
Abstract
Tumor suppressor p53 plays a critical role in the regulation of cell cycle and apoptosis in mammals. Mutations of p53 often cause various cancers. Murine models have improved our understanding on tumorigenesis associated with p53 mutations. However, mice and humans are different in many ways. For example, the short lifespans of mice limit the clinical application of the data obtained from this species. Porcine model could be an alternative as pigs share many anatomical and physiological similarities with humans. Here, we modified the expression levels of p53 messenger RNA (mRNA) and protein in porcine fetal fibroblasts using a combination of gene targeting and RNA interference. First, we disrupted the p53 gene to produce p53 knockout (KO) cells. Second, the p53 shRNA expression vector was introduced into fibroblasts to isolate p53 knockdown (KD) cells. We obtained p53 KO, KD, and KO + KD fibroblasts which involve p53 KO and KD either separately or simultaneously. The mRNA expression of p53 in p53 KO fibroblasts was similar to that in the wild-type control. However, the mRNA expression levels of p53 in KD and KO + KD cells were significantly decreased. The p53 protein level significant reduced in p53 KD. Interestingly, no p53 protein was detected in KO + KD, suggesting a complete reduction of the protein by synergistic effect of KO and KD. This study demonstrated that various expression levels of p53 in porcine fibroblasts could be achieved by gene targeting and RNA interference. Moreover, complete abolishment of protein expression is feasible using a combination of gene targeting and RNA interference.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The tumor suppressor gene p53 plays a central role in genome surveillance, cell cycle control, and apoptosis in response to a variety of intra- and extra-cellular stimulations (Lane 1992). The gene product of p53 is a transcription factor essential for tumor suppression. Mutations of p53 have a pivotal role in diverse step of tumorigenesis and increase susceptibility to many kinds of cancers (Greenblatt et al. 1994; Wang et al. 1998; Fontemaggi et al. 2009; Muller et al. 2009). The murine model of p53 mutation has been used as a powerful tool for genetic studies of cancer, proof-of-principle experiments, and development of new anticancer drugs (Kelland 2004; Sausville and Burger 2006). However, mice and humans have different characteristics such as life span, anatomy, and physiology. Therefore, data obtained from mouse studies might not be applicable to human clinics (Sausville and Burger 2006; Garber 2006; Basel et al. 2012). Previous studies revealed that using a mouse model to predict the effect of anticancer drugs for human patients had severe limits for preclinical research (Vandamme 2014). To overcome the limitation of the mouse model, establishing a more suitable animal model similar to human species for cancer research is gradually increasing (Flisikowska et al. 2013). Pigs could provide one of the most appropriate models to replace the current mouse model because pigs and humans have similar anatomy and physiology (Schook et al. 2005).
Experimental techniques for genetic modification in pigs have been well established. Porcine models of cancer related to p53 regulation have also been developed. Transformed porcine cells exhibited tumorigenic ability by expressing proteins such as a dominant-negative p53 and c-Myc to disturb signaling pathways involved in tumor development (Adam et al. 2007). Reduced expression of p53 in porcine mesenchymal stem cells by RNA interference (RNAi) has also been reported (Merkl et al. 2011). Using somatic cell nuclear transfer of mesenchymal stem cells, p53 gene-targeted pigs have been produced (Leuchs et al. 2012). However, the types of mutated p53 found in human tumor are numerous, resulting in various levels of p53 expression. Hence, the development of porcine models with different levels of p53 expression might be important to study the precise relationship of tumorigenesis with p53 expression. Deficiency of p53 expression could be achieved by gene targeting and reduction of the expression could be induced by RNAi. The combination of the two may give rise to different levels of expression.
In the present study, we investigated alteration of p53 expression in miniature pig fetal fibroblasts after gene targeting and RNAi. Our study will be an alternative to the current rodent model of p53 expression by providing porcine model of p53 mutation with various levels of p53 gene expression. Moreover, the developed cells may be used to produce somatic cell nuclear transfer pigs with different levels of p53 expression ranging from none to the reduced.
Materials and Methods
Chemicals and reagents
All chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise stated.
Preparation of porcine fetal fibroblasts
Porcine fetal fibroblasts were prepared as previously described (Ahn et al. 2011). Briefly, porcine fetuses at 28–39 days of gestation were obtained from specific pathogen-free (SPF) Minnesota miniature pigs. This highly inbred strain of pigs has been used in biomedical research since the 1950s (the earliest development of miniature pigs). The size, anatomy, and physiology of these pigs are similar to humans. The head, the dorsal spine of the medial section, and the tail were removed before collection. Small pieces of the tissue were washed in Dulbecco’s phosphate-buffered saline (DPBS; Invitrogen, Carlsbad, CA) and minced with a surgical blade on a 100-mm Petri dish. Cells were dissociated from the minced tissues in 0.25% Trypsin-EDTA (Invitrogen) for 10 min at 38.5°C. After centrifuging the cell suspension three times at 200×g for 5 min, cell pellets were resuspended and seeded onto 100-mm culture dishes (Falcon, Franklin Lakes, NJ). Cells were cultured in Dulbecco’s modified Eagle medium (DMEM; Invitrogen) supplemented with 15% (v/v) fetal bovine serum (FBS; Hyclone, Logan, UT), 1 mM L-glutamine, 100 units/mL penicillin, and 0.5 mg/mL streptomycin in a humidified atmosphere of 5% CO2 with 95% air for 6 to 8 days. Attached cells were washed with DPBS, cultured until confluent and subcultured at intervals of 5 to 7 days by trypsinization for 5 min using 0.25% Trypsin-EDTA until later use for transfection.
Construction of p53 gene targeting vector
Construction of p53 gene targeting vector was illustrated in Fig. 1A , a targeting vector was constructed from a genomic DNA fragment containing exons 1 and 2 of the porcine p53 gene (GenBank accession no: NC_010454). A neomycin-resistance gene under transcriptional control of the mouse phosphoglycerate kinase-1 promoter (PGK-Neo) was inserted into exon 2 of p53 gene. Approximately 4.6- and 2.1-kb homologous arms amplified by long arm primer set (forward 5ʹ-GCGGCCGCGGCTCAGATCTGGCGTTTCT-3ʹ and reverse 5ʹ-ACGCGTCAGCCTGAGACACACCCAAG-3ʹ) and short arm primer set (forward 5ʹ-GCTAGCCCCTCTGAGTCAGGAGACATTT-3ʹ and reverse 5ʹ-GATATCAGGAAATTTTCTTCCTCTGTGC-3ʹ) were inserted into 5ʹ and 3ʹ region of the selection cassette of PGKneolox2DTA.2 plasmid (Addgene) composed of a neomycin-resistance gene, a polymerase destabilizing signal, a pausing signal for RNA polymerase II, and the diphtheria toxin A fragment (DTA) for negative selection. The Not1-linearized targeting vector was used for introduction into porcine fetal fibroblasts.

p53 gene targeting in miniature pig fetal fibroblasts. (A) p53 gene targeting vector. Arrows PCR primers for detection of homologous recombination, Bar probe for Southern blot analysis. (B) PCR screening and PCR-southern analysis of p53 gene-targeted cells. Lanes 1 p53 gene-targeted fibroblasts, 2 neo (G418)-resistant fibroblasts, 3 normal (non-transfected) fibroblasts, 4 p53 gene targeting vector, 5 distilled water.
Gene targeting in porcine fetal fibroblasts
The linearized 14.8-kb targeting vector was introduced into porcine fetal fibroblasts using Gene Pulser II (Bio-Rad, Hercules, CA) according to the manufacturer’s protocol. Starting from 48 h after transfection, genetically modified cells were selected with the medium containing 500 μg/mL of G418 (Invitrogen) for 14 d. After antibiotic selection, G418-resistant colonies were isolated and transferred onto 0.1% gelatin-coated dishes. Subculture was started with a low density of cells so that clones originated from a single cell could be isolated for later molecular analysis. Cells were continuously cultured at 38.5°C in a humidified atmosphere containing 5% CO2 and 95% air.
Detection of homologous recombination by PCR and PCR-Southern blot
As represented in Fig. 1A , cells from G418-resistant colonies were subjected to analysis for the disrupted porcine p53 allele. Genomic DNA was prepared using DNeasy Tissue Kit (QIAGEN, Valencia, CA) according to the manufacturer’s protocol. For PCR analysis of gene targeting events, genomic DNA was amplified using Maxime PCR Premix Kit (Intron Biotechnology, Seongnam, Korea) in 20-μL reaction volume with the following parameters: 30 cycles of 30 s at 95°C, 30 s at 65°C, 10 min plus 20 s increase per cycle at 68°C; and one final cycle of 15 min at 68°C. The PCR primer set used for amplification includes the forward primer 5ʹ-GGAAGTTGCCAAGCTGATTCCA-3ʹ and the reverse primer 5ʹ-TCCAAGGCGTCATTCAGCTCTC-3ʹ. Amplification products (8.1-kb fragment of the wild-type (WT) allele and 9.9-kb fragment for the targeted allele) were analyzed by running on 0.8% agarose gel electrophoresis. Following electrophoresis, Southern blot hybridization was performed using The DIG high Prime DNA Labeling and Detection Kit 2 (Roche, Indianapolis, IN) according to the manufacturer’s protocol. Amplification products were transferred to a nylon membrane. Then, the membrane was hybridized with a 440-bp DIG-labeled probe specific for neomycin-resistance gene in DIG Easy Hyb solution (Roche) for 16 h at 45°C. After the hybridization, the membrane was washed twice in 0.2 X SSC with 0.1% sodium dodecylsulfate (SDS) at 68°C and exposed to a camera for 30 min.
Porcine p53 shRNA expression vector
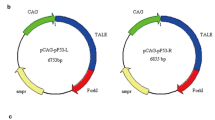
Porcine p53-specific and non-specific control (mock) shRNA expression vector were designed and produced by Genolution (Seoul, Korea). The 25 mer sequence of p53 siRNA was 5ʹ-GACGCCAGUGGCAACUUGCUGGGAC-3ʹ (Top: 5ʹ-cacc GACGCCAGTGGCAACTTGCTGGGAC tctc GTCCCAGCAAGTTGCCACTGGCGTC-3ʹ, Bottom: 5ʹ-aaaa GACGCCAGTGGCAACTTGCTGGGAC gaga GTCCCAGCAAGTTGCCACTGGCGTC-3ʹ). Porcine p53 shRNA was inserted into pENTRTM/H1/TO vector with H1/TO promoter and selectable Zeocin cassette (Invitrogen). The structure of transgene construct was represented in Fig. 2A .

Detection of p53 shRNA expression construct. (A) p53 shRNA expression construct. Size of PCR product with the indicated primers (arrows) is 2.5 kb. (B) PCR analysis of construct integration. WT + mock wild-type fibroblasts transfected with mock vector, KD wild-type fibroblasts transfected with p53 shRNA, KO + mock p53 gene-targeted fibroblasts transfected with mock vector, KO + KD p53 gene-targeted fibroblasts transfected with p53 shRNA, WT wild-type fibroblasts, KO p53 gene-targeted fibroblasts, DW distilled water.
Transfection of porcine fetal fibroblasts with p53 shRNA expression vector
The number of porcine fetal fibroblasts was adjusted to 3 × 105, and cells were transfected with 2–3 μg of p53 or scrambled shRNA constructs using Amaxa Nucleofection Kit (Lonza, Basel, Switzerland) according to the manufacturer’s instructions. After transfection, cells were transferred to 100-mm culture dish. Genetically modified cells were selected with the medium containing 800 μg/mL of Zeocin (Invitrogen) from 48 h after transfection for 21 d. Zeocin-resistant colonies were isolated and transferred onto 0.1% gelatin-coated dishes for clonal culture. Cells were continuously cultured at 38.5°C in a humidified atmosphere containing 5% CO2 and 95% air. Genomic DNA was extracted from cells of Zeocin-resistant colonies, and an integration of shRNA construct was identified by PCR using the forward 5ʹ-CCTGATTCTGTGGATAACCGTATT-3ʹ and reverse 5ʹ-AGTAACCATGCATCATCAGGAGTA-3ʹ primer set specific for pENTRTM/H1/TO vector as showed in Fig. 2. The condition for PCR was 95°C for 10 min; 30 cycles (95°C for 30 s; 62°C for 30 s; 72°C for 3 min); and 72°C for 5 min. Amplification products (2.5kb) were analyzed by 0.8% agarose gel electrophoresis.
Analysis of p53 expression by RT-PCR
Total RNA was extracted from cells of Zeocin-resistant colonies using RNeasy Mini Kit (QIAGEN). Aliquots of 0.5 μg of total RNA were used for complementary DNA (cDNA) synthesis using SuperScript® III First-Strand Synthesis System (Invitrogen) according to the manufacturer’s protocol. Synthesized cDNA was amplified using Maxime PCR Premix Kit in 20-μL reaction volume with the following parameters: 33 cycles of 30 s at 95°C, 30 s at 63°C, and 1 min at 72°C. The sequence of the upstream and downstream primer pairs and length of PCR products were as follows: porcine p53 (Forward: 5ʹ-CCATGGCCATCTACAAGAAGTC-3ʹ and Reverse: 5ʹ-GTCATTCAGCTCTCGGAACATC-3ʹ; 566 bp) and beta-actin for endogenous housekeeping standard (Forward: 5ʹ-TGCGTGACATCAAAGAGAAG-3ʹ and Reverse: 5ʹ-CGGATGTCAACGTCACACTT-3ʹ; 244 bp). Each experiment was repeated three times using cells prepared separately at different times.
Analysis of p53 expression by western blot
Samples of 5 × 105 fetal fibroblasts were washed twice with ice-cold phosphate-buffered saline (PBS), resuspended in 200 μL ice-cold NP-40 Cell Lysis Buffer (Invitrogen), and incubated at 4°C for 30 min. The lysates were centrifuged at 13,000 rpm for 30 min at 4°C. Protein concentrations of cell lysates were determined using Quick Start Bradford Protein Assay (Bio-Rad) according to the manufacturer’s protocol. Equivalent amounts of proteins in each sample were loaded and run on 12% SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Then, the gel was transferred to nitrocellulose membrane (GE Healthcare Bio-Sciences, Piscataway, NJ) and reacted with p53 and beta-actin antibodies (Santa Cruz Biotechnology, Dallas, TX). Immunostaining with antibodies was performed using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific, Rockford, IL) according to the manufacturer’s protocols and detected with ImageQuant LAS4000 mini (GE Healthcare).
Statistical analysis
Statistical analyses were carried out using SPSS version 11.0 for Windows. At least three replicates were conducted for each experiment, and data were presented as means ± SEM, percentage data were subjected to arcsine transformation prior to statistical analysis. All data were analyzed by one-way ANOVA, followed by Fisher’s LSD test. A value of P < 0.05 was considered significant.
Results
After transfection of porcine fetal fibroblasts with p53 gene targeting vector, a total of 48 G418-resistant colonies were obtained and screened by PCR. One colony was identified as p53 gene-targeted (2.08% efficiency). As shown in Fig. 1B , 9.9-kb PCR product indicated that one allele of p53 gene was disrupted by an insertion of selection marker PGK-Neo. Hybridization of Neo probe not with 8.1-kb PCR product (amplified from wild-type allele) but with 9.9-kb product (amplified from p53 gene-targeted allele) confirmed heterozygous knockout in p53 gene (Fig. 1B ).
After introduction of p53 shRNA or mock expression vector into fetal fibroblasts, transfected cells were cultured with Zeocin for 21 d. Then, Zeocin-resistant colonies were analyzed by PCR to confirm the integration of vector into the genome. As shown in Fig. 2A , the integration of vectors was identified by PCR product of 2.5 kb from shRNA cassette across pENTRTM/H1/TO vectors. The integration of vectors was confirmed in all experimental groups including wild-type fibroblasts with mock vector (WT + mock), wild-type fibroblasts with p53 shRNA (KD), p53 gene-targeted fibroblasts with mock (KO + mock), and p53 gene-targeted fibroblasts with p53 shRNA (KO + KD), whereas wild-type (WT) and p53 gene-targeted fibroblasts (KO) were integration-negative (Fig. 2B ).
To evaluate changes of porcine endogenous p53 mRNA by gene targeting and RNAi, RT-PCR was performed. As shown in Fig. 3A , with the mean value of p53 expression in WT as 100%, the p53 expressions in WT + mock and KO (90.6% and 91.8%, respectively) were not different from the control. Unexpectedly, the expression in KO + mock was significantly increased (189.3%), but significant reduction of p53 mRNA was observed in KD and KO + KD (35.5% and 34.8%, respectively). No difference was seen between KD and KO + KD.

Analysis of p53 expression. (A) Expression of p53 mRNA analyzed by RT-PCR. Amount of mRNA was normalized by β-actin expression. (B) Expression of p53 protein analyzed by western blot. WT wild-type fibroblasts, WT + mock wild-type fibroblasts transfected with mock vector, KO p53 gene-targeted fibroblasts, KO + mock p53 gene-targeted fibroblasts transfected with mock vector, KD wild-type fibroblasts transfected with p53 shRNA, KO + KD, p53 gene-targeted fibroblasts transfected with p53 shRNA.
Reduction of porcine p53 protein was evaluated by western blot using lysates from six different fibroblast cell lines, and the results are shown in Fig. 3B . Differences in the expression of p53 protein were not found among WT, WT + mock, KO, and KO + mock. However, a decrease of p53 protein was observed in KD, and the expression was completely abolished in KO + KD.
Discussion
It has been widely known that the amount of p53 expression is closely related to the onset of the tumor in a mouse model (Donehower et al. 1992; Donehower 1996; Yamamoto et al. 2000). Therefore, the control of p53 expression would be important in the study of tumorigenesis caused by p53 mutation. We investigated the expression level of p53 mRNA and protein in porcine fetal fibroblasts that could be altered by two different genetic modification techniques, such as p53 gene knockout (KO) by gene targeting and knockdown (KD) by RNAi.
First, porcine p53 gene was targeted by disruption in exon 2 of the gene where the initiation codon is located (Fig. 1A, B ). However, compared with wild-type control, no reduction in the expression of p53 messenger RNA (mRNA) and protein was observed (Fig. 3A, B ). This could be expected since the compensation of p53 gene expression might occur. In biallelic genes, such as p53, loss of gene expression from one allele could be compensated by the expression from the other. Transcriptional compensation in heterozygous disruption of a tumor suppressor gene has been reported (Guidi et al. 2004). However, it has not been clear to what extent a wild-type allele of p53 gene can compensate for a mutated or lost allele. Such compensatory mechanisms could be gene amplification, transcription upregulation of the remaining allele, or simply facilitated catalytic activation of the native p53 protein (Forslund et al. 2001). Since the exon 2 of p53 gene was targeted in the present study, there might be a possibility of alternative splicing that could affect the accuracy of the experiment. An isoform of p53 protein (Δ40p53) does not include exon 2. The primers used in the experiment were on exon 5 (forward) and exon 10 (reverse). Thus, the primer set used in this study will not amplify Δ40p53. Since Δ40p53 does not have transacting domain (MDM2 binding domain), MDM2 (p53-specific E3 ubiquitin ligase)-dependent negative regulation of p53 will be hampered. If this is the case, an increase of p53 protein could have seen in western blot. The increase of p53 protein by MDM2 inhibitor has been reported (Tovar et al. 2013). In our study, the increase of p53 protein was not seen in p53 gene-targeted fibroblasts compared to the wild-type control. It may suggest that the stability of p53 was not affected. A reduction of p53 mRNA and protein in monoallelic KO was not observed because there might be a compensatory expression of p53 from the non-targeted wild-type allele.
Since the heterozygous disruption had little effect on expression levels of p53 mRNA and protein, we investigated the effects of additional knockdown on transcription and translation of p53 gene. To reduce p53 mRNA expressed from intact p53 allele, we assessed knockdown effects of RNAi for porcine p53. It has been reported that approximately 70%–80% reduction of p53 protein in murine and human fibroblasts was achieved by shRNA (Tovar et al. 2013). We compared sequence homology among the species including mice, humans, and pigs, and chose the RNAi sequence from 756 to 774 of porcine p53 (GenBank accession no: AF098067) containing close homology with the sequence used in the previous report (Kawamura et al. 2009). Our study using shRNA with the chosen sequence resulted in significant reduction of p53 mRNA levels to approximately one third in both wild-type and p53 gene-targeted porcine fibroblasts compared with the control (Fig. 3A ). Expression of p53 protein was also reduced by shRNA with similar manner to the mRNA expression (Fig. 3B ). More drastic effect of p53 shRNA was observed in additional knockdown (KD) on heterozygous knockout (KO) fibroblasts. Despite remaining mRNA expression, there was a complete abolition of p53 protein expression by KD in addition to KO (Fig. 3B ). This result suggests that a synergistic effect of KO and KD occurred. Additional KD might have overridden the compensation of p53 expression from the intact wild-type allele in heterozygous KO fibroblasts. Since the downregulation of p53 mRNA was the same level in KD and KO + KD (Fig. 3A ), the abolishment of compensatory protein expression could be post-transcriptional (Fig. 3B ). Relative levels of p53 protein, KO + mock in particular, were not completely matched to mRNA level (Fig. 3A, B ). Under the normal condition without DNA damage, the p53 protein is very unstable and exists in low concentration. This is because the binding of MDM2 induces degradation of p53 via proteasomes. Once DNA is damaged, phosphorylation of p53 separates MDM3. Then, p53 could be increased. Since the concentration of p53 protein is regulated by its degradation as well as translation, increase of p53 mRNA due to other reasons than DNA damage may not faithfully reflect the concentration of p53 protein.
In rat model of p53 KO, tumorigenesis in monoallelic mutation by loss of heterozygosity was occurred, although the tumorigenesis was delayed than biallelic mutation (van Boxtel et al. 2011). The result of the present study may be useful in filling the gap between the two. Recently, a similar model of cloned pigs with p53 mutation by somatic cell nuclear transfer was reported (Sieren et al. 2014).
Overall, our results demonstrated that in porcine fetal fibroblast, (i) gene targeting alone did not show any effects of p53 expression level in mRNA and protein, (ii) KD gave rise to a significant reduction in p53 mRNA expression, and (iii) when both KO and KD were applied simultaneously, p53 mRNA was significantly deceased, and expression of p53 protein was completely diminished. The results suggest that the combination of genetic modification techniques could modify the expression level of endogenous p53 in porcine fetal fibroblasts. These cells with different levels of p53 expression may provide useful tools to study relations between development of cancer and p53 expression in large animal model, as an alternative to the current mouse model, with close anatomical and physiological and similarities to humans..
References
Adam SJ, Rund LA, Kuzmuk KN, Zachary JF, Schook LB, Counter CM (2007) Genetic induction of tumorigenesis in swine. Oncogene 26:1038–1045
Ahn KS, Kim YJ, Kim M, Lee BH, Heo SY, Kang MJ, Kang YK, Lee JW, Lee KK, Kim JH, Nho WG, Hwang SS, Woo JS, Park JK, Park SB, Shim H (2011) Resurrection of an alpha-1,3-galactosyltransferase gene-targeted miniature pig by recloning using postmortem ear skin fibroblasts. Theriogenology 75:933–939
Basel MT, Balivada S, Beck AP, Kerrigan MA, Pyle MM, Dekkers JC, Wyatt CR, Rowland RR, Anderson DE, Bossmann SH, Troyer DL (2012) Human xenografts are not rejected in a naturally occurring immunodeficient porcine line: a human tumor model in pigs. Biores Open Access 1:63–68
Donehower LA (1996) The p53-deficient mouse: a model for basic and applied cancer studies. Semin Cancer Biol 7:269–278
Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr, Butel JS, Bradley A (1992) Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356:215–221
Flisikowska T, Kind A, Schnieke A (2013) The new pig on the block: modelling cancer in pigs. Transgenic Res 22:673–680
Fontemaggi G, Dell’Orso S, Trisciuoglio D, Shay T, Melucci E, Fazi F, Terrenato I, Mottolese M, Muti P, Domany E, Del Bufalo D, Strano S, Blandino G (2009) The execution of the transcriptional axis mutant p53, E2F1 and ID4 promotes tumor neo-angiogenesis. Nat Struct Mol Biol 16:1086–1093
Forslund A, Lönnroth C, Andersson M, Brevinge H, Lundholm K (2001) Mutations and allelic loss of p53 in primary tumor DNA from potentially cured patients with colorectal carcinoma. J Clin Oncol 19:2829–2836
Garber K (2006) Realistic rodents? Debate grows over new mouse models of cancer. J Natl Cancer Inst 98:1176–1178
Greenblatt MS, Bennett WP, Hollstein M, Harris CC (1994) Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res 54:4855–4878
Guidi CJ, Veal TM, Jones SN, Imbalzano AN (2004) Transcriptional compensation for loss of an allele of the Ini1 tumor suppressor. J Biol Chem 279:4180–4185
Kawamura T, Suzuki J, Wang YV, Menendez S, Morera LB, Raya A, Wahl GM, Izpisúa Belmonte JC (2009) Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 460:1140–1144
Kelland LR (2004) Of mice and men: values and liabilities of the athymic nude mouse model in anticancer drug development. Eur J Cancer 40:827–836
Lane DP (1992) Cancer. p53, guardian of the genome. Nature 358:15–16
Leuchs S, Saalfrank A, Merkl C, Flisikowska T, Edlinger M, Durkovic M, Rezaei N, Kurome M, Zakhartchenko V, Kessler B, Flisikowski K, Kind A, Wolf E, Schnieke A (2012) Inactivation and inducible oncogenic mutation of p53 in gene targeted pigs. PLoS One 7:e43323
Merkl C, Leuchs S, Saalfrank A, Kind A, Schnieke A (2011) RNA interference in pigs: comparison of RNAi test systems and expression vectors. Mol Biotechnol 48:38–48
Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, Lukashchuk N, Gillespie DA, Ludwig RL, Gosselin P, Cromer A, Brugge JS, Sansom OJ, Norman JC, Vousden KH (2009) Mutant p53 drives invasion by promoting integrin recycling. Cell 139:1327–1341
Sausville EA, Burger AM (2006) Contributions of human tumor xenografts to anticancer drug development. Cancer Res 66:3351–3354
Schook L, Beattie C, Beever J, Donovan S, Jamison R, Zuckermann F, Niemi S, Rothschild M, Rutherford M, Smith D (2005) Swine in biomedical research: creating the building blocks of animal models. Anim Biotechnol 16:183–190
Sieren JC, Meyerholz DK, Wang XJ, Davis BT, Newell JD Jr, Hammond E, Rohret JA, Rohret FA, Struzynski JT, Goeken JA, Naumann PW, Leidinger MR, Taghiyev A, Van Rheeden R, Hagen J, Darbro BW, Quelle DE, Rogers CS (2014) Development and translational imaging of a TP53 porcine tumorigenesis model. J Clin Invest 124:4052–4066
Tovar C, Graves B, Packman K, Filipovic Z, Higgins B, Xia M, Tardell C, Garrido R, Lee E, Kolinsky K, To KH, Linn M, Podlaski F, Wovkulich P, Vu B, Vassilev LT (2013) MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res 73:2587–2597
van Boxtel R, Kuiper RV, Toonen PW, van Heesch S, Hermsen R, de Bruin A, Cuppen E (2011) Homozygous and heterozygous p53 knockout rats develop metastasizing sarcomas with high frequency. Am J Pathol 179:1616–1622
Vandamme TF (2014) Use of rodents as models of human diseases. J Pharm Bioallied Sci 6:2–9
Wang XJ, Greenhalgh DA, Jiang A, He D, Zhong L, Medina D, Brinkley BR, Roop DR (1998) Expression of a p53 mutant in the epidermis of transgenic mice accelerates chemical carcinogenesis. Oncogene 17:35–45
Yamamoto M, Tsukamoto T, Sakai H, Shirai N, Ohgaki H, Furihata C, Donehower LA, Yoshida K, Tatematsu M (2000) p53 knockout mice (−/−) are more susceptible than (+/−) or (+/+) mice to N-methyl-N-nitrosourea stomach carcinogenesis. Carcinogenesis 21:1891–1897
Acknowledgments
This research was supported by a grant (PJ011375) from the Next-Generation BioGreen 21 Program, Rural Development Administration, a grant (2014034046) supported by the Bio & Medical Technology Development Program, and a grant (2009–0093829) supported by the Priority Research Centers Program through National Research Foundation (NRF) funded by the Ministry of Science, ICT and Future Planning.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editor: Tetsuji Okamoto
Young June Kim and Tae-Hyun Kim contributed equally to this work.
Rights and permissions
About this article

Cite this article
Kim, Y.J., Kim, TH., Kim, M. et al. Complete reduction of p53 expression by RNA interference following heterozygous knockout in porcine fibroblasts. In Vitro Cell.Dev.Biol.-Animal 52, 736–741 (2016). https://doi.org/10.1007/s11626-016-0026-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11626-016-0026-0