
Abstract
Background
The aims of this study are to evaluate the effect of Speckle-type POZ protein (SPOP) in colorectal cancer (CRC) patients and explore its significance in the prognosis.
Methods
We used immunohistochemistry to detect the expression of SPOP in CRC. Moreover, this result was further confirmed at the protein and messenger RNA (mRNA) level in paired CRC specimens and matched adjacent noncancerous colon tissues by Western blotting and real-time quantitative PCR (qRT-PCR), respectively. Furthermore, we evaluate the effects of SPOP on CRC cell proliferation and migration in vitro. The Kaplan–Meier method and log-rank test were employed to compare the overall survival between SPOP low expression group and SPOP high expression group. Correlation of survival with clinicopathologic parameters, including SPOP level, was investigated with multivariate analyses.
Results
We confirmed frequent SPOP downregulation in both mRNA (P = 0.0286) and protein (P = 0.004) levels in CRC tissues as compared to matched adjacent nontumorous tissues. Besides, the downregulated SPOP expression in CRC tissues was significantly correlated to poor differentiation (P = 0.013), distant metastasis (P = 0.003), gross type (P < 0.001), and high TNM stage (P = 0.002). Kaplan–Meier survival analysis showed that low SPOP expression exhibited a significant correlation with poor prognosis for CRC patients. Overexpression of SPOP in CRC cell lines significantly suppressed cell proliferation, migration, and clone formation. In contrast, SPOP knockdown dramatically promoted cell proliferation, migration, and clone formation in vitro. In addition, overexpression of SPOP increased E-cadherin and suppressed vimentin in HCT116 cells and silencing of SPOP reversed all these biomarkers. Furthermore, SPOP significantly downregulated MMP2 and MMP7 protein levels in HCT116 cell lines.
Conclusion
Our results suggest that SPOP plays a pivotal role in colorectal cancer (CRC) through mesenchymal–epithelial transition and MMPs, and it may be a potential therapeutic target in colorectal cancer.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Speckle-type POZ protein (SPOP) is a TRAF domain (TD) and POZ-containing nuclear speckle-associated protein, and as an E3 ubiquitin ligase adaptor, it contains three domains: an N-terminal MATH domain that recruits substrates, an internal BTB domain that binds Cul3, and a C-terminal nuclear localization sequence.1 It is a member of the MATH-BTB protein family which constitutes Cul3-based ubiquitin ligases and functions as a CRL adaptor protein in complex with Cul3.2 SPOP binds with Cul3 ubiquitin ligase through the POZ domain and interacts with substrate proteins via the TD domain to form an ubiquitin ligase complex, and it can form heteromeric species with the closely related paralog SPOP-like (SPOPL). In combination, SPOP and SPOPL form a molecular rheostat that can fine-tune E3 ubiquitin ligase activity by affecting the oligomeric state of the E3 complex.3, 4 Mounting evidence indicates that dysregulation of the ubiquitin-proteasome pathway is involved in cancer pathogenesis. A recent report has been shown that SPOP involve in mediating the polyubiquitylation and subsequent proteolysis of the oncogenic steroid receptor SRC-3, which indicated a tumor suppressor role.5
Recent genome-wide somatic mutation analyses identified that SPOP gene encoding the speckle-type POZ protein was frequently mutated in some human cancers, including 8 % of serous endometrial cancers,6 2 % of lung cancers,7 2.2 % CRC, and 4.4 % prostate cancer harbored SPOP somatic mutations. However, SPOP mutation was rare and may not play an important role in gastric cancer and CRC pathogenesis, which suggested that somatic mutation and loss of expression in SPOP gene might be separate events in the cancers.8 In addition to the somatic mutation, altered expression of ubiquitination-related genes is observed in many cancers9, 10 and SPOP protein expression is often downregulated in prostate tumors.8 An earlier study described analyzed SPOP expression in a wide range of normal and neoplastic tissues. They found that gastric and colon cancers expressed weak or negative SPOP expression. Also, normal tissue of stomach and colon showed weak or negative SPOP expression as well.11 However, how this contributes to colorectal cancer pathogenesis and progression remains to be unknown. It is well known that the development of CRC is a long and complicated process, including the activation of multiple oncogenes and the inactivation of tumor suppressor genes. In this study, we sought to determine SPOP expression in human CRC patient specimens and evaluate the clinicopathologic implications of it in CRC, in an attempt to discover the potential influence of SPOP in the development of CRC.
Material and Methods
Patients, Tissue Samples, and CRC Cell Lines
A total of 126 paraffin-fixed colorectal cancer and matched normal tissue samples from patients who underwent surgery were obtained between 2007 and 2009 at the Department of General Surgery, Affiliated Hospital of Nantong University, were formalin-fixed and paraffin-embedded for histopathologic diagnosis and immunohistochemical study. The matched nontumor tissues were taken more than 3 cm from the tumor margin and confirmed by pathology. Fresh CRC and surrounding nontumor tissue samples were randomly obtained from 40 CRC patients who underwent surgical resection at the Affiliated Hospital of Nantong University between 2012 and 2013. Furthermore, fresh samples were frozen in liquid nitrogen immediately after surgical removal and maintained at −80 °C until use for the real-time RT-PCR and Western blot analysis. The clinical features of the patients, including age, histologic grade, tumor size, lymph node status, TNM stage, and histology were also collected. The TNM system of tumor staging and histological grade were performed according to the World Health Organization guidelines. The follow-up data from the CRC patients in this study were available and complete. Overall survival, which was defined as the time from the operation to the time of patient death or the last follow-up, was used as a measure of prognosis. All human tissue was collected using protocols approved by the Ethics Committee of Affiliated Hospital of Nantong University.
Five human CRC cell lines, Caco-2, SW480, HCT116, SW620, and LoVo were all obtained from the Cell Bank of the Committee on Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). HCT116 and Caco-2 cells were grown in DMEM medium (Gibco) with 10 % fetal bovine serum (FBS, HyClone, UT). SW480 and SW620 cells were grown in Leibovitz’s L-15 medium (Gibco) with 10 % FBS. LoVo cells were grown in RPMI-1640 medium (Gibco) with 10 % FBS. All cell lines were cultured in humidified incubator at 37 °C with 5 % CO2.
Transfection of siRNA
For small interfering RNA (siRNA) silencing of SPOP, RNA interference was performed by using synthetic siRNA duplexes. According to sequences of human SPOP gene sequence (NM_001007226.1), we designed, synthesized four siRNAs, and then transfected into HCT-116 cells, respectively, in our preliminary experiments, and both siRNA1 and siRNA2 could effectively reduce endogenous SPOP expression by Western blotting assay. Therefore, siRNA1 and siRNA2 were chosen for downstream experiment. SPOP siRNAs and scrambled siRNA (NC-siRNA) were purchased from GenePharma (Shanghai, China). The targeting sequences were as follows: si-SPOP1: 5′-GGGTTAGATGAAGAAAGCA-3′, si-SPOP2: 5′-CCTCGACAAAATGGCTGAT-3′. The target sequence for scrambled siRNA was 5′-GCAGATAGGTAGGCGTTAT-3′. Si-SPOP or scramble siRNA were transfected into HCT-116 cells with 400 pmol, respectively, using Lipofectamine RNAiMAX transfection reagent (Invitrogen Life Technologies) according to the manufacturer’s instructions. After 24 h of transfection, cells were harvested for cell proliferation, migration, and colony formation assays.
Plasmid Construction and Transfection
SPOP overexpression vector pEGFP-N1-SPOP (p-SPOP) was constructed using the overlap PCR or PCR method. The PCR products were confirmed by direct DNA sequencing and cloned into the mammalian expression vector pEGFP-N1. HCT116 cell lines were used for the overexpression studies. We obtained stably transfected clones by G418 selection (Promega). A stable transfectant of the pEGFP-N1 empty vector was used as a control. For transfection, complexes of Lipofectamine 2000 (Invitrogen, Carlsbad, USA) and one of the plasmids mentioned above was prepared according to the manufacturer’s instructions. The level of SPOP expression after transfection was assayed by Western blot assay.
Immunohistochemistry
The expression of SPOP protein was determined by immunohistochemical (IHC) analysis. The sections were deparaffinized using a graded ethanol series, and endogenous peroxidase activity was blocked by soaking in 0.3 % hydrogen peroxide. Antigen retrieval was performed by heating (100 °C) each section for 10 min in 0.01 mol/L sodium citrate buffer (pH = 6.0). After three rinses (each for 5 min in phosphate-buffered saline (PBS)), sections were incubated for 2 h at room temperature with goat polyclonal anti-human SPOP antibody (1:100, sc-66649, Santa Cruz Biotechnology, Inc., USA) diluted (1:200) in PBS. After three washes (each for 5 min in PBS), the sections were then incubated with a donkey anti-goat second antibody conjugated horseradish peroxidase (1:5000; Abcam, Cambridge, UK) at 4 °C overnight. After washing in PBS, the visualization signal was developed with 3,3′-diaminobenzidine (DAB) solution, and all of the slides were counterstained with hematoxylin, dehydrated, and coverslipped.
For visual assessment, the assessment of immunostaining was performed independently by two pathologists who were blinded to clinical data of patients. The score for the percentage of positive tumor cells was graded as the immunohistochemical staining scores (ISS): no staining is scored as 0, 1 ~ 25 % of cells stained scored as 1, 25 ~ 50 % as 2, 51 ~ 75 % as 3, and >75 % as 4. Moreover, staining intensity is rated on a scale of 0 to 3, with 0, negative; 1, weak; 2, moderate; and 3, strong. We created an immunoreactive score by multiplying the score for the percent of positive cells and the score for staining intensity. Theoretically, the ISS could range from 0 to 12. An ISS of 9 ~ 12 was considered strong immunoreactivity (+++), 5 ~ 8 was considered moderate (++), 1 ~ 4 was considered weak (+), and 0 was scored as negative (−). Sections were considered positive for SPOP when more than 25 % of tumor cells were stained in the cell cytoplasm.
Western Blotting
Coupled tumor and their adjacent nontumor tissues were taken for Western blot analysis. Total protein was extracted by lysis buffer containing protease inhibitors (Promega, Madison, WI). Equal amounts of protein separated by 10 % sulfate polyacrylamide gel electrophoresis and then transferred to a polyvinylidene fluoride (PVDF) membrane. Nonspecific binding was blocked for 2 h with 5 % nonfat milk in Tris-buffered saline containing 0.1 % Tween 20 (TBST). After incubation with anti-SPOP antibody (sc-66649, Santa Cruz Biotechnology, USA), anti-E-cadherin antibody (ab1416, Abcam, Cambridge, MA, USA), anti-vimentin antibody (ab92547, Abcam), anti-N-cadherin antibody (ab18203, Abcam), anti-Snail antibody (ab180714), anti-Slug antibody (ab27568, Abcam), and anti-Twist antibody (ab49254, Abcam) overnight at 4 °C, membranes were washed three times in TBST for 5 min and subsequently incubated secondary antibody with conjugated IRDye800 (1:5000, Rockland Gilbertsville, CA) at room temperature for 2 h, followed by scanning with an Odyssey infrared imaging system (LI-COR, Lincoln, NE), and analyzed with PDQuest 7.2.0 software (Bio-Rad).
Real-Time RT-PCR
The messenger RNA (mRNA) expression of SPOP was analyzed by quantitative real-time PCR. Total RNAs were extracted using the Trizol (Gibco). Quantitative real-time PCR was performed using HotStart-IT SYBR Green QPCR Master Mix (2×; USB Corporation, Cleveland, OH 75762, USA). According to the HotStart-IT protocol, 25-μl reactions were run of with 2 μl cDNA. RT-PCR experiments were performed in a LightCycler 480 system (Roche Applied Sciences). Cycling parameters were as follows: hot start at 95 °C for 10 min; 40 cycles of amplification/quantification at 95 °C for 10 s, 60 °C for 30 s, and 72 °C for 30 s during which time fluorescence was measured. Melting curve analysis was performed using continuous fluorescence acquisition from 65 to 97 °C. These cycling parameters generated single amplicons for both primer sets used according to the presence of a single melt peak. The β-actin was selected as the internal reference. All quantitative real-time PCRs were repeated three times for each gene, and each sample was done in triplicate. Primer sequences were as follows: SPOP forward 5′-TGACCACCAGGTAGACAGCG-3′, SPOP reverse 5′-CCCGTTTCCCCCAAGTTA-3′. The β-actin gene served as an internal control. The sequences of the primers for β-actin were as follows: β-actin forward 5′-AACTTCCGTTGCTGCCAT-3′, β-actin reverse 5′-TTTCTTCCACAGGGCTTTG-3′. Relative expression level for each target gene was normalized by the Ct value of β-actin (internal control) using a 2ΔΔCt relative quantification method.
Cell Proliferation Assay
Cell viability and proliferation was assessed using an MTT assay. Following transfection, HCT116 and LoVo cells were plated in 96-well plates at 1 × 103 cells/well and incubated for 24, 48, and 72 h. A total of 20 μl MTT (5 mg/ml, Sigma-Aldrich) was added to each corresponding test well and incubated for 4 h at 37 °C. The supernatant was then discarded, and 200 μl dimethyl sulfoxide (DMSO) (Sigma, St. Louis, MO, USA) was added to each well to dissolve the formazan. Optical density was assessed by measuring the absorbance of each well at 490 nm using a spectrophotometer (SpectraMax Plus384; Molecular Devices, Sunnyvale, CA, USA). All experiments were performed in triplicate.
Colony Formation Assay
After transfection of SPOP-siRNA/p-SPOP or control vector, HCT116 and LoVo cells (5 × 104 cells/well) were separately plated in a 24-well plate. After 24 h, the cells collected and seeded (1000–1500/well) in a fresh six-well plate for 14 days. Surviving colonies (>50 cells per colony) were counted after fixed with methanol/acetone (1:1) and stained with 5 % Gentian Violet (ICM Pharma, Singapore, Singapore), and then rinsed three times with PBS to remove excess dye, photographed, and counted. The experiment was carried out in triplicate wells for three times.
Cell Migration Assay
The cell migratory capacity was determined using transwell chambers with 8-mm diameters (Corning, Corning, NY, USA). Briefly, HCT116 and LoVo cells (1 × 105/well) were transfected and suspended in 100-μl serum-free medium and then added to the upper chamber of the inserts; DMEM medium (Gibco) containing 10 % FBS (500 μl) was added to the lower chamber as a chemoattractant. After culture for 24 h at 37 °C, nonmigrated cells on the upper surface were removed gently with a cotton swab and cells that migrated to the lower side of the department were fixed with methanol and dyed with 0.1 % crystal violet. Then, cells that migrated through the upper chamber were counted in five random fields under a light microscope, and the average value of five fields was expressed. All assays were performed in triplicate.
Wound Healing Assay
Cells were plated in six-well plates and incubated overnight; when the cells had grown by 90–100 %, they were scraped with a pipette tip to generate straight wounds. To eliminate dislodged cells, culture medium was removed and wells were washed with PBS. We measured the closure or filling of the wounds at 0, 24, 48, and 72 h using the phase-contrast microscope with a magnification × 100. To ensure documentation of the same region, the wells were marked across the wounded area. The untreated cells served as controls. Each experiment was repeated in triplicate.
Statistical Analysis
The SPSS 17.0 statistical software (SPSS Inc., Chicago, IL, USA) was applied for statistical analysis. The association between SPOP expression and clinicopathologic features was analyzed using χ 2 test. Spearman rank correlation analysis was used to analyze the relationship between SPOP expression in CRC and TNM stage. Survival was calculated by the Kaplan–Meier method, and differences in survival were determined by the log-rank analysis. A multivariable analysis of several independent prognostic factors was carried out using Cox’s proportional hazards regression model. The results are expressed as the mean ± SD of at least three independent experiments. P < 0.05 was considered statistically significant.
Results
Downregulation of SPOP in CRC Tissue Samples
To investigate SPOP gene expression profile, a total of 126 paraffin-embedded colorectal tissue blocks were evaluated by IHC analysis. We found that SPOP was expressed at various levels in the CRC tissues and the adjacent noncancerous tissue samples (Fig. 1). Overall, 78 of 126 (61.90 %) cases showed low SPOP expression (SPOP − or SPOP +) in CRC tissues, whereas the remaining 48 (38.10 %) cases displayed high SPOP expression (SPOP ++ or SPOP +++) (Fig. 1, Table 1). The adjacent noncancerous colorectal tissues showed the strongest SPOP-positive staining (Fig. 1a).

SPOP protein expression in CRC and surrounding nontumorous tissues detected by immunohistochemistry. a Normal CRC tissues distant from the tumor were scored as SPOP (+++); b well-differentiated CRC was scored as SPOP (++); c moderately differentiated CRC was scored as SPOP (+); and d Poorly differentiated CRC was scored as SPOP (−). Original magnification × 200
The protein expression of SPOP in CRC patients was forward examined by Western blotting analysis in 24 randomly selected pairs of CRC and their matched nontumor colorectal tissues. Five representative cases of the Western blotting result are shown in Fig. 2a. The relative quantity of SPOP protein expression was normalized to the β-actin of the same samples. SPOP protein was downregulated in the CRC tissues compared with their adjacent nontumor colorectal tissues in most of CRC patients (75 %, 18 of 24 patients), and the average SPOP protein level in 24 CRC tissues was significantly lower than that in their adjacent nontumor colorectal tissues (P = 0.004) (Fig. 2b).

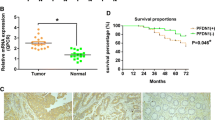
Expression of SPOP in CRC tissues as evaluated by Western blotting and real-time quantitative RT-PCR. a Representative result of SPOP protein expression in five paired CRC and the matched adjacent nontumorous tissues (T, CRC; N, matched nontumorous colon mucosa). b Relative SPOP protein expression level was remarkably decreased in 18 of 24 (75 %) CRC tissues compared with the corresponding adjacent nontumorous tissues (n = 24, P = 0.004). c The relative mRNA expression of SPOP was significantly decreased in CRC tissues compared to matched adjacent nontumor tissues (n = 42) assessed by qRT-PCR (P = 0.0286)
To check SPOP mRNA level in CRC tissues, we performed real-time RT-PCR analysis in the 42 colorectal tumor tissues and their corresponding nontumor mucosal samples. As shown in Fig. 2c, the SPOP expression was significantly reduced in colorectal cancer samples as compared with that in their corresponding nontumor mucosal samples (P = 0.0286).
Correlation Between SPOP Relative Expression and Clinicopathologic Factors in CRC
The degree of SPOP immunohistochemical staining correlates with clinicopathologic characteristics. Clinicopathologic factors were compared between two SPOP expression groups (Table 1). We used χ 2 test to analyze the correlation between immunohistochemical SPOP expression and various clinicopathologic characteristics of CRC patients. In colorectal tumor tissues from 126 CRC patients, decreased SPOP expression was found to be significantly correlated to poor differentiation (P = 0.013), distant metastasis (P = 0.003), gross type (P < 0.001), and high TNM stage (P = 0.002). Nevertheless, there was no significant correlation between SPOP expression and age, gender, location, lymph node metastasis, invasive depth, and tumor size (P > 0.05; Table 1). To further clarify the relationship between SPOP expression and TNM stage, Spearman rank correlation analysis was employed. The result showed that SPOP expression in CRC was negative correlation with TNM stage, which suggested the more advanced clinical TNM stage corresponding to the lower expression level of SPOP in CRC (r s = 0.306, P < 0.001; Table 2). The above results were in line with its role as the tumor suppressor in other cancers.
To further estimate the correlation between SPOP expression and prognosis of patients with CRC, Kaplan–Meier survival analysis and log-rank tests were performed using patients’ postoperative survival time. For the purpose of finding the further effect of SPOP, we analyzed survival based on SPOP positivity by separating stages I–II to stages III–IV. In early stage (stages I and II) CRC, the patients with the low levels of SPOP expression (n = 37) had a poorer prognosis than the patients with high levels of SPOP expression (n = 36) (χ 2 = 5.074, P = 0.024, Fig. 7a). Meanwhile, in late stage (stages III and IV) CRC, the patients with the low levels of SEMA3A expression (n = 41) also had a poorer prognosis than the patients with high levels of SEMA3A expression (n = 12) (χ 2 = 6.506, P = 0.011, Fig. 7b). With the above, the overall survival of the low level expression group was significantly longer than that of the high level expression group, no matter in the stages I–II or stages III–IV. The results indicated that patients with low SPOP expression had an obviously poorer prognosis than those with high SPOP expression.
A multivariate analysis was performed to evaluate the independent prognostic roles of SPOP after adjusting for other significant covariates. All variables that significantly affected survival in univariate analysis were introduced into a Cox proportional hazard model (Table 3). We can know that TNM stage(P < 0.001), histological differentiation (P = 0.027), distant metastasis (P = 0.032), and SPOP expression (P = 0.001) influenced overall survival in CRC patients.
Decreased SPOP Expression in Colorectal Cancer Cell Lines
To determine the expression level of the SPOP protein in cell lines, five colorectal cancer cell lines were analyzed using Western blotting. The expression of SPOP was detected in all cell lines at levels that higher SPOP expression was detected in LoVo cell line, and lower SPOP expression was detected in HCT-116 cell line when compared with the other colorectal cancer cell lines (Fig. 3).

Expression of SPOP in CRC cells. a, b The SPOP protein level was remarkably decreased in CRC cell lines Caco-2, SW480, HCT116, SW620, and LoVo using Western blotting analysis. β-Actin level was considered loading control
Downregulation of SPOP Enhanced the Growth and Tumorigenicity of Colorectal Cancer Cells In Vitro
To analyze the effects of SPOP on the malignant phenotype of colorectal cancer cells, LoVo cells were transfected with two SPOP siRNA-containing plasmids (SPOP siRNA1 and SPOP siRNA2). After G418 selection, Western blotting analysis confirmed that the stable clones expressing the SPOP siRNA showed lower expression levels of the SPOP protein compared with LoVo/pSilencer cells transfected with the empty vector control. In addition, SPOP siRNA2 decreased the level of endogenous SPOP protein more effectively than SPOP siRNA1, but the difference is not statistically significant (Fig. 4a). The results from the MTT assay showed that from the fourth day onward, SPOP siRNA significantly stimulated colorectal cancer cell proliferation compared with the control cells (Fig. 4b). Next, the colony formation ability of transfected and control cells was measured on plates. LoVo/siRNA1 and LoVo/siRNA2 lines resulted in more colonies than the control cells plate (Fig. 4c, d) assays, demonstrating that suppression of SPOP could increase the transformation phenotype of colorectal cancer cells in vitro.

Knockdown of SPOP expression by siRNA stimulated growth potential in vitro. a Western blotting analysis of SPOP protein expression levels. β-Actin level was considered loading control. The two different SPOP siRNA transfectants are indicated by “siRNA1” and “siRNA2,” and the empty vector transfectant is indicated by “Vector.” b Growth rates of the SPOP siRNA-transfected CRC cells and control cells in vitro. Cell numbers were evaluated with the MTT assay using absorbance readings at 490 nm at the indicated times. The values shown are the mean of three determinations. c, d Colony numbers of the LoVo/siRNA1, LoVo/siRNA2, and control cells in plate assays. The data represent the means ± SEM of three independent experiments. e, f Knockdown of SPOP expression promotes cell migration in LoVo cells as demonstrated by transwell assays (*P < 0.05). Representative photos of stained cells are shown with the original magnification of × 100. g, h LoVo cells were transfected with siRNA or empty vector for 0, 24, 48, and 72 h, respectively (*P < 0.05 compared to empty vector)
Furthermore, the impact of SPOP on cellular migration was further confirmed by transwell assays after SPOP knockdown in LoVo cells. The results showed knockdown of SPOP significantly promoted the migratory capacity of LoVo cells compared with that of control cells (Fig. 4e, f, P < 0.05). These findings were further confirmed by the wound healing assay. The knockdown of SPOP significantly increased the migration of LoVo cells at 48 h after transfection (Fig. 4g, h, P < 0.05). Taken together, these data suggest that suppression of SPOP by target siRNA may enhance the malignant growth potential of colorectal cancer cells in vitro.
Upregulation of SPOP Inhibited the Growth and Tumorigenicity of Colorectal Cancer Cells In Vitro
Because the downregulation of SPOP enhanced the growth and tumorigenicity of colorectal cancer cells, the possibility that its upregulation could inhibit the growth and tumorigenicity of colorectal cancer cells was investigated. HCT-116 cells were transfected with recombinant plasmids containing the full ORF of the wild-type SPOP gene. After G418 selection, Western blotting analysis confirmed that the stable clone transfected with the SPOP recombinant plasmid showed higher expression levels of the SPOP protein compared with HCT-116/pEGFP-N1 cells transfected with the empty vector control (Fig. 5a). The effect of SPOP overexpression on the growth of the colorectal cancer cell line HCT-116 was investigated using an MTT assay. After transfection with the gene encoding SPOP, the HCT-116/SPOP cells grew at a significantly slower rate than the control cells (Fig. 5b). Colony formation assays are an effective method to evaluate the proliferative ability and tumorigenicity of single cells in vitro. Therefore, colony formation assays were used to determine whether the upregulation of SPOP expression inhibited colorectal cancer cell growth. There was a substantial reduction in the number of colonies produced by HCT-116/SPOP cells when compared with control cells in plate (Fig. 5c, d) assays, demonstrating that SPOP can suppress the transformation phenotype of colorectal cancer cells in vitro.

Exogenous overexpression of SPOP inhibited the growth potential of CRC cells in vitro. a Western blotting analysis of SPOP protein expression levels. β-Actin level was considered loading control. The SPOP sense expression transfectant is indicated by “SPOP,” and the empty vector transfectant is indicated by “Vector.” b Growth rates of the HCT-116/SPOP and control cells in vitro. Cell numbers were evaluated with the MTT assay using absorbance readings at 490 nm at the indicated times. The values shown are the means of three determinations. c, d Colony numbers of the HCT-116/SPOP and control cells in plate assays. The data represent the means ± SEM of three independent experiments. e, f Overexpression of SPOP inhibits cell migration in HCT-116 cells as demonstrated by transwell assays (*P < 0.05). Representative photos of stained cells are shown with the original magnification of × 100. g, h HCT-116 cells were transfected with p-SPOP or empty vector for 0, 24, 48, and 72 h, respectively (*P < 0.05 compared to empty vector)
As SPOP expression is associated with distant metastasis in CRC patients, we evaluated the potential role of SPOP on cellular migration by transwell assays. HCT-116 cells were transfected with SPOP overexpressing or control plasmid and seeded in the chamber, and their migratory abilities were determined 24 h later. The results showed overexpression of SPOP significantly decreased the migratory capacity of HCT-116 cells (Fig. 5e, f, P < 0.05). These findings were further confirmed by the wound healing assay. The overexpression of SPOP significantly inhibited the migration of HCT-116 cells at 48 h after transfection (Fig. 5g, h, P < 0.05). Taken together, these data suggest that SPOP may have a suppressive effect and upregulation can partly reverse the malignant growth potential of colorectal cancer cells both in vitro.
SPOP Induces EMT Phenotypes and Inhibition of Matrix MMPs
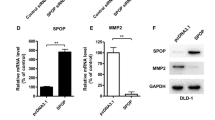
Epithelial–mesenchymal transition (EMT) is an important process during tumor progression by which the epithelial cells acquire mesenchymal properties and show reduced intercellular adhesion and increased motility.12 E-cadherin and vimentin are the markers of epithelial cells and mesenchymal cells, respectively.13 To define the role of SPOP in the progression of cell migration within CRC cells, the changes in expression of epithelial and mesenchymal markers were detected following modulation of the SPOP expression level. The overexpression of the SPOP levels within the HCT116 cells was found to be associated with the upregulated E-cadherin and downregulated vimentin expression at the protein levels. Vimentin expression was upregulated and E-cadherin was downregulated in the SPOP-downregulated LoVo cells (Fig. 6a–d). Overexpression of SPOP also downregulated the expression of other EMT markers, such as N-cadherin, Slug, and Twist in HCT116 cells (Fig. 6g, h). However, inhibition of SPOP expression reversed all these EMT markers expression in LoVo cells by Western blot analysis (Fig. 6i, j).

a, b SPOP expression increased E-cadherin expression but suppressed vimentin expression in HCT116 cells transfected with SPOP overexpression plasmid compared with the control group by Western blot analysis. However, SPOP knockdown reversed all these changes in LoVo cells by Western blot analysis (c, d). e, f SPOP significantly downregulated MMP2 and MMP7 protein levels in HCT116 cell lines. g, h Overexpression of SPOP downregulated the expression of N-cadherin, Slug, and Twist in HCT116 cells. However, inhibition of SPOP expression reversed all these changes in LoVo cells by Western blot analysis (i, j). (*P < 0.05 compared to empty vector)
The matrix metalloproteinases (MMPs) are family members of extracellular proteinases that regulate basic cellular processes including survival, migration and morphogenesis and degradation extracellular matrix (ECM) during the cancer metastatic process.14, 15 Among the MMPs members, MMP2 and MMP7 play a prominent role in CRC cell invasion and dissemination.16 Therefore, we examined the expression levels of these two proteinases and found that ectopic expression of SPOP markedly suppressed protein expression of MMP2 and MMP7, concomitant with the inhibition of cell migration and invasion (Fig. 6e, f).
Discussion
CRC is the third most prevalent cancer worldwide.17 Death of these patients results from uncontrolled metastatic disease, including peritoneum metastases, lymph nodes metastases, and liver metastases.18 Many risk factors, such as inflammatory bowel disease, inherited syndrome, diets low in fiber, smoke, obesity, and heavy alcohol use, are related to intestinal mucosa injury and dysplasia and may result in carcinogenesis of CRC.19, 20 CRC frequently migrates through the lymphatic route, depositing tumor cells into local lymph nodes, namely lymph node metastasis (LNM). Tumor metastasis is responsible for approximately 90 % of all cancer-related deaths.21 Despite substantial progress achieved in diagnosis and treatment for CRC in recent years, the overall 5-year survival rate remains unsatisfactory due to metastasis leading to poor outcomes22 and the underlying molecular mechanisms for the organ-oriented metastasis of CRC are not fully understood. Therefore, it is a necessary to establish novel prognostic markers for cancer recurrence and survival to improve treatment for individual patients.
The SPOP protein has been previously localized to nuclear bodies in a speckled pattern, thus prompting the acronym SPOP for Speckle-type POZ-domain protein and was initially identified as an autoantigen of scleroderma patients.23 SPOP has roles in maintaining normal cellular growth and development as indicated by the high incidence of both SPOP gene overexpression and SPOP gene copy loss in a number of human cancers. The SPOP genomic locus has also been shown to harbor a high percentage of genomic loss or loss of heterozygosity in breast cancers.7 These reports further link SPOP to the carcinogenesis process. Furthermore, we demonstrate that restoration of SPOP expression inhibited SRC-3-mediated oncogenic signaling and tumorigenesis, thus positioning SPOP as a tumor suppressor.5 As a component of a Cul3-based ubiquitin ligase complex, SPOP affects a variety of substrates that impact multiple pathways, including hedgehog, JNK, and steroid receptor signaling cascades24–26 and has been shown to directly target several proteins for ubiquitylation via its MATH domain, including the polycomb protein Bmi1,27 the Jun-kinase phosphatase Puckered,11 the pancreatic and duodenal homeobox protein Pdx1,28 the inositol kinase PIPKIIb,29 and the Hedgehog signaling transcription factors Gli2 and Gli3,30 the variant histone MacroH2A,24 the death domain-associated protein Daxx.3 Daxx has been reported to function as a pro-apoptotic as well as anti-apoptotic protein,25 and SPOP/Cul3-mediated degradation of Daxx reversed Daxx-mediated repression of transactivation function of ETS1 and p53, both of which are known to induce the expression of pro-apoptotic genes, such as MMP1 and Bax, respectively,26, 31, 32 which induces apoptosis. Our finding that HIB/SPOP also regulates Gli stability raises the exciting possibility that SPOP and its paralogs may act as tumor suppressors whose loss of function could synergize with aberrant Gli activation to induce tumor formation.33
Given that CRC is one of the most malignant cancers in the world and that tumor recurrence and metastases are the major causes of death in patients with CRC, resulting in a poor prognosis of the disease among the digestive tract tumors. The incidence, development, invasion, and metastasis of CRC are a multistep and multifactor complex process. It is regulated by many genes and involves a variety of gene activation, inactivation, or regulated disorder. Despite many therapy methods such as combination chemotherapy and radiation therapy had developed, little improvement has been achieved within the last decade in terms of prognosis and quality of life for CRC patients. Due to the frequent failure of conventional treatment strategies, many cancer-related molecules have been characterized with the goal of developing novel anticancer therapies, including targeted drugs or antibodies and cancer vaccines.35 Currently, identification of some biological markers to determine the risk of poor prognosis is important for designing treatment strategies. In this study, it is the first to demonstrate the potential role of SPOP in detection of CRC and we sought to determine SPOP expression in human CRC patient specimens and evaluate the clinicopathologic implications of SPOP expression in CRC, in an attempt to discover the potential influence of SPOP in the development of CRC. In our previous study, we found, using qRT-PCR and Western blotting analysis, that SPOP expression was decreased at the mRNA and protein levels, respectively, in most tumor tissues compared to their adjacent nontumorous tissues. Immunohistochemical staining analysis also exhibited that SPOP expression was significantly lower in the tumor tissues than other colorectal tissues. Moreover, SPOP was previously shown to be expressed in high levels in renal cell carcinomas; SPOP overexpression and cotreatment with tumor necrosis factor (TNF) of HEK293 embryonic kidney cells increased the expression of the oncogenic P-JNK and P-c-Jun proteins, suggesting a role for SPOP in regulating tumor growth.11 Herein, our study is consistent with previous research which revealed that overexpression of SPOP in HCT-116 cells significantly inhibits cell proliferation and migration both in vitro. Furthermore, LoVo cells stably transfected with the SPOP siRNA significantly stimulated the proliferation and migration of LoVo. Functionally, knockdown of SPOP gene in cancer cells enhanced growth and invasion of the cells, whereas its restoration inhibited oncogenic signaling and tumorigenesis,5 suggesting that SPOP gene functions as a tumor suppressor gene. Furthermore, a decreased expression of SPOP was significantly associated with advanced TNM stage, poor differentiation, depth of invasion, lymph node metastasis, vascular invasion, and distance metastasis, suggesting that abnormal SPOP expression might be involved in CRC progression and metastasis and that SPOP could also play a tumor suppressor role in CRC. A Kaplan–Meier survival analysis showed that low SPOP expression significantly correlated with shorter survival time of CRC patients (Fig. 7). Spearman rank correlation analysis suggested the more advanced clinical TNM stage corresponding to the lower expression level of SPOP in CRC. Cox hazard ratio regression analyses further demonstrated that the SPOP expression level was an independent risk factor for survival, suggesting that it may serve as a valuable prognostic biomarker for CRC patients after surgery and a potential target for gene therapy in the treatment of CRC. These results are consistent with other reports indicating that some experimental results from breast cancer cell proliferation, invasion, anchorage-independent growth, and tumor growth in nude mice indicate that SPOP inhibits cancer cell functions.5

Kaplan–Meier overall survival (OS) curve of SPOP expression in all patients and subgroup analysis of SPOP according to TNM. a, b Stage-specific survival curves showed that patients with low SPOP expression had poor survival to those with high SPOP expression in stages I–II and stages III–IV CRC patients (P = 0.024 and P = 0.011, respectively)
Many CRCs are characterized by infiltrative growth which is associated with EMT. EMT has a pivotal role in the initiation of metastasis, a process in which epithelial cells lose adhesion and cytoskeletal components concomitant with a gain of mesenchymal components and the initiation of a migratory phenotype.34, 35 EMT is frequently seen at the invasive front of CRCs and is accompanied by an increased expression of mesenchymal marker genes like vimentin.36, 37 Western blot showed that exogenous SPOP overexpression resulted in the increase of epithelial markers E-cadherin and decrease of mesenchymal marker vimentin. In addition, it has been proposed that MMPs might play a part not only in tumor invasion and initiation of metastasis but also in carcinogenesis from CRC. Increased plasma MMP2 expression was observed in lymph-node-positive patients with CRC compared to those without lymph node metastasis.38 As well as overexpression of MMP7 is seen in ~80 % of CRC,39 serum levels of MMP7 are associated with decreased survival in advanced CRC and correlate with cancer progression.40 By transfecting SPOP into human colon cancer cells, the expression of both MMP2 and MMP7 was decreased substantially and ultimately led to inhibition of human colon cancer cell migration and invasion.
Conclusion
In summary, we have identified SPOP as a novel conditional tumor suppressor gene in the CRC. We have provided evidence that SPOP can trigger cell growth, migration progression, and inhibit tumor growth. We also showed that suppression of SPOP can induce CRC migration by inducing EMT and that expression of SPOP can inhibit CRC migration through decreasing MMPs. By integrating clinical, molecular, and cellular data, our study has demonstrated that SPOP may be an index of prognosis in CRC patients and may offer new therapeutic strategies.
References
Huang, C. J.; Chen, C. Y.; Chen, H. H.; Tsai, S. F.; Choo, K. B., TDPOZ, a family of bipartite animal and plant proteins that contain the TRAF (TD) and POZ/BTB domains. Gene 2004, 324, 117–27.
Berger, M. F.; Lawrence, M. S.; Demichelis, F.; Drier, Y.; Cibulskis, K.; Sivachenko, A. Y.; Sboner, A.; Esgueva, R.; Pflueger, D.; Sougnez, C.; Onofrio, R.; Carter, S. L.; Park, K.; Habegger, L.; Ambrogio, L.; Fennell, T.; Parkin, M.; Saksena, G.; Voet, D.; Ramos, A. H.; Pugh, T. J.; Wilkinson, J.; Fisher, S.; Winckler, W.; Mahan, S.; Ardlie, K.; Baldwin, J.; Simons, J. W.; Kitabayashi, N.; MacDonald, T. Y.; Kantoff, P. W.; Chin, L.; Gabriel, S. B.; Gerstein, M. B.; Golub, T. R.; Meyerson, M.; Tewari, A.; Lander, E. S.; Getz, G.; Rubin, M. A.; Garraway, L. A., The genomic complexity of primary human prostate cancer. Nature 2011, 470, (7333), 214–20.
Kwon, J. E.; La, M.; Oh, K. H.; Oh, Y. M.; Kim, G. R.; Seol, J. H.; Baek, S. H.; Chiba, T.; Tanaka, K.; Bang, O. S.; Joe, C. O.; Chung, C. H., BTB domain-containing speckle-type POZ protein (SPOP) serves as an adaptor of Daxx for ubiquitination by Cul3-based ubiquitin ligase. J Biol Chem 2006, 281, (18), 12664–72.
Errington, W. J.; Khan, M. Q.; Bueler, S. A.; Rubinstein, J. L.; Chakrabartty, A.; Prive, G. G., Adaptor protein self-assembly drives the control of a cullin-RING ubiquitin ligase. Structure 2012, 20, (7), 1141–53.
Li, C.; Ao, J.; Fu, J.; Lee, D. F.; Xu, J.; Lonard, D.; O'Malley, B. W., Tumor-suppressor role for the SPOP ubiquitin ligase in signal-dependent proteolysis of the oncogenic co-activator SRC-3/AIB1. Oncogene 2011, 30, (42), 4350–64.
Le Gallo, M.; O'Hara, A. J.; Rudd, M. L.; Urick, M. E.; Hansen, N. F.; O'Neil, N. J.; Price, J. C.; Zhang, S.; England, B. M.; Godwin, A. K.; Sgroi, D. C.; Hieter, P.; Mullikin, J. C.; Merino, M. J.; Bell, D. W., Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat Genet 2012, 44, (12), 1310–5.
Kan, Z.; Jaiswal, B. S.; Stinson, J.; Janakiraman, V.; Bhatt, D.; Stern, H. M.; Yue, P.; Haverty, P. M.; Bourgon, R.; Zheng, J.; Moorhead, M.; Chaudhuri, S.; Tomsho, L. P.; Peters, B. A.; Pujara, K.; Cordes, S.; Davis, D. P.; Carlton, V. E.; Yuan, W.; Li, L.; Wang, W.; Eigenbrot, C.; Kaminker, J. S.; Eberhard, D. A.; Waring, P.; Schuster, S. C.; Modrusan, Z.; Zhang, Z.; Stokoe, D.; de Sauvage, F. J.; Faham, M.; Seshagiri, S., Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 2010, 466, (7308), 869–73.
Kim, M. S.; Je, E. M.; Oh, J. E.; Yoo, N. J.; Lee, S. H., Mutational and expressional analyses of SPOP, a candidate tumor suppressor gene, in prostate, gastric and colorectal cancers. APMIS 2013, 121, (7), 626–33.
Nakayama, K. I.; Nakayama, K., Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer 2006, 6, (5), 369–81.
Bode, A. M.; Dong, Z., Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer 2004, 4, (10), 793–805.
Liu, J.; Ghanim, M.; Xue, L.; Brown, C. D.; Iossifov, I.; Angeletti, C.; Hua, S.; Negre, N.; Ludwig, M.; Stricker, T.; Al-Ahmadie, H. A.; Tretiakova, M.; Camp, R. L.; Perera-Alberto, M.; Rimm, D. L.; Xu, T.; Rzhetsky, A.; White, K. P., Analysis of Drosophila segmentation network identifies a JNK pathway factor overexpressed in kidney cancer. Science 2009, 323, (5918), 1218–22.
Larue, L.; Bellacosa, A., Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3ʹ kinase/AKT pathways. Oncogene 2005, 24, (50), 7443–54.
Thiery, J. P.; Sleeman, J. P., Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol 2006, 7, (2), 131–42.
Vu, T. H.; Werb, Z., Matrix metalloproteinases: effectors of development and normal physiology. Genes Dev 2000, 14, (17), 2123–33.
Zucker, S.; Vacirca, J., Role of matrix metalloproteinases (MMPs) in colorectal cancer. Cancer Metastasis Rev 2004, 23, (1–2), 101–17.
Said, A. H.; Raufman, J. P.; Xie, G., The role of matrix metalloproteinases in colorectal cancer. Cancers (Basel) 2014, 6, (1), 366–75.
Jemal, A.; Siegel, R.; Xu, J.; Ward, E., Cancer statistics, 2010. CA Cancer J Clin 2010, 60, (5), 277–300.
Ishizuka, M.; Kita, J.; Shimoda, M.; Kato, M.; Sawada, T.; Kubota, K., Impact of grading of liver metastasis on postoperative outcome in patients with liver metastases from colorectal cancer. Hepatogastroenterology 2012, 59, (113), 54–8.
Harpaz, N.; Polydorides, A. D., Colorectal dysplasia in chronic inflammatory bowel disease: pathology, clinical implications, and pathogenesis. Arch Pathol Lab Med 2010, 134, (6), 876–95.
Campos, F. G.; Logullo Waitzberg, A. G.; Kiss, D. R.; Waitzberg, D. L.; Habr-Gama, A.; Gama-Rodrigues, J., Diet and colorectal cancer: current evidence for etiology and prevention. Nutr Hosp 2005, 20, (1), 18–25.
Roessler, S.; Jia, H. L.; Budhu, A.; Forgues, M.; Ye, Q. H.; Lee, J. S.; Thorgeirsson, S. S.; Sun, Z.; Tang, Z. Y.; Qin, L. X.; Wang, X. W., A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Res 2010, 70, (24), 10202–12.
Kim, J.; Huynh, R.; Abraham, I.; Kim, E.; Kumar, R. R., Number of lymph nodes examined and its impact on colorectal cancer staging. Am Surg 2006, 72, (10), 902–5.
Nagai, Y.; Kojima, T.; Muro, Y.; Hachiya, T.; Nishizawa, Y.; Wakabayashi, T.; Hagiwara, M., Identification of a novel nuclear speckle-type protein, SPOP. FEBS Lett 1997, 418, (1–2), 23–6.
Zhuang, M.; Calabrese, M. F.; Liu, J.; Waddell, M. B.; Nourse, A.; Hammel, M.; Miller, D. J.; Walden, H.; Duda, D. M.; Seyedin, S. N.; Hoggard, T.; Harper, J. W.; White, K. P.; Schulman, B. A., Structures of SPOP-substrate complexes: insights into molecular architectures of BTB-Cul3 ubiquitin ligases. Mol Cell 2009, 36, (1), 39–50.
Michaelson, J. S., The Daxx enigma. Apoptosis 2000, 5, (3), 217–20.
Buttice, G.; Duterque-Coquillaud, M.; Basuyaux, J. P.; Carrere, S.; Kurkinen, M.; Stehelin, D., Erg, an Ets-family member, differentially regulates human collagenase1 (MMP1) and stromelysin1 (MMP3) gene expression by physically interacting with the Fos/Jun complex. Oncogene 1996, 13, (11), 2297–306.
Hernandez-Munoz, I.; Lund, A. H.; van der Stoop, P.; Boutsma, E.; Muijrers, I.; Verhoeven, E.; Nusinow, D. A.; Panning, B.; Marahrens, Y.; van Lohuizen, M., Stable X chromosome inactivation involves the PRC1 Polycomb complex and requires histone MACROH2A1 and the CULLIN3/SPOP ubiquitin E3 ligase. Proc Natl Acad Sci U S A 2005, 102, (21), 7635–40.
Liu, A.; Desai, B. M.; Stoffers, D. A., Identification of PCIF1, a POZ domain protein that inhibits PDX-1 (MODY4) transcriptional activity. Mol Cell Biol 2004, 24, (10), 4372–83.
Bunce, M. W.; Boronenkov, I. V.; Anderson, R. A., Coordinated activation of the nuclear ubiquitin ligase Cul3-SPOP by the generation of phosphatidylinositol 5-phosphate. J Biol Chem 2008, 283, (13), 8678–86.
Wang, C.; Pan, Y.; Wang, B., Suppressor of fused and Spop regulate the stability, processing and function of Gli2 and Gli3 full-length activators but not their repressors. Development 2010, 137, (12), 2001–9.
Miyashita, T.; Reed, J. C., Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, (2), 293–9.
Ala-aho, R.; Grenman, R.; Seth, P.; Kahari, V. M., Adenoviral delivery of p53 gene suppresses expression of collagenase-3 (MMP-13) in squamous carcinoma cells. Oncogene 2002, 21, (8), 1187–95.
Zhang, Q.; Zhang, L.; Wang, B.; Ou, C. Y.; Chien, C. T.; Jiang, J., A hedgehog-induced BTB protein modulates hedgehog signaling by degrading Ci/Gli transcription factor. Dev Cell 2006, 10, (6), 719–29.
Hugo, H.; Ackland, M. L.; Blick, T.; Lawrence, M. G.; Clements, J. A.; Williams, E. D.; Thompson, E. W., Epithelial–mesenchymal and mesenchymal–epithelial transitions in carcinoma progression. J Cell Physiol 2007, 213, (2), 374–83.
Iwatsuki, M.; Mimori, K.; Yokobori, T.; Ishi, H.; Beppu, T.; Nakamori, S.; Baba, H.; Mori, M., Epithelial-mesenchymal transition in cancer development and its clinical significance. Cancer Sci 2010, 101, (2), 293–9.
Thiery, J. P.; Acloque, H.; Huang, R. Y.; Nieto, M. A., Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, (5), 871–90.
Brabletz, T.; Jung, A.; Reu, S.; Porzner, M.; Hlubek, F.; Kunz-Schughart, L. A.; Knuechel, R.; Kirchner, T., Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci U S A 2001, 98, (18), 10356–61.
Langenskiold, M.; Holmdahl, L.; Falk, P.; Ivarsson, M. L., Increased plasma MMP-2 protein expression in lymph node-positive patients with colorectal cancer. Int J Colorectal Dis 2005, 20, (3), 245–52.
Brabletz, T.; Jung, A.; Dag, S.; Hlubek, F.; Kirchner, T., beta-catenin regulates the expression of the matrix metalloproteinase-7 in human colorectal cancer. Am J Pathol 1999, 155, (4), 1033–8.
Maurel, J.; Nadal, C.; Garcia-Albeniz, X.; Gallego, R.; Carcereny, E.; Almendro, V.; Marmol, M.; Gallardo, E.; Maria Auge, J.; Longaron, R.; Martinez-Fernandez, A.; Molina, R.; Castells, A.; Gascon, P., Serum matrix metalloproteinase 7 levels identifies poor prognosis advanced colorectal cancer patients. Int J Cancer 2007, 121, (5), 1066–71.
Acknowledgments
This work is supported by the grants from the China Postdoctoral Science Foundation and the Department of Health of Jiangsu Province of China (no. H201214).
Conflict of Interest
None
Author information
Authors and Affiliations
Corresponding authors
Additional information
Junfei Xu and Feiran Wang contributed equally to this work.
Rights and permissions
About this article

Cite this article
Xu, J., Wang, F., Jiang, H. et al. Properties and Clinical Relevance of Speckle-Type POZ Protein in Human Colorectal Cancer. J Gastrointest Surg 19, 1484–1496 (2015). https://doi.org/10.1007/s11605-015-2767-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11605-015-2767-6