
Abstract
The objective of this study was to establish and evaluate a new model for studying lipogenesis in grass carp preadipocytes. The morphology characteristic from preadipocytes to mature adipocytes was observed with the microscopic morphology, and the proliferation kinetics of cells was tested by cell counting. In addition, the nature and differentiation degree of cells were evaluated using Oil Red O staining, lipase activity determination, and reverse transcription PCR (RT-PCR) assay. Morphologically, grass carp preadipocytes started to attach and grow on day 3, then they resembled fibroblasts, and most underwent attachment, proliferation, and growth arrest with subsequent accumulation of intracellular lipid droplets before becoming mature adipocytes. Glycerol-3-phosphate dehydrogenase (GPDH) activity was increased gradually during the progress of culture. Analysis of RT-PCR confirmed that peroxisome proliferator-activated receptor-γ expression patterns were consistent with my observations regarding GPDH activity. In summary, grass carp preadipocytes cultured with 10% FBS at 28°C in a humidified 5% CO2 atmosphere have high proliferation potential. Furthermore, the cells synthesize a range of markers that are consistent with this cell type. We conclude therefore that the grass carp preadipocytes described here have high capacity for lipogenesis and may, therefore, represent a unique tool for studying fish fat cell development and metabolism.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Excess deposition of fat in the carcass and liver of farmed fish has increased in the last decade as a result of the rapid expansion of intensive aquaculture, which affects the growth of the animal and decreases the quality of fish flesh, especially in grass carp. Grass carp is one of the four major Chinese carps in freshwater aquaculture, and excess fat is stored in the form of triglycerides in visceral, intramuscular, and subcutaneous adipocyte depots that strongly restricts the development of aquaculture. However, how the excess fat in the fish should be managed is not yet clear. In mammals, the cellular and molecular mechanisms of adipose tissue biology have been studied extensively over the last two decades. Relative to mammals, little is known about energy homeostasis in fish, and even less is known about adipose development in grass carp adipocytes. Thus, it is important to study lipogenesis in grass carp adipose tissue.
The degree of fat deposition is characterized by the volume and number of adipocytes, which is regulated in the so-called adipocyte life cycle (Rayalam et al. 2008) that involves both an increase in fat cell size and the formation of mature adipocytes from precursor cells. A large number of investigations have been done on the development of adipose tissue using murine preadipocyte cell lines (Kristen et al. 2004; Cartwright et al. 2010; Nakatani et al. 2010) and primary adipocytes in rodents (Kim et al. 1998; Palmer et al. 2002). Although differentiated cell lines have been widely used for studying adipocyte development and metabolism, they often are not as responsive as primary adipocytes, and these cell lines are immortalized aneuploid cells in which genetic regulation might be distorted. In primary culture, adipose tissue is removed from their in vivo environment where cells may exhibit differences in factors of circulation, and any differences obtained in primary culture would represent intrinsic properties of these cells independently of the in vivo conditions. Therefore, the use of primary cultured grass carp adipocytes is necessary for physiological validation of the data obtained.
No literature about the culture system of grass carp preadipocytes has been reported. The objective of this study was to establish a new model of grass carp preadipocyte and evaluate the proliferation and differentiation capacity of fish preadipocytes in vitro. The establishment of the culture system for grass carp will make the cells a novel and valuable tool for studying lipogenesis in fish.
Materials and Methods
Chemicals.
Bovine serum albumin (BSA), N-2-hydroxyethylpiperazine-N′-2′-ethane sulfonic acid (HEPES), ethylenediaminetetraaceticacid (EDTA), phosphate-buffered saline (PBS), streptomycin/penicillin, trypsase, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, Oil Red O, and nicotinamide-adenine dinucleotide (NADH) were purchased from Sigma Company (St. Louis, MO). Dulbecco's modified eagle medium/F12 (DMEM/F12), collagenase, fetal bovine serum (FBS), and cell culture plates were ordered from Gibco Company (Carlsbad, CA). TRIZOL and cDNA Synthesis Kit were obtained from Fermentas Life Science Company (Glen Burnie, MD). The primers of PPARγ and β-actin were synthesized by Shanghai Sangon Biological Engineering Technology & Services Co., Ltd. (Shanghai, China).
Adipose tissue collection, grass carp preadipocyte isolation, and primary culture.
Six healthy (average weight, 300 g) grass carps were bought from local fisheries. All the fish were anesthetized with metacain (50 mg/L) and killed by a blow to the head. White adipose tissue surrounding the intestinal tract was obtained from the abdominal cavity of the fish. Primary culture of grass carp preadipocytes was conducted according to the methods of Li et al. (2006), with several modifications. Briefly, adipose tissue was rinsed with Krebs-Ringer bicarbonate (containing 3% BSA, 10 mM glucose, 50 U penicillin/ml, and streptomycin) buffer. The tissue was cut with scissors into approximately 1-mm3 sections and then incubated in a digestion buffer comprised of DMEM/F12 (a 50:50 mixture of DMEM/F12), 100 mM HEPES, and 20 g/L bovine serum albumin, pH 7.4, containing 1 g/L collagenase (type I). A fivefold excess of digestion buffer (room temperature, excluding collagenase) was added to the digestion flask after incubating for 60 min at 28°C in a shaking water bath. To remove undigested tissue and large cell aggregates, flask contents were mixed and filtered through a 200-μm nylon mesh filter. The filtered cells were centrifuged at 800×g for 5 min to separate the floating adipocytes from the pellet of stromal-vascular cells. The stromal-vascular cells were then incubated with erythrocyte lysis buffer (0.154 M NH4Cl, 10 mM KHCO3, 0.1 mM EDTA) at room temperature for 10 min, followed by centrifugation. The stromal-vascular cell pellet was washed twice with DMEM/F12 medium supplemented with 15 mM NaHCO3, 50 U penicillin/ml, and streptomycin. After washing, the cells were resuspended in DMEM/F12 medium containing 10% FBS. Finally, aliquots were seeded in culture flasks, 96-well plates, 24-well plates, or 6-well plates at a density of 5.0 × 104 cells/cm2 and maintained at 28°C in a humidified 5% CO2 atmosphere. The medium was changed every other day.
Morphology observation.
The cell morphology was observed and photographed with an Olympus microscope every day.
Cell counting.
Cell proliferation was evaluated with cell counting. Briefly, cells were maintained in the medium until days 2, 4, 6, 8, 10, and 12 of culture at which time they were washed three times with PBS to remove the rudimental medium, digested with 0.25% trypsase, and then centrifuged and resuspended in DMEM/F12 medium containing 10% FBS. The total cell number was counted using the hemocytometer (n = 6).
Oil Red O staining.
Adipocyte differentiation was identified by Oil Red O staining after 20 d of culture according to the methods of Ramirez-Zacarias et al. (1992), with a minor modification. The Oil Red O working solution was prepared by dissolving 4.2 g of Oil Red O in 1,200 mL of absolute isopropanol. The solution was allowed to sit overnight at room temperature without stirring. On the second day, the solution was filtered through qualitative filter paper, mixed with 900 mL of deionized water, and allowed to sit overnight at 4°C without stirring. The final working solution was filtered twice and stored at room temperature until use. Cell culture plates for staining were taken out, the medium was removed, and cells were washed three times with PBS, fixed for a minimum of 30 min with 10% neutral buffered formalin, and drained. After washing with PBS, the cells were covered with the Oil Red O working solution for 60 min. The Oil Red O was removed, and the cells were subsequently destained with 60% propylene glycol using gentle agitation for 30 s. Finally, the cells were rinsed thoroughly with PBS, and then cell morphology was examined and photographed with a microscope.
GPDH activity analysis.
Glycerol-3-phosphate dehydrogenase (GPDH) activity was determined according to the methods of Hauner et al. (1989). At days 5, 10, 15, and 20 of the culture period, the medium was removed from the 24-well plates. The cells were rinsed three times with PBS, digested with 0.25% trypsase, centrifuged, resuspended in PBS, and sonicated. The samples were centrifuged at 7,000×g for 15 min at 4°C; then the infranate was assayed for GPDH activity in 100 mM triethanolamine/HCl, pH 7.5, 2.5 mM EDTA, 0.12 mM NADH, 0.2 mM dihydroxyacetone phosphate, and 0.1 mM mercaptoethanol. The change in absorbance at 340 nm with time was used to calculate the rate. One unit of activity was expressed as the amount causing the oxidation of 1 nmol of NADH per minute. The protein content of the extracts was measured with a modification of the method of Peterson (1977).
Extraction of total RNA and RT-PCR analysis.
The PPARγ expression was normalized by the following method: at days 5, 10, 15, and 20 of the culture period, the total cell numbers were counted using the hemocytometer. Equal cell numbers (8 × 105 cells) from different culture period were used to extract total cellular RNA by using TRIZOL reagent following the manufacturer's protocol. After DNase treatment, RNA quality and quantification were measured by electrophoresis and spectrophotometry at 260 and 280 nm, respectively (Peterson 1977).
Reverse transcription was performed to synthesize cDNA with a first-strand cDNA synthesis kit. The synthesized cDNA were then amplified by PCR using paired sense and antisense primers of both β-actin (as the internal control) and PPARγ (Table 1) in the same sample according to the methods of Maria et al. (2001). The PCR conditions included an initial denaturation at 95°C for 10 min, followed by 30 cycles of 95°C for 1 min, 50.5°C for 1 min, and 72°C for 1 min, and a final extension step at 72°C for 10 min. The amplified fragments were separated with electrophoresis by using 1% agarose gel.
Images of the reverse transcription (RT)-PCR ethidium bromide-stained agarose gels were acquired with a CCD camera, and quantification of the bands was performed using Dolphin-1D software (Wealtec International Ltd.). Band intensity was expressed as relative absorbance units. The ratio of band intensity between the mRNA of interest and β-actin was calculated to normalize for initial variations in sample concentration.
Statistical analysis.
A single batch of fish preadipocytes from an individual fish was used for each replicate. All data were obtained from one independent experiment carried out with six replicates. Each morphology observation, cell counting, Oil Red O staining experiments, GPDH activity, and RT-PCR analysis within an experiment was replicated six times. Treatments were compared by ANOVA (SPSS 13.0). Differences were considered statistically significant at P < 0.05. Data were presented as means ± S.E. calculated from six replicates.
Results
Morphological change.
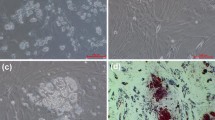
Morphology of the cells were observed in primary culture over a period of 20 d. Morphological change from preadipocytes to mature adipocytes of primary (A–F) cells are shown in Fig. 1. After seeding, majority of the cells were small and round (Fig. 1A ); then they started to attach and grow on day 3. Cells grow exponentially and resembled fibroblasts (Fig. 1B ) on day 5. There was a large increase in the number of cells between days 6 and 8, and the cells gradually arrived at a monolayer confluence by day 10 (Fig. 1C ). From days 10 to 15, many small intracellular lipid droplets began to accumulate around the nucleolus (Fig. 1D ). At day 20, larger lipid droplets appeared, and the cells tended to be round (Fig. 1E ), suggesting that the cells had fully differentiated. Oil Red O staining indicated that numerous large cytoplasmic lipids had formed in the cells, which confirmed that the fish preadipocytes had fully differentiated by day 20 (Fig. 1F ).

Morphological change from preadipocyte to mature adipocyte. A, Cells were photographed at day 1. B, Cells were photographed at day 5. C, Cells were photographed at day 10. D, Cells were photographed at day 15. E, Cells were photographed at day 20. F, Cells were stained by Oil Red O and photographed at day 20. Lipid droplets were stained bright red. (Bars = 20 μm).
Cell proliferation evaluation.
The proliferation capacity of grass carp preadipocytes was tested by cell counting. The results (Fig. 2) indicated that it showed a unique S-shaped growth curve and had a 10-d growth period. No significant difference was found in the proliferation capacity of cells (P = 0.283) between seeding at 2 and 4 d, while there was a very significant (P < 0.001) increase in mitogenic activity of cells from 6 to 8 d, which corresponded well with an increase in cell number. At day 10, the total cell number reached the peak, and then cells underwent growth arrest; subsequently, the cell number began to decline gradually by day 12.

Growth curve of grass carp adipocytes. Grass carp preadipocytes were seeded and maintained in 24-well plates. At 2, 4, 6, 8, 10, and 12 d, proliferation kinetics of cells was evaluated by cell counting. Data are expressed as means ± SE of six independent determinations.
GPDH activity.
I assayed GPDH activity, a late marker for adipocyte differentiation, at days 5, 10, 15, and 20 of the culture. The GPDH activities of cells were increased during the differentiation of preadipocytes to adipocytes (Fig. 3).

GPDH activity in grass carp adipocytes. Grass carp preadipocytes were seeded and maintained in 24-well plates. At days 5, 10, 15, and 20, the differentiation degree of cells was determined through GPDH activity. Columns and error bars indicate means ± SE of six independent determinations.
PPARγ mRNA expression.
The PPARγ expression patterns were consistent with our observations regarding cell morphology and GDPH activity (Fig. 4). PPARγ is the master regulator of adipocyte gene transcription. Adipocytes clearly demonstrate adipose-specific characteristics with the expression of PPARγ. In my study, the expression level of PPARγ was relatively low at day 5; with the prolonging of the culture time, the mRNA level of PPARγ increased gradually during the cells' differentiation.

The time-spatial expression of PPARγ mRNA levels in grass carp adipocytes. At days 5, 10, 15, and 20, total RNA was isolated and subjected to RT-PCR analysis. PPARγ mRNA abundance assigned as a ratio to β -actin mRNA. Columns and error bars are represented as means ± SE of six independent determinations.
Discussion
Our current understanding of adipocyte biology is largely derived from rodent models and the cell culture system. Although these model systems provide abundant knowledge with revealing cellular and molecular mechanisms governing adipocyte differentiation and physiology, many important aspects of adipocyte development and adipose depot formation remain poorly understood, and species and cell type specificity often become a point of debate, especially when precursor cells that originated from adipose tissue from various species and were maintained in primary cultures behave in some ways different from established mouse cell lines (Boone et al. 1999; Cartwright et al. 2010; Poulos et al. 2010). Short-term primary cultures have been performed in several studies using rat (Wardzala et al. 1978), mouse (Barbaras et al. 1985), porcine (Ding and Mersmann 2001; Li et al. 2006), human (Hauner et al. 1989; Wabitsch et al. 2001), and Atlantic salmon (Vegusdal et al. 2003) preadipocytes, but little is known about primary culture of adipocytes in fish. Thus, I established a long-term culture system of grass carp preadipocytes to study the development of adipocytes.
In this study, cells were seeded and maintained at 28°C in a humidified 5% CO2 atmosphere, and they started to attach and grow on day 3, while Atlantic salmon preadipocytes were incubated at 13°C with 3% CO2, and most cells were attached on the next day (Vegusdal et al. 2003). The differences of culture temperature between the two cell types are related to the living environment that the fish live in. Atlantic salmon lives in cold water, while grass carp lives in warm water. This was probably the reason why there was the temperature difference between the two culture systems. In addition, the attachment time of grass carp cells was later when compared with other preadipocyte types. This may be due to the species difference.
The development of adipose tissue is the result of the increase not only in adipocyte number but also in adipocyte size. I studied the morphological change, proliferation kinetics, and differentiation degree from preadipocyte to mature adipocyte. Morphologically, the majority of cells underwent attachment, proliferation, and growth arrest with subsequent accumulation of intracellular lipid droplets, before finally developing into mature adipocytes. These changes are consistent with the morphology characteristic of adipocytes in vitro (Mills 2000; Brandebourg and Hu 2005), but the intracellular lipid droplets around the nuclei were smaller and more numerous in the differentiating fish adipocyte when compared with the rat, porcine, and human adipocyte. Likewise, the proliferation progress of grass carp fat cells showed a unique S-shaped growth curve that is the common growth pattern of cells and had a 10-d growth period. Furthermore, adipocytes clearly demonstrated adipose-specific characteristics. GPDH activity, which is a late marker for adipocyte differentiation (Hauner et al. 1989), was low in undifferentiated preadipocytes and increased as differentiation progressed and the cells accumulated lipid droplets. PPARγ is the master regulator of adipocyte gene transcription (Hauner et al. 1989; Ding and Mersmann 2001; Brandebourg and Hu 2005). Analysis of RT-PCR confirmed that PPARγ expression patterns were consistent with my observations regarding GPDH activity from days 5 to 20 of the culture period. The morphological and biochemical data presented here show that fish preadipocytes have properties that are similar to those of preadipocytes in mammals.
In summary, the data described here represent, to the best of our knowledge, an attempt to develop an understanding of the fundamental culture system of grass carp preadipocytes. The results demonstrated that I provided a convenient and efficient culture system for grass carp preadipocytes, which will be of great value for studying the lipogenesis events of fish adipose tissue. The profound studies on the 1ipogenesis in adipocyte adipogenesis will be explored in future work.
References
Barbaras R.; Grimaldi P.; Negrel R.; Ailhaud G. Binding of lipoproteins and regulation of cholesterol synthesis in cultured mouse adipose cells. BBA–Mol. Cell. Res. 845: 492–501; 1985.
Boone C.; Gregoire F.; Remacle C. Regulation of porcine adipogenesis in vitro, as compared with other species. Domest. Anim. Endocrin. 17: 257–267; 1999.
Brandebourg T. D.; Hu C. Y. Regulation of differentiating pig preadipocytes by retinoic acid. J. Anim. Sci. 83: 98–108; 2005.
Cartwright M. J.; Schlauch K.; Lenburg M. E.; Tchkonia T.; Pirtskhalava T.; Cartwright A.; Thomou T.; Kirkland J. L. Aging, depot origin, and preadipocyte gene expression. J. Gerontol. A-Biol. 65: 242–251; 2010.
Ding S. T.; Mersmann H. J. Fatty acids modulate porcine adipocyte differentiation and transcripts for transcription factors and adipocyte-characteristic proteins. J. Nutr. Biochem. 12: 101–108; 2001.
Hauner H.; Entenmann G.; Wabitsch M.; Gaillard D.; Ailhaud G.; Negrel R.; Pfeiffer E. F. Promoting effect of glucocorticoids on the differentiation of human adipocyte precursor cells cultured in a chemically defined medium. J. Clin. Invest. 84: 1663–1670; 1989.
Kim J.; Sarraf P.; Wright M.; Yao K. M.; Meuller E.; Solanes G.; Lowell B. B.; Spiegelman B. M. Nutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1. J. Clin. Invest. 101: 1–9; 1998.
Kristen J.; Nadeau J.; Wayne L.; Inga G.; Boris D. Insulin regulation of sterol regulatory element-binding protein-1 expression in L-6 muscle cells and 3T3L1 adipocytes. J. Biol. Chem. 279: 34380–34387; 2004.
Li Y.; Lu R. H.; Luo G. F.; Pang W. J.; Yang G. S. Effects of different cryoprotectants on the viability and biological characteristics of porcine preadipocyte. Cryobiology 53: 240–247; 2006.
Maria M.; Simona M.; Daniela D. R.; Luca P.; Giovanni S. Semiquantitative RT-PCR analysis to assess the expression levels of multiple transcripts from the same sample. Biol. Proced. Online 3: 19–25; 2001.
Mills S. Beta-adrenergic receptor subtypes mediating lipolysis in porcine adipocytes. Studies with BRL-37344, a putativeβ3-adrenergic agonist. Comp. Biochem. Phys. C 126: 11–20; 2000.
Nakatani H.; Aoki N.; Okajima T.; Nadano D.; Flint D. J.; Matsuda T. Establishment of a mammary stromal fibroblastic cell line for in vitro studies of mammary adipocyte differentiation. Biol. Reprod. 82: 44–53; 2010.
Palmer D. G.; Rutter G. A.; Tavare J. M. Insulin-stimulated fatty acid synthase gene expression does not require increased sterol response element binding protein 1 transcription in primary adipocytes. Biochem. Bioph. Res. Co. 291: 439–443; 2002.
Peterson G. L. A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal. Biochem. 83: 346–356; 1977.
Poulos S. P.; Dodson M. V.; Hausman G. J. Cell line models for differentiation: preadipocytes and adipocytes. Exp. Biol. Med. 235: 1185–1193; 2010.
Ramirez-Zacarias J. L.; Castro-Munozledo F.; Kuri-Harcuch W. Quantitation of adipose conversion and triglycerides by staining intracytoplasmic lipids with oil red o. Histochem. Cell Biol. 97: 493–497; 1992.
Rayalam S.; Della-Fera M. A.; Baile C. A. Phytochemicals and regulation of the adipocyte life cycle. J. Nutr. Biochem. 19: 717–726; 2008.
Vegusdal A.; Sundvold H.; Gjøen T.; Ruyter B. An in vitro method for studying the proliferation and differentiation of Atlantic salmon preadipocytes. Lipids 38: 289–296; 2003.
Wabitsch M.; Brenner R. E.; Melzner I.; Braun M.; Möller P.; Heinze E.; Debatin K. M.; Hauner H. Characterization of a human preadipocyte cell strain with high capacity for adipose differentiation. Int. J. Obesity 25: 8–15; 2001.
Wardzala L. J.; Cushman S. W.; Salans L. B. Mechanism of insulin action on glucose transport in the isolated rat adipose cell. Enhancement of the number of functional transport systems. J. Biol. Chem. 253: 8002–8005; 1978.
Acknowledgments
This research was sponsored by the National Natural Science Foundation of China (30800843) and Chongqing Natural Science Foundation Project (cstc2011jjA80016).
Author information
Authors and Affiliations
Corresponding author
Additional information
Editor: T. Okamoto
Rights and permissions
About this article
Cite this article
Li, Y. Establishment and evaluation of a new model for studying lipogenesis in grass carp (Ctenopharyngodon idella) preadipocytes. In Vitro Cell.Dev.Biol.-Animal 48, 37–42 (2012). https://doi.org/10.1007/s11626-011-9474-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11626-011-9474-8