
Abstract
Constructing non-noble metal-based catalysts has attracted more attention owing to these materials owning high abundance, low cost, and expected to take the place of Pt/C as a candidate commercial the oxygen reduction reaction (ORR) catalyst. Herein, hollow microspheres self-assembled by a layer of interweaving Fe2O3 nanorods are first fabricated by a one-step hydrothermal process, which are then covered by dopamine. Finally, annealing treatment at the assigned temperature in N2 is conducted to synthesize Fe/FexOy nanorod-based hollow microspheres (~ 900 nm in diameter) covered by N-doped carbon. Electrochemical measurements demonstrate that, by the side of the commercial 20% Pt/C, the as-obtained T-650 has a higher onset potential (83-mV positive shift), more positive half wave potential (55-mV positive shift), and larger limiting current density (increased by 0.9 mA cm-2). It is found that the simultaneous presence of zero-valence Fe and nitrogen atoms is the key to the high catalytic activity of the as-synthesized Fe–N-C material, and these unique hierarchical hollow porous microspheres make the electrocatalyst possess excellent catalytic performance, long-time stability, and superior methanol tolerance.

Graphical Abstract
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The key to promote actual application of fuel cells is to accelerate efficient electrocatalyst with low price and high performance for oxygen reduction reaction (ORR) [1, 2]. So far, Pt-based catalysts are the optimum selection to expedite the sluggish dynamic process of ORR [3, 4]. However, high cost, scarcity, and poor CO poison resistance hinder the large-scale practical application of Pt-based catalysts. Valuable exertions have been dedicated to exploiting carbon-based materials and noble-metal-free-based materials (such as non-precious metal oxides, nitrides, carbides) [5].
Recently, many non-noble-metal compounds cladded by carbon displayed enhanced ORR catalytic activity according to theoretical and experimental works [6, 7]. Among these non-precious-metal oxides, Fe compounds are attracting more and more attention [8]. As Zang Y et al. reported, Fe/Fe2O3 nano-sized particle loading on Fe-N co-doped carbon nanosheets showed high ORR performance [9]. Benzigar M R et al. designed a facile strategy to implant small Fe2O3 nanoparticles into the channels of highly ordered mesoporous fullerene (C60) for ORR [10]. Nevertheless, the inherent drawbacks of the FexOy@NC catalysts, such as the oxidation agglomeration of iron, small surface area, and limited available active sites, indicate that more works should be done to improve the ORR properties.
Compared to post-growth transition metal nanoparticles on the pre-synthesized N-CNTs or N-C [11], the surface coated carbon with doping N to form unique packaging structure on the FexOy framework could effectively resist acid etching and attenuate oxidation aggregation of transition metal oxide nanoparticles in service involving the ORR process [12], resulting in outstanding durability in all kinds of electrolyte [13]. More importantly, the existence of transition metal nanoparticles during the carbonization process was beneficial to the graphitization degree of carbon, which is also helpful to electron transfer within the surface carbon layer during ORR [14]. According to previous research [6], the iron content in the Fe-N-C system by post-growing FexOy nanoparticles was usually less than 3%; hence, the low active site density caused a poor durability and ORR performance.
Fe-N carbon materials are deemed as promising non-precious-metal ORR catalysts because the iron atoms and N atoms combine to form the Fe-NX group (regarded as the main active sites in the Fe-N-C system) [15]. Meanwhile the introduced N atoms break the electrical neutrality of the carbon surface, bring on uneven distribution of charge density, positively charged adjacent C atoms, and increased spin density, thereby promoting the adsorption of O2 and further reduction reactions [16, 17]. In addition, by preparing hierarchical hollow microspheres Fe-N-C materials, the inherent superiority of porous in high-throughput mass transfer and the advantage of effective utilization of active catalytic sites could be exploited [18]. Nevertheless, the common method constructing hierarchical hollow microspheres highly depended on SiO2 or other hard templates [19, 20], wherein dangerous processes of strong acid or alkali solution was required to wash away the silica template, which implied excess time-consuming and was not friendly to the environment. Meanwhile, hierarchical hollow microspheres possess high specific surface area, stable open frameworks, and high conductivity comparable to other porous structures. These result in shortening the length of the charge (ions and electrons) diffusion and avoiding accumulation on the electrode surface to guarantee efficient quality transmission, thus exhibiting great potential for advanced ORR catalysts [21]. Meanwhile, a facile preparation of hierarchical hollow microspheres iron-nitrogen-doped electrocatalysts with efficient activity for ORR is still an important challenge to accelerate the practical utilization of M@NC.
Herein, an electrocatalyst of highly uniform hierarchical hollow microspheres Fe/FexOy@NC consisting of ultrathin nitrogen-doped carbon nanorod encapsulated with Fe/FexOy was synthesized using a simple and friendly environment approach. The electrochemical tests on the as-obtained catalysts manifested that the obtained sample at a pyrolysis temperature of 650 °C revealed an outstanding ORR activity under alkaline conditions, which was superior to the commercial 20% Pt/C ORR catalysts. The superior ORR activity of the as-obtained Fe/FexOy@NC was discussed by comparing the samples obtained at different pyrolysis temperatures and different pyrolysis durations at 650 °C. The stability and methanol tolerance of the as-obtained Fe/FexOy@NC were also tested. In-depth study on the relationship between the electrochemical properties and morphology, composition, as well as microstructure of the as-obtained catalysts indicated that zero-valence Fe in Fe-N-C materials can significantly enhance the ORR activity.
Experimental
Synthesis of Fe2O3 hollow spheres assembled thin nanorods
The uniform hierarchical Fe-glycerate precursor hollow spheres were first prepared by a one-pot solvothermal method, where 4 mmol Fe (NO3)3·9H2O and 60 ml glycerol were stirringly dispersed into 520 ml isopropanol. Ten minutes later, dropped 6 ml of deionized (DI) water into the above solution at room temperature with continuous stirring; 15 min later, the obtained mixtures were poured into a Teflon-lined autoclave and heated at 190 ℃ for 12 h. After naturally cooling to room temperature, the obtained exudates were washed with ethanol and DI water several times and then dried at 60 ℃ for 12 h. The resulting products were transferred into a quartz boat and annealed in air at 400 ℃ for 3 h with a heating velocity of 1 ℃ min-1 obtaining hollow Fe2O3.
Synthesis of Fe/FexOy@NC
One hundred milligram of Fe2O3 was first stirringly separated in 75 ml ultrapure water for 1 h, and then 45 mg dopamine hydrochloride (DA) and 25 mg Tris (hydroxymethyl) aminomethane were added under magnetic stirring. After 5 h later, the resulting mixtures were collected via centrifugation, washed with DI water three times, and then dried at 60 ℃ for 12 h. To prepare series of Fe/FexOy@NC-T (T = 600, 650, 700, and 750℃) catalysts, the resulting products were heated to a specific temperature (600, 650, 700, and 750) for 3 h with a heating velocity of 2 ℃ min-1. The final products were called as T-x (x = 600, 650, 700, and 750 ℃).
Characterization
Powder X-ray diffraction (XRD) measurement was detected with a DMAX-Ultima+ diffraction meter with Co Kα (λ = 1.7902Å) radiation (40 kV, 40 mA) from Rigaku. Scanning electron microscopy (SEM, SUPRA 55, SAPPHIRE) and transmission electron microscopy (TEM, JEOL JEM 2100F) equipped with an energy-dispersive X-ray spectrometer (EDS, Oxford, Link ISIS) were applied to examine the micromorphology and microstructure of the prepared materials. The composition of element and its chemical valence states were assayed by X-ray photoelectron spectroscopy (XPS, THERMO ESCALAB250 system, Al Kα radiation). Raman spectra analysis was executed by a Labram-010 Raman microspectroscopy (JY, FRA) system. The as-prepared product specific surface area was performed with the Brunauer-Emmett-Teller (BET) and their pore diameter was measured on a Barrett-Joyner-Halenda (BJH) model.
Electrochemical measurement
The ORR activities of the prepared catalysts were carried out in a typical three-electrode system in 0.1 M KOH electrolyte saturated with N2-O2 by VMP3 electrochemical workstation. A Pt wire served as a counter electrode, a Hg/HgO (1.0 M KOH) as a reference electrode, a rotating disk (glassy carbon, GC) as a working electrode, and a 0.1 M KOH solution as an electrolyte. The prepared catalysts (10.00 mg) were first dispersed in isopropanol (2.5 ml)/Nafion solution (40 μl) and sonicated for 30 min to obtain the ink. A certain volume of the catalyst ink was dropped onto the surface of GC with Fe/FexOy@NC loading of 0.160 mg cm-2 and drying naturally. In the process of the cyclic voltammogram (CV) measurement, the KOH electrolyte should be bubbled with N2 or O2 flow realized in an oxygen-free or oxygen-saturated environment. For each as-prepared working electrode test, 50 cycle CVs with a scan rate 200 mV s-1 were achieved to make it stable, recorded between −0.8 and 0.3 V. The polarization curves (linear sweep voltammetry, LSV) were scanned at 10 mV s-1 in 0.1 M KOH saturated with O2 at the rotational speed of 400–2500 rpm, in which the potential window was −0.8 to 0.3 V. Conversion of all recorded potentials to standard reversible hydrogen electrode (RHE) for expression:
The Koutecky-Levich (KL) diagrams of all catalysts were generated based on the LSV curves to calculate the number of electron transfers (n) and the dynamic current density (JK), where n and JK for each O2 molecule in the ORR could be evaluated according to the following equation:
In which J (mA cm-2) is the measured current density, which was related to the diffusion-limiting current (JL) and the kinetic current (JK), n is the electron transfer number per O2 molecule, F is the Faraday constant (96,485 C mol-1), C0 is the volume concentration of oxygen in oxygen-saturated 0.1 mol l-1 (1.2 × 10-6 mol cm-3), D0 is the diffusion coefficient of oxygen in 0.1 mol l-1 KOH (1.9 × 10-5 cm-2 s-1), ν is the kinematic viscosity of water (0.01 cm-2 s-1), and ω is the angular velocity of the disk (ω = 2πN, N (r s-1) is the linear rotation speed). Methanol tolerance evaluation study of the as-prepared products was executed in 0.1 M KOH electrolyte without or with the addition of 3 M methanol.
Calculation method
The calculation details about mass activity and turnover frequency (TOF) are listed as below. In consideration of the catalyst, mass loading (m) was 0.16 mg cm-2, the mass activity (A g-1) could be calculated at E1/2 (the half-wave potential of catalysts, V) and j (current density, mA cm-2) measured at E1/2 by the following formula [22]:
The TOF values of as-obtained catalysts can be estimated by presuming that every corresponding atom participated in the catalytic processes [3]:
where j and S (0.196 cm-2) represent the measured current density at E1/2 and the surface area of glassy carbon disk, respectively; the 4 implies that 4 electrons were involved in reacting with each mole of O2; F denotes Faraday’s constant (96,485.3 C mol-1), and ɳ is the molecular weight of the corresponding atom calculated from the catalyst loading m.
Results and discussion
Structure and composition characterization
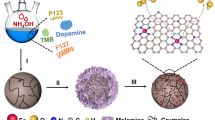
The Fe/FexOy@NC catalysts were prepared following the procedure shown in Fig. 1a. Briefly, the formation of Fe-glycerate was prepared by a self-template mechanism, which was then subjected to pyrolysis at 400 ℃ in air for 3 h. In the heat solution process, Fe-glycerate was gradually oxidized by oxygen generated from the decomposition of O2, forming Fe2O3 species with a layer of the interweaving nanorods. Then, it was subsequently coated with dopamine by self-polymerization at room temperature; after pyrolysis, a part of the Fe2O3 was reduced to zero-valence Fe and FeO by carbon and obtained Fe/FexOy@NC. To investigate the phase compositions of as-synthesized T-x species, XRD patterns were recorded and displayed in Fig. 1b. The XRD measurement of the Fe2O3 precursor is shown in Fig. S1. The XRD pattern of T-x presents crystal diffraction peaks of three phases. For the 600 ℃ catalysts, the characteristic peaks at ca. 35.1, 41.4, 50.5, 62.9, 67.3, and 74.2° pertained to the cubic maghemite (Fe3O4, JCPDF: 85-1436), and at 41.9, 48.78, and 71.47° attribute to the crystal planes of the wustite (FeO, JCPDF: 74-1886); no crystal diffraction peaks of zero-valence Fe appeared owing to the decomposition temperature. Raising the pyrolysis temperature to 650 ℃ induced the emergence of diffraction peaks at 52.5° and 77.4°, manifesting the formation of zero-valence Fe nanocrystals (iron, JCPDF: 87-0722) [23]. The peak intensity of zero-valence Fe and FeO increased with the rising of temperature to 700 and 750 °C, while the peak intensity of Fe3O4 decreased. In short, increasing the temperature from 600 to 750 ℃, the peak intensity of zero-valence Fe and FeO increasingly enhanced. No diffraction peaks of C were discovered, perhaps indicating the carbon was in amorphous state [24].

(a) schematic diagram of forming course of Fe/FexOy@NC hollow spheres assembled. (b) X-ray diffraction patterns of T-x at different temperatures. (c) Raman spectra of T-650. (d) Nitrogen adsorption–desorption isotherms of T. (e) The pore-size distribution of T-650
The XRD results manifested that iron oxides were reduced by carbon at high temperature and the reduced Fe atoms are easy to react readily with N atoms to form the Fe-NX coordination; the spare Fe will assemble to generate Fe and FeO nanocrystalline [25]. The Fe-NX species are known as the active site for ORR [25,26,27]. Besides, the presence of zero-valence Fe can improve the conductivity of the catalyst. The relative content of Fe3O4, FeO, and zero-valence Fe phases can be calculated through XRD fine scanning; the results are shown in Table 1. As the pyrolysis temperature increased, the content of Fe3O4 decreased, zero-valence Fe appeared, and the content increased first and then decreased. In the Raman spectra of T-650 (Fig. 1c), the typical bands for Fe3O4 at 212, 275, 390, 474, and 586 cm-1, as well as carbon bands at 1357 (the D band) and 1591cm-1-2 (the G band), are recognized. For FeO, iron exists in a simple hexa-coordinate forming with the same bond length. And the simulation calculation certified the Fe-O bond without Raman activity. In terms of carbon band, the D peak stands for deficiency carbon in graphitic carbon, and the G peak delegates to the degree of crystallinity of the sp2 hybrid carbon atoms [28]. Consequently, the degree of the structure disorder and the number of defects can be calculated quantitatively by the intensity ratio of the D to G bands (ID/IG). The value of ID/IG was 1.26 for T-650, indicating that the T-650 involved both amorphous and graphitic carbon, and formed the covalent bond by the presence of nitrogen and a certain degree of defect carbon.
To understand the specific surface area and porosity of T-650 catalysts, a N2 adsorption–desorption isotherm curve is shown in Fig. 1d and e. The isotherms of T-650 were found to be typical type IV isotherms with a distinct hysteresis loop, indicating that both micropores and mesopores existed due to the carbonization of polydopamine [29, 30]. The BET specific surface area, pore volume, and pore size of T-650 are approximately 649.7 m2 g-1, 0.408 m3 g-1, and 4.7 nm, respectively. From Fig. 1e, it can be seen that mesopores (3 ~ 20 nm) dominated the pore volume, which coincided with the interspaces formed by the interweaving nanorods (referring to Fig. 2). The high surface area and porosity of the as-synthesized Fe/FexOy@NC are derived from the entanglement between nanorods in iron oxide templates and carbonization of polydopamine.

SEM image of (a) Fe2O3 template and (b) Fe/FexOy@NC-650. (c–f) T-x (x = 600, 650, 700, and 750 ℃). (g–j) TEM images of T-650. (i) High-resolution TEM image of T-650. (k) Corresponding EDS element mapping images of C (red), Fe (green), N (brown), and O (yellow). (m) The mapping image of the overlaid iron and nitrogen
SEM, TEM, and HRTEM images of T-x are shown in Fig. 2. Figure 2a shows that Fe2O3 hollow microspheres with a ca. diameter of 800 nm were self-assembled by a layer of interweaving nanorods with a diameter about 16 nm. As displayed in Fig. 2b, after wrapping with DA and pyrolysis treatment, the as-synthesized Fe/FexOy@NC inherited the hollow microspherical structure from Fe2O3 precursor with about 100-nm increase in diameter. The core-shell microspheres were well dispersed with no obvious agglomeration. From Fig. 2c and d, the interspaces formed by the interweaving nanorods were well kept forming a porous spherical surface for T-600 and T-650. In contrast, the shrinkage of nanorods at higher temperature (T-700, Fig. 2e) compressed the interspaces among the nanorods, accompanied with a growth in nanorod diameter. As a result, the as-synthesized T-700 presented in hollow nanospheres in a smaller diameter (~ 500 nm) with a nearly closed spherical surface. However, for T-750, the fast reduction reaction between FexOy and carbon due to high temperature led to an early close in the spherical surface (refer to Fig. S3). Consequently, T-750 had a spherical diameter close to that of T-600 and T-650, while its surface was almost fully closed and had no micro- or mesopores (Fig. 2f).
Figure 2g exhibits that the microspheres were secondary hierarchical structures composed by nanorods. The core-shell structure of T-650, of which a few layers of graphitic carbon encapsulated with Fe/FexOy nanorods, can be clearly observed from Fig. 2h. HRTEM image of the T-650 sample is exhibited in Fig. 2i; the distinct crystal-plane distance of 0.20 and 0.25 nm is indexed to the (110) plane of zero-valence Fe and (311) crystal plane of Fe3O4, respectively. The porous structure of the as-synthesized T-650 was further observed by TEM image (Fig. 2j).
The composition and elemental distribution of the T-650 samples were then identified by EDS. Figure 2k demonstrated the symmetrical distribution of C, Fe, N, and O atoms within microsphere in T-650. From Fig. 2k, C mainly distributed on the surface of a microsphere, while Fe, N, and O are uniformly distributed. The distribution area of Fe was smaller than that of C and N, which indicated a core–shell structure of the as-synthesized catalysts. It was noticed that N was uniformly distributed throughout the microsphere, which meant that it combined with carbon and iron to form C-N and Fe-NX. The superposition of the Fe and N signals in the EDS image is shown in Fig. 2m, which could reveal that the Fe atoms were contiguous to the N atoms on the level of atomic, indicating that Fe and N atoms form Fe-NX coordination.
The surface chemical composition and primary bonding configuration of Fe/FexOy@NC samples were evaluated by XPS (Fig. S2). The detailed XPS analyses of T-650 are revealed in Fig. 3. From Fig. S2 and Fig. 3a, the peaks of C, N, Fe, and O were all presented in the survey spectrum for all samples. A certain strong C 1s peaks discovered were in keeping with the EDS analysis, suggesting carbon successfully coated on the surface of Fe/FexOy. The C 1s spectra can be assigned to C=C, C–C/C–N, C=N, and C–O at 284.0, 285.1, 286.3 eV, and 289.3 eV, respectively (Fig. 3b). This result demonstrated that the nitrogen atoms are successfully incorporated into the carbon skeleton. The N 1s spectra were deconvoluted into 397.8, 399.6, 400.6, and 401.5 eV, which could be allocated to pyridinic N, Fe-NX, pyrrolic N, and graphitic N, respectively (Fig. 3c). Numerous studies have discovered that in the Fe-N-C system, Fe atoms coordinate with N atoms to form Fe-NX sites for catalytic activity at high temperature [31]. Figure 3d reveals the high-resolution Fe 2p XPS spectra. When the pyrolysis temperature goes up to 650 ℃, the Fe 2p spectrum could be volumed into five peaks, and the banding energies at around 709.9 and 723.5 eV could be due to the banding energies of 2p3/2 and 2p1/2 bands of Fe2+, respectively. The banding energies at 717.6 eV could be ascribed to satellite peaks. The banding energy at 711.3 eV could be attributed to the banding energy of 2p2/3 band of Fe3+, and the singles at around 708.7 eV can be attributed to the Fe0. According to Fig. S2, no obvious singles Fe0 were detected in the T-600 sample, which was identified with the XRD and TEM results. To understand the effect of pyrolysis temperature on element components and primary bonding configuration of all the Fe/FexOy@NC samples, the relative content of N and Fe according to XPS is shown in Fig. 3e and f. From Fig. 3e, it clearly showed that the total content of N decreased as pyrolysis temperature increased. For the Fe-N-C system, most of ORR active sites were related to Fe-N or C-N bonds [24]. Therefore, its ORR property is sensitive to the content of N. It was noticed that there was a significant N content drop (~ 50%) from 650 to 700 ℃, which implied a possible degradation in the ORR properties. Unfortunately, the total content of N decreased from 4.67 to 3.3 at% as pyrolysis temperature increased from 600 to 650 ℃. However, the total contents of Fe-NX in these two samples were still very close to each other (1.58 at% at 600 °C, 1.42 at% at 650 °C). From Fig. 3f, the total content of Fe on the surface of the as-prepared materials gradually increased as pyrolysis temperature increased, indicating more and more lose in C and N at higher temperature. It should be pointed out the high content of Fe and low content of N shown in Fig. 3e and f, which implied that Fe was more than enough to form Fe-NX on the surface of the as-synthesized catalysts, only part of the detected Fe was bonding to the adjacent N atoms.

(a) XPS spectrum of T-650. (b–d) XPS C 1 s, N 1 s, and Fe 2p profiles for T-650. (e, f) The relative content of N, function-N, and Fe according to XPS
Electrochemical measurement analysis
On the basis of the above analysis, as-obtained Fe/FexOy@NC catalysts were expected to develop as promising high-performance ORR catalysts. The ORR activities were preliminarily appraised by CV measurements in N2-saturated/O2-saturated 0.1 M KOH electrolyte. For all the samples, oxygen reduction processes were revealed by the vivid reduction peaks in O2-saturated 0.1 M KOH electrolyte (Fig. 4a and Fig. S4). In particular, the higher reduction peak current density of T-650 indicated that the ORR process was easier compared to other catalysts pyrolyzed under different temperatures. In addition, the reduction peak current density of T-650 (4.23 mA cm-2) sample at 20 mV s-1 was superior to 20% Pt/C catalyst (2.79 mA cm-2), hinting an outstanding ORR performance of T-650 sample in 0.1 M KOH electrolyte [29].

(a) CV curves of T-x at scan rate of 20 mV s-1 in O2-saturated 0.1 M KOH solution. (b) LSV curves of T-x in O2-saturated 0.1 M KOH solution with a rotation rate of 1600 rpm. (c) LSV curves of T-650 in O2-saturated 0.1 M KOH at different rotation rates. (d) The corresponding Koutecky-Levich plots of T-x at different potentials. (e) The summary of Eonset, E1/2, and JL. (f) Corresponding Tafel plots of T-650 and Pt/C
To get quantitative review and deep understanding of the ORR process on different temperature catalysts, linear sweep voltammetry (LSV) was acquired on a rotating disk electrode (RDE). Figure 4b presents the typical LSV curves of five different temperature samples at 1600 rpm in 0.1 M KOH electrolyte. To clearly evaluate the ORR properties of the as-obtained catalysts, the onset-potential (Eonset), half-wave potential (E1/2), and limiting current density (JL) of all the samples together with the referenced 20% Pt/C are summarized in Fig. 4e and Table S2. From Fig. 4b and e, it can be seen that, among all the samples, the T-650 sample exhibited the best ORR properties with a Eonset = 0.935 V, E1/2 = 0.806 V along with a JL = 6.26 mA cm-2 at 1600 rpm. Correspondingly, 20% Pt/C exhibited Eonset = 0.852, E1/2 = 0.751 V, and JL = 5.36 mA cm-2 under the same condition. However, for pure nitrogen-doped carbon, in other words, without Fe2O3 as a template and iron source, its performance was far lower than T-650 (Fig. S5). According to XPS analysis in Fig. 3d, T-600 had the most content of Fe-NX. However, T-600 showed the ORR properties closed to that of 20% Pt/C, which was lower than that of T-650. In fact, the content of Fe-NX in T-650 was slightly less than that in T-600. Therefore, the much better ORR properties of T-650, compared to those of T-600, can be attributed to the appearance of zero-valence Fe at 650 ℃ according to Fig. 1a and S2. On the other hand, T-700 and T-750 had significant amounts of zero-valence iron detected, with a large reduction in N content. As a result, the ORR properties of T-700 and T-750 were much lower than T-650. Consequently, the simultaneous presence of zero-valence Fe and nitrogen atoms was crucial to further advance ORR performance for the prepared Fe-N-C material.
The outstanding ORR kinetics of the T-650 catalyst was also appraised by the RDE test at different rotation speeds (Fig. 4c). These LSV curves showed a typical phenomenon that the current densities improved with increasing of the rotating rates forasmuch as the availability of oxygen [30]. In Fig. 4d, the corresponding K-L plots exhibited excellent linearity, indicating the first-order kinetic reaction related to the concentration of O2 in the electrolyte [32]. The average value of n calculated by K-L formula in the potential window from 0.2 to 0.4 V was 3.87, displaying a nearly four-electron transfer process [33]. In order to research the effect of overpotential on current density (Fig. 4f), Tafel plots were drawn in view of LSV curves in Fig. 4b. For T-650, the Tafel slope was calculated to be 87.1 mV dec-1 in low current density region, less than the slope of 20% Pt/C catalyst (92.3 mV dec-1). This result adequately demonstrates a beneficial kinetics mechanism towards ORR for T-650 sample, implying that T-650 catalyst can reach higher current density at a lower overpotential [34].
To preferably acquaint the ORR catalyst activity, the mass activity and turnover frequency (TOF) of Fe/FexOy@NC samples at the potential value of E1/2 are calculated and listed in Table 2. The mass activity of T-650 was 15.65 A g-1, overmatching that of other pyrolysis temperatures. The intrinsic catalytic active sites of the Fe/FexOy@NC samples were further evaluated by TOF values. When hypothesizing iron atoms and N atoms centers were as catalytically active sites, T-650 exhibited the highest TOF values of 0.023 s-1 and 0.045 s-1, respectively, representing the best ORR activity (Table 2). This result manifested that either Fe or N atom centers (even Fe-NX) were the primary active sites for ORR in the as-obtained catalysts.
On the other hand, durability and methanol tolerance were deemed to other pivotal factors to appraise the catalyst performances [35]. i-t curves at 1600 rpm under a constant cathodic voltage of 0.5 V are displayed in Fig. 5a. After 18,000 s, the current density of 20% Pt/C gone down 30.1%, possibly attributed to the damage and inactivation of Pt nanoparticles [36]. In striking comparison, the current density of the T-650 sample only displays a slight loss of about 1.5%, indicating an outstanding durability for ORR. As shown in Fig. 5a inset, after 18,000 s, the morphology and structure of T-650 hardly changed. This could be concerned in the settled hierarchical hollow structure to offered various paths for mass transfer and well-protected active sites of metallic Fe coated by carbon. Furthermore, the T-650 sample possessed excellent tolerance for methanol poisoning, showing overlapped CV curves with/without the presence of 3 M methanol. In contrast, the addition of 3 M methanol to the KOH electrolyte resulted in a clear oxidation peak on the CV curve for the 20% Pt/C catalyst (Fig. 5b), manifesting the presence of a methanol oxidation reaction.

(a) The i-t curves of T-650 and commercial Pt/C at 0.5 V at 1600 rpm and the inset shows SEM image of T-650 after 18,000 s. (b) Methanol tolerance tests of T-650 and commercial Pt/C
Understanding the origin of the enhanced activity
Different pyrolysis time of T-650
It was noticed that there was a significant improvement on ORR activity from T-600 to T-650 due to the appearance of zero-valence Fe at 650 ℃. In order to comprehend the impact of zero-valence Fe on the electrocatalytic activity, T-650 at different pyrolysis times (1 h, 2 h, and 3 h) was tested. In Fig. 6a, the CV curve of T-650-1, T-650-2, and T-650 all had significant oxygen reduction peaks. Their LSV results in Fig. 6b revealed that T-650-1 and T-650-2 had a very similar Eonset and E1/2 (Eonset = 0.814, E1/2 = 0.652 V), obviously decayed in comparison to T-650. Notably, the limiting current density of T-650-2 was greater than T-650-1 and lower than T-650 (Fig. 7b).

(a) CV curves of different pyrolysis times of T-650 at 20 mV s-1 in O2-saturated 0.1 M KOH. (b) LSV curves of different pyrolysis times of T-650 in O2-saturated 0.1 M KOH at 1600 rpm

(a, b) The relative content of Fe and N, function-N according to XPS
According to the XRD results of T-650 at different pyrolysis times shown in Fig. S6, the peaks of FeO increased in the early 2 h (T-650-1 vs. T-650-2) due to reduction of Fe3O4 by carbon. Then, zero-valence Fe appeared at 3 h with a decrease in FeO peaks, implying that the zero-valence Fe could be obtained by reduction of FeO. The high-resolution Fe 2p spectrum of T-650-1 and T-650-2 by XPS analysis can only be deconvoluted into Fe3+, Fe2+, and satellite peak, and no Fe0 peak appeared which was consistent with XRD (Fig. S7). Correspondingly, Fe content gradually increased as prolonging pyrolysis time (Fig. 7c). The detected Fe-NX in T-650-1 and T-650-2 was higher than that of T-650 according to the comparative content of N and function-N in Fig. 7d and Table S1. Meanwhile, the morphology of T-650 during pyrolysis almost had no change from 1 to 3 h (Fig. S8). Therefore, the reinforced ORR performance of T-650 can only ascribe to the appearance of zero-valence Fe.
Conclusions
In conclusion, utilizing the superior catalytic activity of M@NC and the rapid mass transfer in hollow nanosphere structure, Fe/FexOy@NC hollow spheres (~900 nm) composed of interweaved nanorods (~16 nm) were successfully constructed via an original in situ reproducibility and polymerization method, where the Fe2O3 hierarchical hollow microspheres (~800 nm) were employed as the multifunctional template and the iron source for the Fe/FexOy@NC. T-600 presented ORR properties comparable to those of Pt/C. Especially, T-650 exhibited much better catalytic performances than T-700, T-750, and commercial 20% Pt/C catalyst, with more positive Eonset (83-mV positive shift), JL (increased by 0.9 mA cm-2), and E1/2 (55-mV positive shift), as well as a smaller Tafel slope (87.1 mv dec-1), in keeping with TOF values (TOFFe = 0.023 s-1, and TOFN = 0.045 s-1). T-650-1 and T-650-2 had similar onset and half-wave potential (Eonset = 0.814, E1/2 = 0.652 V), but both were lower than T-650. It was found that the stable hierarchical hollow structure and the coexistence of Fe0 and nitrogen atoms in the Fe/FexOy@NC were essential to improve its catalytic activity. Moreover, the as-obtained T-650 was also extremely stable in long-time service and high tolerance of methanol. T-650 of superior ORR activity was synthesized by an environmentally friendly method. Therefore, the proposed Fe/FexOy@NC electrocatalysts possess bright future for the fuel cell application.
References
Lyu DD, Mollamahale YB, Huang SL, Zhu PC, Zhang XR, Du YH, Wang SB, Qing M, Tian ZQ, Shen PK (2018) Ultra-high surface area graphitic Fe-N-C nanospheres with single-atom iron sites as highly efficient non-precious metal bifunctional catalysts towards oxygen redox reactions. J Catal 368:279–290. https://doi.org/10.1016/j.jcat.2018.10.025
Han JX, Meng XY, Lu L, Bian JJ, Li ZP, Sun CW (2019) Single-atom Fe-N-x-C as an efficient electrocatalyst for zinc-air batteries. Adv Funct Mater 29(41):8. https://doi.org/10.1002/adfm.201808872
Zhan TR, Liu XL, Lu SS, Hou WG (2017) Nitrogen doped NiFe layered double hydroxide/reduced graphene oxide mesoporous nanosphere as an effective bifunctional electrocatalyst for oxygen reduction and evolution reactions. Appl Catal B Environ 205:551–558. https://doi.org/10.1016/j.apcatb.2017.01.010
Chen C, Yang XD, Zhou ZY, Lai YJ, Rauf M, Wang Y, Pan J, Zhuang L, Wang Q, Wang YC, Tian N, Zhang XS, Sun SG (2015) Aminothiazole-derived N, S, Fe-doped graphene nanosheets as high performance electrocatalysts for oxygen reduction. Chem Commun 51(96):17092–17095. https://doi.org/10.1039/c5cc06562c
Feng W, Liu M, Liu JJ, Song Y, Wang F (2018) Well-defined Fe, Fe3C, and Fe2O3 heterostructures on carbon black: a synergistic catalyst for oxygen reduction reaction. Catal Sci Technol 8(19):4900–4906. https://doi.org/10.1039/c8cy01223g
He YH, Liu SW, Priest C, Shi QR, Wu G (2020) Atomically dispersed metal-nitrogen-carbon catalysts for fuel cells: advances in catalyst design, electrode performance, and durability improvement. Chem Soc Rev 49(11):3484–3524. https://doi.org/10.1039/c9cs00903e
Sun T, Tian BB, Lu J, Su CL (2017) Recent advances in Fe (or Co)/N/C electrocatalysts for the oxygen reduction reaction in polymer electrolyte membrane fuel cells. J Mater Chem A 5(36):18933–18950. https://doi.org/10.1039/c7ta04915c
Wang J, Wang GX, Miao S, Jiang XL, Li JY, Bao XH (2014) Synthesis of Fe/Fe3C nanoparticles encapsulated in nitrogen-doped carbon with single-source molecular precursor for the oxygen reduction reaction. Carbon 75:381–389. https://doi.org/10.1016/j.carbon.2014.04.017
Zang YP, Zhang HM, Zhang X, Liu RR, Liu SW, Wang GZ, Zhang YX, Zhao HJ (2016) Fe/Fe2O3 nanoparticles anchored on Fe-N-doped carbon nanosheets as bifunctional oxygen electrocatalysts for rechargeable zinc-air batteries. Nano Res 9(7):2123–2137. https://doi.org/10.1007/s12274-016-1102-1
Benzigar MR, Joseph S, Saianand G, Gopalan AI, Sarkar S, Srinivasan S, Park DH, Kim S, Talapaneni SN, Ramadass K, Vinu A (2019) Highly ordered iron oxide-mesoporous fullerene nanocomposites for oxygen reduction reaction and supercapacitor applications. Microporous Mesoporous Mat 285:21–31. https://doi.org/10.1016/j.micromeso.2019.04.071
Fan H, Mao K, Liu M, Zhuo O, Zhao J, Sun T, Jiang YF, Du X, Zhang XL, Wu QS, Che RC, Yang LJ, Wu Q, Wang XZ, Hu Z (2018) Tailoring the nano heterointerface of hematite/magnetite on hierarchical nitrogen-doped carbon nanocages for superb oxygen reduction. J Mater Chem A 6(43):21313–21319. https://doi.org/10.1039/c8ta06442c
Xiao ZR, Hou F, Li YT, Zhang RR, Shen GQ, Wang L, Zhang XW, Wang QF, Li GZ (2019) Confinement of Fe2O3 nanoparticles in the shell of N-doped carbon hollow microsphere for efficient oxygen reduction reaction. Chem Eng Sci 207:235–246. https://doi.org/10.1016/j.ces.2019.06.029
Zhang GJ, Li CX, Liu J, Zhou L, Liu RH, Han X, Huang H, Hu HL, Liu Y, Kang ZH (2014) One- step conversion from metal-organic frameworks to Co3O4@N-doped carbon nanocomposites towards highly efficient oxygen reduction catalysts. J Mater Chem A 2(22):8184–8189. https://doi.org/10.1039/c4ta00677a
Li CP, Hu Q, Li Y, Zhou H, Lv ZL, Yang XJ, Liu LX, Guo H (2016) Hierarchical hollow Fe2O3@MIL-101(Fe)/C derived from metal-organic frameworks for superior sodium storage. Sci Rep 6:8. https://doi.org/10.1038/srep25556
Wang J, Wu HH, Gao DF, Miao S, Wang GX, Bao XH (2015) High-density iron nanoparticles encapsulated within nitrogen-doped carbon nanoshell as efficient oxygen electrocatalyst for zinc-air battery. Nano Energy 13:387–396. https://doi.org/10.1016/j.nanoen.2015.02.025
Ramaswamy N, Tylus U, Jia QY, Mukerjee S (2013) Activity descriptor identification for oxygen reduction on nonprecious electrocatalysts: linking surface science to coordination chemistry. J Am Chem Soc 135(41):15443–15449. https://doi.org/10.1021/ja405149m
Feng X, Bai Y, Liu MQ, Li Y, Yang HY, Wang XR, Wu C (2021) Untangling the respective effects of heteroatom-doped carbon materials in batteries, supercapacitors and the ORR to design high performance materials. Energy Environ Sci 14(4):2036–2089. https://doi.org/10.1039/d1ee00166c
Wang SY, Chen LL, Liu XJ, Long L, Liu HH, Liu CY, Dong SJ, Jia JB (2020) Fe/N-doped hollow porous carbon spheres for oxygen reduction reaction. Nanotechnology 31(12):9. https://doi.org/10.1088/1361-6528/ab5b56
Yang J, Xiang F, Guo H, Wang LP, Niu XB (2020) Honeycomb-like porous carbon with N and S dual-doping as metal-free catalyst for the oxygen reduction reaction. Carbon 156:514–522. https://doi.org/10.1016/j.carbon.2019.09.087
Chen XX, Zhen XJ, Gong HY, Li L, Xiao JW, Xu Z, Yan DY, Xiao GY, Yang RZ (2019) Cobalt and nitrogen codoped porous carbon as superior bifunctional electrocatalyst for oxygen reduction and hydrogen evolution reaction in alkaline medium. Chin Chem Lett 30(3):681–685. https://doi.org/10.1016/j.cclet.2018.09.017
Zhao J, Lai HW, Lyu ZY, Jiang YF, Xie K, Wang XZ, Wu Q, Yang LJ, Jin Z, Ma YW, Liu J, Hu Z (2015) Hydrophilic hierarchical nitrogen-doped carbon nanocages for ultrahigh supercapacitive performance. Adv Mater 27(23):3541–3545. https://doi.org/10.1002/adma.201500945
Sharma M, Jang JH, Shin DY, Kwon JA, Lim DH, Choi D, Sung H, Jang J, Lee SY, Lee KY, Park HY, Jung N, Yoo SJ (2019) Work function-tailored graphene via transition metal encapsulation as a highly active and durable catalyst for the oxygen reduction reaction. Energy Environ Sci 12(7):2200–2211. https://doi.org/10.1039/c9ee00381a
Meskin PE, Ivanov VK, Barantchikov AE, Churagulov BR, Tretyakov YD (2006) Ultrasonically assisted hydrothermal synthesis of nanocrystalline ZrO2, TiO2, NiFe2O4 and Ni0.5Zn0.5Fe2O4 powders. Ultrason Sonochem 13(1):47–53. https://doi.org/10.1016/j.ultsonch.2004.12.002
Fan XL, Shao J, Xiao XZ, Chen LX, Wang XH, Li SQ, Ge HW (2014) Carbon encapsulated 3D hierarchical Fe3O4 spheres as advanced anode materials with long cycle lifetimes for lithium-ion batteries. J Mater Chem A 2(35):14641–14648. https://doi.org/10.1039/c4ta01511h
Jiang WJ, Gu L, Li L, Zhang Y, Zhang X, Zhang LJ, Wang JQ, Hu JS, Wei ZD, Wan LJ (2016) Understanding the high activity of Fe-N-C electrocatalysts in oxygen reduction: Fe/Fe3C nanoparticles boost the activity of Fe-N-x. J Am Chem Soc 138(10):3570–3578. https://doi.org/10.1021/jacs.6b00757
Zhu AQ, Qiao LL, Tan PF, Ma YJ, Zeng WX, Dong R, Ma C, Pan J (2019) Iron-nitrogen-carbon species for oxygen electro-reduction and Zn-air battery: surface engineering and experimental probe into active sites. Appl Catal B Environ 254:601–611. https://doi.org/10.1016/j.apcatb.2019.05.037
Wang Y, Wu MM, Wang K, Chen JW, Yu TW, Song SQ (2020) Fe3O4@N-doped interconnected hierarchical porous carbon and its 3D integrated electrode for oxygen reduction in acidic media. Adv Sci 7(14):8. https://doi.org/10.1002/advs.202000407
Liu SW, Wang XB, Zhao HJ, Cai WP (2015) Micro/nano-scaled carbon spheres based on hydrothermal carbonization of agarose. Colloids Surf A Physicochem Eng Asp 484:386–393. https://doi.org/10.1016/j.colsurfa.2015.08.019
Meng FL, Wang ZL, Zhong HX, Wang J, Yan JM, Zhang XB (2016) Reactive multifunctional template-induced preparation of Fe-N-doped mesoporous carbon microspheres towards highly efficient electrocatalysts for oxygen reduction. Adv Mater 28(36):7948–7955. https://doi.org/10.1002/adma.201602490
Singh DK, Jenjeti RN, Sampath S, Eswaramoorthy M (2017) Two in one: N-doped tubular carbon nanostructure as an efficient metal-free dual electrocatalyst for hydrogen evolution and oxygen reduction reactions. J Mater Chem A 5(13):6025–6031. https://doi.org/10.1039/c6ta11057f
Chen YF, Li ZJ, Zhu YB, Sun DM, Liu XE, Xu L, Tang YW (2019) Atomic Fe dispersed on N- doped carbon hollow nanospheres for high-efficiency electrocatalytic oxygen reduction. Adv Mater 31(8):8. https://doi.org/10.1002/adma.201806312
Pei ZX, Li HF, Huang Y, Xue Q, Huang Y, Zhu MS, Wang ZF, Zhi CY (2017) Texturing in situ: N, S-enriched hierarchically porous carbon as a highly active reversible oxygen electrocatalyst. Energy Environ Sci 10(3):742–749. https://doi.org/10.1039/c6ee03265f
Yang LJ, Yu JY, Wei ZQ, Li GX, Cao LD, Zhou WJ, Chen SW (2017) Co-N-doped MoO2 nanowires as efficient electrocatalysts for the oxygen reduction reaction and hydrogen evolution reaction. Nano Energy 41:772–779. https://doi.org/10.1016/j.nanoen.2017.03.032
Lin YM, Zhu YS, Zhang BS, Kim YA, Endo M, Su DS (2015) Boron-doped onion-like carbon with enriched substitutional boron: the relationship between electronic properties and catalytic performance. J Mater Chem A 3(43):21805–21814. https://doi.org/10.1039/c5ta03141a
Sa YJ, Seo DJ, Woo J, Lim JT, Cheon JY, Yang SY, Lee JM, Kang D, Shin TJ, Shin HS, Jeong HY, Kim CS, Kim MG, Kim TY, Joo SH (2016) A general approach to preferential formation of active Fe- N-x sites in Fe-N/C electrocatalysts for efficient oxygen reduction reaction. J Am Chem Soc 138(45):15046–15056. https://doi.org/10.1021/jacs.6b09470
Srivastava R, Mani P, Hahn N, Strasser P (2007) Efficient oxygen reduction fuel cell electrocatalysis on voltammetrically dealloyed Pt-Cu-Co nanoparticles. Angew Chem Int Ed 46(47):8988–8991. https://doi.org/10.1002/anie.200703331
Funding
This work was supported by the National Natural Science Foundation of China (No. 21676040, 51879018), National Key Research and Development Program of China (2016YFB0101206), Joint Research Fund Liaoning-Shenyang National Laboratory for Materials Science (2019JH3/30100009), Dalian Science and Technology Innovation Funds (2018J12GX053), and the Fundamental Research Funds for the Central Universities (3132019327, 3132019328).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Dong, W., Zhang, J., Li, W. et al. Zero-valence Fe boosts the activity of Fe–N-C electrocatalyst in oxygen reduction reaction. Ionics 28, 879–891 (2022). https://doi.org/10.1007/s11581-021-04318-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-021-04318-0