, the rates of demanganization were lower in stage 1 than in stage 3. This observation is similar to previously reported data by the authors[33] for Fe–15Mn–0.42C alloy. However, for Fe–15Mn–0.05C and Fe–15Mn–0.18C the rates of demanganization were higher in stage 1. It has been shown that with decreasing carbon concentration in the alloy, the contribution of evaporative loss to overall demanganization increased. To facilitate discussion demanganization data from the authors’ previous work[33] are shown in Figure 7 along with the equivalent data from the current study.

The rate of demanganization of Fe–15Mn–1C alloys in stages 1 and 3 versus percent oxygen in the gas mixture normalized for oxygen flow rate

Manganese concentration in the bath versus time for Fe–15Mn–C alloys, Gas flow rate: 300 Nml/min and gas composition Ar–6.7 pct O2
In stage 1, the data show that for a higher carbon concentration in the melt the rate of demanganization is decreased. In stage 3, for lower carbon alloys (< 0.42 pct), the rate of demanganization is the same regardless of carbon content. However, at 1 pct carbon, demanganization in stage 3 is much slower than that for the lower carbon alloys. These observations suggest that the role of carbon in competing for oxygen is more significant during stage 1, and nonexistent in stage 3 for lower carbon alloys, but for 1 pct C alloy is very significant. In order to understand the factors in play, it is worth analyzing how oxygen is partitioned between the species in the system.
Oxygen Partitioning
The objective of this section is to determine the way in which oxygen partitions among CO, CO2, MnO, and FeO. It is reasonable to assume that the products of oxidation are primarily CO and MnO, so the efficiencies of oxygen for decarburization and manganese removal are the fraction of oxygen consumed for CO and MnO formation to the total supplied oxygen. While this assumption is consistent with previous findings of the authors,[33] examination of Table II shows that based on this assumption, a considerable amount of oxygen would remain unconsumed for the case of Fe–15Mn–1C alloy. This is especially true in stage 1 of the process. In Fruehan’s work[24] on Fe–Cr–C alloys, 50 pct of the oxygen was consumed for the oxidation of Cr and C. He stated that the rest of the oxygen might be consumed for the oxidation of Fe or leave the system unreacted. The latter does not seem to be realistic. To understand how the available oxygen is distributed to each species in this process, a more refined calculation is conducted below. It employs a mass balance, some assumptions about the relative rates of mass transfer of carbon and manganese, and some assumptions of local equilibrium at the metal-gas interface. The following detailed assumptions are made in calculations:
-
1.
Given the excess oxygen relative to carbon and manganese in the bubble it is assumed that all the manganese is lost as MnO. (Note: approximately 3 to 5 pct of the manganese loss is likely from Mn vapor, however, for the convenience of the calculation, it is assumed that the only mechanism responsible for manganese loss in the current work is oxidation to MnO)
-
2.
All oxygen is consumed for oxidizing C, Mn, and Fe and the products are CO, CO2, MnO, and FeO.
-
3.
During the period in which carbon and manganese are actively being removed at the bubble surface, the following is true:
-
(a)
The competition between Mn and C is controlled by their relative rates of mass transport in the metal; there is neither gas-phase nor slag-phase control.
-
(b)
In keeping with the assumption of control by mass transport in the metal, all reactions are assumed to be in local equilibrium at the gas-metal interface.
-
(c)
MnO and FeO form slag at the gas-metal interface which according to Reference 34 is an ideal solution, therefore the sum of their activities is equal to 1.
-
(d)
Again, during the active removal of manganese and carbon, the interfacial concentrations of manganese and carbon will be negligible in comparison to iron so at the gas-metal interface aFe \( \cong \) 1.
-
(a)
-
4.
After the active reaction is over it is expected that the melt composition at the gas-melt interface would recover to the bulk composition. It is worth noting that experimental observation suggests no change in the composition in the bubble in response to the change in the composition at the surface.
Based on the preceding assumptions, the primary equations required to describe the oxidation reactions are:
By simultaneously solving Equations 1 to 6, the values of \( n_{\text{CO}} \), \( n_{{{\text{CO}}_{2} }} \), \( a_{\text{MnO}} \), \( a_{\text{FeO}} \), \( a_{\text{C}}^{i} \), and \( a_{\text{Mn}}^{i} \) are obtained (see Tables III and IV for stages 1 and 3). The calculated partitioned oxygen among CO, CO2, MnO, and FeO is provided in Figure 8. The total partitioning is 100 pct because Fe is assumed to react with the oxygen that did not react with Mn or C. There is much more CO than CO2 in the gas, reflective of equilibrium with the melt interface. The various conditions in Figure 8 demonstrate that increasing oxygen in the gas mixture resulted in less CO and more FeO, CO2, and MnO in both stages 1 and 3. Although the overall rate of decarburization increased with increasing oxygen in the gas mixture, gas mixtures containing lower oxygen were more efficient in decarburization and resulted in less FeO and MnO. The trend for CO is in agreement with the work of You et al.[28] for the bottom injection of Ar–O2 into high carbon ferromanganese and Dey et al.[35] for O2–N2 and Ohno et al.[36] for O2–Ar blowing into Fe–Cr–C melt.

The calculated partitioned oxygen among MnO, CO, CO2, and FeO for Fe–15Mn–1C alloy in (a) stage 1-total gas flow rate 200 Nml/min, (b) stage 1-total gas flow rate 300 Nml/min, (c) stage 3-total gas flow rate 200 Nml/min, (d) stage 3-total gas flow rate 300 Nml/min
Mass Transfer Coefficients for Manganese and Carbon
The mass transfer coefficients for C and Mn in the liquid were investigated using an approach suggested by Fruehan[24] where the ratio of rates of removal was almost equal to the concentration ratios. Likewise, in this work, it is assumed that the molar ratio of the rates of demanganization to decarburization is equal to the ratio of the flux of Mn to C in the liquid (Eq. [7]). The concentrations are written based on activities. The activity coefficient for carbon and manganese are calculated by Lee’s[37] thermodynamic model for the liquid Fe–Mn–C system. For manganese, the activity coefficient is approximately 1 regardless of the carbon content. For carbon, only the activity coefficient of the bulk matters (0.71 and 0.67 for stages 1 and 3, respectively). Therefore, the values obtained for \( a_{\text{Mn}}^{i} \) and \( a_{\text{C}}^{i} \) from Section IV–C, were substituted in Eq. [7], and the ratio of mass transfer coefficients \( k_{\text{Mn}} /k_{\text{C}} \) calculated for each gas flow rate and composition. This ratio and the interfacial concentrations of Mn and C are listed in Tables III and IV for stages 1 and 3 of the process.
JMn and JC are the flux of Mn and C in the liquid in units of mol/cm2 s. The mass transfer coefficients of Mn and C are represented as kMn and kC in units of cm/s. The superscript “i” and “b” denote the interface and bulk, respectively.
Compared to the concentration of Mn and C in the bulk liquid, \( 2 \times 10^{ - 2} \) and \( 6 \times 10^{ - 3} \) mol/cm3, the gas-liquid interface concentrations can be considered negligible. This means that there is a steep concentration gradient in the liquid boundary layer. This observation confirms the assumption that at the point where the partitioning of oxygen to the different species is defined, the gas bubble is in equilibrium with the gas-metal interface but far from equilibrium with the bulk metal. This seems quite likely during the period where manganese and carbon are being transported to the bubble to react with oxygen. However, as will be discussed in the next section, the period during which this is occurring appears to be a short fraction of the overall bubble lifetime, further suggesting that the concentrations of manganese and carbon at the bubble-melt interface will recover to their bulk values within the lifetime of the bubble. This in turn might lead one to expect a redistribution of elements in the bubble approaching equilibrium with the melt. Clearly, this did not happen as shown in Section IV–A. The authors are not able to offer a definitive explanation for the initial partitioning in the bubble not reverting to equilibrium but believe if the MnO had initially formed as a fume inside the bubble, it might not be in contact with the melt for reduction by carbon.
According to Tables III and IV, the ratio of mass transfer coefficients for C and Mn was on average 3. This can be explained by the dependency of the mass transfer coefficient on the diffusivity presented as \( k \propto D^{n} \) where power n typically varies from 0.5 to 1.[38,39] Different researchers in the past have reported various diffusion coefficients for Mn in molten Fe or Fe–Csat alloys as given in Table V. According to Yagi et al.[40] and Ono et al.,[41] the effect of C on the diffusion coefficient of Mn is very small. Hence, the diffusion coefficient of Mn in liquid steel at 1823 K is taken by average as \( 4.4 \times 10^{ - 5} \) cm2/s which is 3 times smaller than that of C according to the literature. As a result, the calculated ratios of mass transfer coefficients in this work are in the expected range.
To estimate the fraction of bubble lifetime over which active reaction is occurring, the individual mass transfer coefficients for manganese and carbon were assessed. It is assumed that kC is 0.035 cm/s which is the average of reported values for carbon mass transfer to bubbles in liquid steel in the temperature range 1823 K to 1873 K[11,15,21,49,50]; the mass transfer coefficient for manganese is assumed to be 3 times smaller. Based on these estimates the time required for decarburization and demanganization can be calculated as follows. Knowing the amount of carbon or manganese removed per bubble and using the calculated mass transfer coefficients, the reaction time is estimated to be approximately 0.001 seconds. This is about 1 pct of the residence time of the bubble in the liquid. In the present work, determination of the actual mass transfer coefficient is not possible because the reactions of manganese and carbon with oxygen are over within times considerably less than the residence time of the bubble in the liquid. This suggests that in this system the ability for carbon to reduce MnO back into melt is not very strong. The authors cannot offer a definitive explanation for this but if MnO forms as fume inside the bubble the contact between MnO and carbon in the liquid would be minimal. In other work by the authors[51] the concentration of manganese and carbon versus time were not affected by the depth of submergence of the lance. This supports the findings of the current work that the time for the reaction of manganese and carbon with oxygen is much shorter than the residence time of the bubble in the liquid.
It is worth reflecting on one of the major assumptions in the forgoing calculations, that all manganese is oxidized to MnO in the bubble. This assumption is contrary to the authors’ previous work[33] where to justify the total loss the authors had to assume a significant contribution from manganese vapor and an evaporation-condensation mechanism. To understand why such a mechanism did not operate in the current work one must consider the role of carbon. In the current work, the carbon concentration is 1 pct whereas in the previous work carbon was less than 0.42 pct. In this paper, it has been established that the competition for oxygen is controlled by the relative rates of mass transport of carbon and manganese in the steel. Hence, one might expect that for the same manganese concentrations a higher carbon concentration would lead to less MnO formation. Figure 9 shows the contribution to the rate of manganese loss by a combination of vapor and by evaporation-condensation. This value is plotted against the rate of heat production based on the oxide species formed. From this figure, it is clear that the heat available for evaporation-based mechanisms is less for higher carbon alloys and negligible for 1 pct C alloy considered in the current work. The effect of carbon on manganese loss by mechanisms involving vapor is qualitatively consistent with the effect of carbon on decreasing the heat generated by the oxidation of manganese. The detailed behavior differs from stage 1 to stage 3, but the overall effect of carbon is similar. The authors are not currently able to offer an explanation for the difference between the stages.

Rate of manganese losses as metallic manganese due to evaporation and evaporation-condensation versus the rate of heat generation for Fe–15Mn–C alloys
For two experiments in this work, the final composition of slag was analyzed by ICP for their Mn and Fe content. Assuming that the oxides in the slag are present as MnO and FeO, their corresponding weight percent is calculated and given in Table IV. The measured amounts of MnO in the final slag are in fair agreement with the calculated activities of MnO. However, the amounts of measured FeO are different from their calculated activities.
Conclusions
The effect of gas composition and flow rate on the rate of decarburization and demanganization of Fe–15Mn–1C alloy at 1823 K was studied experimentally and thermodynamically.
-
1.
The competition between manganese and carbon for oxygen is controlled by the ratio of mass transfer coefficients for these elements in the metal.
-
2.
For the 1 pct C alloy considered in this work, manganese loss occurred almost entirely as MnO dust rather than as a combination of MnO and mechanisms involving vapor as seen in previous work for lower carbon alloys. The absence of evaporation-related mechanisms appears to be related to the higher carbon content decreasing the amount of MnO formed thereby decreasing the heat generated and therefore decreasing the amount of vapor that could be produced.
-
3.
Based on the estimated mass transfer coefficient for either manganese or carbon, the reaction time for each bubble was 0.001 seconds which is about 1 pct of the residence time of the bubble in the liquid.
-
4.
The AOD mechanism did not play a major role in the current work where the competition for oxygen was dominated by mass transport. This mechanism is similar to that proposed by Fruehan for chromium, however, the repartitioning of species towards the equilibrium suggested by Fruehan does not seem to have operated in this case. This would appear to be in contradiction to the very short reaction time highlighted in conclusion 3 which would have suggested a fairly rapid repartitioning, but if the form of manganese oxide was as fine dust there might be insufficient contact with the melt for rapid repartitioning. It is still possible that in a deep bath there would be sufficient time for MnO to be reduced back into the melt.
References
B. C. De Cooman, K. Chin, and J. Kim: in New Trends and Developments in Automotive System Engineering, InTech, Rijeka, 2011, pp. 101–28.
2 R. Elliott, K. Coley, S. Mostaghel, and M. Barati: JOM, 2018, vol. 70, pp. 680–90.
3 O. Bouaziz, S. Allain, C.P. Scott, P. Cugy, and D. Barbier: Curr. Opin. Solid State Mater. Sci., 2011, vol. 15, pp. 141–68.
POSCO, POSCO Steel Plates, Pohang, South Korea, unpublished research, 2014.
POSCO, New Materials: High Mn Steels, Pohang, South Korea, unpublished research, 2015.
Thyssenkrupp, Precision Steel, Precidur ® X40MnCrAIV 19-2 HY, Product Information for Hot-Rolled Precision Strip Made in Hohenlimburg, Germany, Hohenlimburg, unpublished research, 2019.
W. Dresler: in Steelmaking Conference Proceedings, 1989, pp. 13–20.
D.S. Kozak and L.R. Matricardi.: Iron Steelmak., 1981, vol. 8, pp. 28–31.
9 T. Germershausen, J. Bader, J. Reichel, U. Gerike, and S.M.S.S. Ag: in The thirteenth International Ferroalloys Congress Efficient technologies in ferroalloy industry, Almaty, Kazakhstan, 2013, pp. 335–46.
10 T. Ohno and T. Nishida: Tetsu-to-Hagane, 1977, vol. 63, pp. 2094–9.
11 R.J. Fruehan: Ironmak. Steelmak., 1976, vol. 3, pp. 153–8.
12 N. Andersson, A. Tilliander, L. Jonsson, and P. Jönsson: Steel Res. Int., 2012, vol. 83, pp. 1039–52.
13 J. Wei, H. Zhu, Q. Jiang, G. Shi, H. Chi, and H. Wang: ISIJ Int., 2010, vol. 50, pp. 1347–56.
G.S. Rao and D.G.C. Robertson: in Advanced Processing of Metals And Minerals, The Minerals, Metals & Materials Society (TMS), 2006, pp. 325–42.
15 T.D. Roy and D.G.C. Robertson: Ironmak. Steelmak., 1978, vol. 5, pp. 198–206.
16 T.D. Roy, D.G.C. Robertson, and J.C.C. Leach: Ironmak. Steelmak., 1978, vol. 5, pp. 207–10.
17 J.-H. Wei and D.-P. Zhu: Metall. Mater. Trans. B, 2002, vol. 33, pp. 111–19.
18 S. Asai and J. Szekely: Metall. Trans., 1974, vol. 5, pp. 651–7.
19 J. Szekely and S. Asai: Metall. Trans., 1974, vol. 5, pp. 1573–80.
20 J. Reichel and J. Szekely: Iron Steelmak., 1995, vol. 22, pp. 41–8.
21 R.J. Fruehan: Ironmak. Steelmak., 1976, vol. 3, pp. 33–37.
22 W.A. Krivsky: Metall. Trans., 1973, vol. 4, pp. 1439–47.
23 J.M. Saccomano, R.J. Choulet, and J.D. Ellis: J. Met., 1969, vol. 21, pp. 59–64.
24 R.J. Fruehan: Met. Trans. B, 1975, vol. 6, pp. 573–8.
25 K. Yamamoto, T. Mimura, S. Ito, and T. Onoye: Tetsu-to-Hagane, 1986, vol. 72, pp. 1034–46.
26 Y.E. Lee and L. Kolbeinsen: ISIJ Int., 2005, vol. 45, pp. 1282–90.
27 B.-D. You: J. Korean Institue Met. Mater., 1995, vol. 33, pp. 1508–13.
28 B.-D. You, J. Han, and J. Pak: Steel Res., 2000, vol. 71, pp. 22–6.
29 B.-D. You, K.-Y. Park, J.-J. Pak, and J.-W. Han: Met. Mater., 1999, vol. 5, pp. 395–9.
30 B.-D. You, B.-W. Lee, and J.-J. Pak: Met. Mater., 1999, vol. 5, pp. 497–502.
31 E.T. Turkdogan, P. Grieveson, and L.S. Darken: J. Am. Chem. Soc., 1963, vol. 67, pp. 1647–54.
32 H. Liu, J. Liu, J. Schenk, F.M. Penz, L. Sun, R. Zhang, and Z. An: Metall. Mater. Trans. B, 2020, vol. 51, pp. 756–62.
A. Rafiei, G.A. Irons, and K.S. Coley: Steel Res. Int., https://doi.org/10.1002/srin.202000480.
34 F. Oeters, K. Koch, R. Scheel, and U. Noelle: Arch Eisenhuettenwes, 1977, vol. 48, pp. 475–80.
35 S.K. Dey and D.N. Ghosh: Metall. Mater. Trans. B, 1976, vol. 7, pp. 43–8.
R. Tsujin, M. Hirai, T. Ohno, N. Ishiwata, and T. Inoshita: ISIJ Int., 1989, vol. 29, pp. 291–9.
37 Y.E. Lee: Metall. Mater. Trans. B, 1998, vol. 29, pp. 397–403.
38 G.F. Versteeg, P.M.M. Blauwhoff, and W.P.M. VAN Swaaij: Chem. Eng. Sci., 1987, vol. 42, pp. 1103–19.
39 J. Wei and H. Zuo: Steel Res. Int., 2007, vol. 78, pp. 863–75.
40 T. Yagi, Y. Ono, and M. Ushijima: Tetsu-to-Hagane, 1970, vol. 56, pp. 1640–45.
41 Y. Ono and S. Matsumoto: Trans. JIM, 1975, vol. 16, pp. 415–22.
42 H. Nomura and K. Mori: Tetsu-to-Hagane, 1969, vol. 55, pp. 1134–41.
43 D. Goldberg and G.R. Belton: Metall. Trans., 1974, vol. 5, pp. 1643–8.
Y. Ono: Tetsu-to-Hagane, 1977, vol. 63, 1229–34.
45 Y. Wanibe, S. Takai, T. Kojima, and H. Sakao: Trans. Iron Steel Inst. Japan, 1980, vol. 20, pp. 783–89.
46 F.P. Calderon, N. Sano, and Y. Matsushita: Metall. Mater. Trans. B, 1971, vol. 2, pp. 3325–32.
T. Saito, Y. Kawai, K. Maruya, and M. Maki: Sci. Rep. Res. Inst. Tohoku Univ. Ser. A Phys. Chem. Metall., 1959, vol. 11, pp. 401–10.
48 A. Majdic, D. Graf, and H. Schenck: Arch. für das Eisenhüttenwes, 1969, vol. 40, pp. 627–30.
49 J.H. Park, D.S. Kim, and S. Lee: 2005, vol. 36, pp. 67–73.
50 O.K. Tokovoi, A. V Tokarev, A.N. Volkadaev, S.N. Prokofev, and A.N. Komarov: Izvvestiya Ross. Akad. Nauk. Met., 1995, vol. 3, pp. 10–4.
A. Rafiei, G.A. Irons, and K.S. Coley: Metall. Mater. Trans. B, Accepted for publication.
Acknowledgments
The authors gratefully acknowledge financial support from the Natural Sciences and Engineering Research Council of Canada. Additional thanks go to ArcelorMittal Dofasco, Stelco, Praxair, and Hatch Ltd. for in-kind support and technical expertise.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Manuscript submitted December 5, 2020; accepted April 6, 2021.
Rights and permissions
About this article

Cite this article
Rafiei, A., Irons, G.A. & Coley, K.S. Kinetics of Decarburization and Manganese Loss from Fe–15Mn–1C Alloy by Bubbling of Argon–Oxygen Gas Mixtures. Metall Mater Trans B 52, 2375–2385 (2021). https://doi.org/10.1007/s11663-021-02180-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-021-02180-z