
Abstract
Manganous oxide (MnO) nanoparticles strongly anchored on reduced graphene oxide (RGO) have been synthesized by precipitating MnO precursor on RGO surface followed by an annealing process. The MnO/RGO composite has a homogeneous distribution of MnO nanoparticles on electrically conductive RGO, showing more uniform and reduced nanoparticle sizes of MnO than bare MnO owing to the dispersion effect of RGO. The RGO prevents the aggregation and thus increases the contact area of MnO with the electrolyte; The RGO also provides electrically conductive networks for fast charge transfer and buffers the volume change of MnO during lithium intercalation; as a result, the MnO/RGO electrode shows improved performances than the bare MnO electrode in terms of higher specific capacity, superior cycling stability, and enhanced rate capability. The MnO/RGO electrode exhibits an initial discharge/charge capacity of 919/552 mA h g−1 at 0.3 A g−1 and retains a discharge capacity of 544 mA h g−1 after 60 cycles as compared to the bare MnO electrode which shows a rapidly declined specific capacity and only retains 249 mA h g−1 after 60 cycles. The MnO/RGO electrode also displays enhanced specific capacity after testing at different rates. The improved performance of MnO/RGO can be attributed to the strong interphase interaction between MnO and RGO flakes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Advanced lithium-ion batteries (LIBs) with higher energy/power densities and longer lifespan are being intensively pursued to satisfy their increased applications in electric vehicles and portable electronic devices [1,2,3,4]. The charge storage performance of electroactive materials is determinant for the whole property of LIBs, and thus new types of transition metal oxide-based anode candidates with higher capacity and desired stability are actively investigated to replace the graphite anode with a limited theoretical capacity of 372 mA h g−1 [5,6,7,8,9]. Among the transition metal oxide anodes for LIBs, MnO is receiving more and more research attentions owing to its high theoretical capacity of 756 mA h g−1, high bulk density of 5.43 g cm−3, low operational potential, and environmental affability [10,11,12]. However, as a result of the clipping capacity fading from the large volume expansion during the repeated discharge/charge process and the low rate capability due to the kinetic limitations, the pure MnO was restricted from practical application.
Graphene is monolayer carbon atoms with a tight packing honeycomb lattice structure. Graphene has been widespread used to composite with transition metal compounds for applications in fields of energy storage owing to its specific 2D nanostructure, outstanding electrical conductivity, large surface area, and high chemical stability [13,14,15,16]. With respect to the research progress about MnO, graphene can serve as electrically conductive networks to dramatically improve the electrical conductivity of MnO [10, 17], resulting in a significantly improved rate performance. Graphene can also serve as template for the MnO growing [18, 19], which reduces the aggregation of MnO. In addition, graphene can act as flexible buffer layer for alleviating the volume variation of the MnO particles with cycling. Unfortunately, the exposed MnO nanoparticles on graphene surface are still prone to pulverization and aggregation during the cycling owing to the low dispersion of MnO on graphene surface. To date, MnO with morphologies of nanorods [20], nanowires [11], nanosheets, [21], and interconnected particles [22, 23] has been synthesized on graphene surface, and the large size and low dispersion of MnO cause large fluctuated specific capacity during the repeated cycling because of the structure conversion of MnO on graphene surface. Therefore, new method is also needed to prepare excellently dispersed MnO on graphene surface.
In this work, we present a simple and controllable method for the synthesis of uniform MnO nanoparticles on reduced graphene oxide (RGO) surface. The MnO nanoparticles are strongly anchored on graphene oxide (GO) surface, showing reduced particle sizes than the bare MnO. Owing to the well distribution of MnO nanoparticles on RGO surface, the RGO prevents the aggregation of MnO and thus buffers the volume change of MnO during the lithium intercalation, which also provides high electrically conductive networks for fast charge transfer. As a result, the MnO/RGO electrode achieves improved performance of higher specific capacity, superior cycling stability, and enhanced rate capability than the bare MnO electrode.
Experimental
Fabrication of MnO/RGO and bare MnO
First, the GO aqueous solution with a concentration of 0.5 mg mL−1 was prepared by dispersing 50 mg of GO into 100 mL of deionized water under ultrasonic treatment. Then, manganous nitrate 50 wt.% aqueous solution containing 2 mmol of manganous nitrate was added into above solution under magnetic stirring for 30 min. After that, excess ammonia solution was added into the solution and then rested overnight. The product was collected by the centrifugation method and washed repeatedly using deionized water. The product was dried using the freeze-drying method and then calcined at 600 °C for 6 h under H2/Ar gas (H2:10% by volume). For comparison, bare MnO was also prepared by the same procedures without the addition of GO.
Materials characterization
The crystal structures of the products were measured using the X-ray diffraction (XRD; Rigaku, Ultima IV; Cu Kα radiation λ = 0.15418 nm) technique with diffraction angles 2θ from 10 to 80°. The morphology of samples was characterized by field-emission scanning electron microscope (FESEM; JEOL, JSM-7800F) and transmission electron microscope (TEM; JEOL, JEM-2100 plus). The element compositions of samples were tested by X-ray photoelectron spectroscopy (XPS; Kratos, AXIS-ULTRA, DLD-600 W) and energy dispersive X-ray spectroscopy (EDS) mapping equipped on FESEM. The Raman spectra were tested using a Raman spectroscopy (Renishaw inVia Raman spectrometer) at a laser wavelength of 532 nm and the Raman spectra were fitted using the Xpspeak software. Thermal gravimetric analyzer (TGA; Mettler Toledo SMP/ PF7548/ MET/ 600 W) was used to characterize the weight variation of sample at different temperatures under air atmosphere.
Electrochemical measurements
For the method preparing the working electrode, the active material, acetylene black (Super-P), and polyvinylidene fluoride (PVDF) with a mass ratio of 7.5:1.5:1 were mixed in N-methyl-2-pyrrolidinone to form a slurry, and the slurry was coated on copper foil and dried at 110 °C in vacuum overnight; then, the coated copper foil was cut into disk electrodes (8 mm in diameter) and pressed at 10 MPa. The areal density for the mass loading of active material is about 2 mg cm−1 in each electrode. The lithium storage performance of the electrode was tested by assembling 2016-type coin half cells, where a pure lithium foil was used as the counter electrode, Celgard 2300 as the separator, and 1 M LiPF6 in a mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) (1:1 by volume) was used as the electrolyte. The test cells were assembled in an argon-filled glovebox with concentrations of moisture and oxygen below 1.0 ppm. The CV curves were measured using a CHI 660E electrochemical workstation. The specific capacity of the cells was measured by a multichannel battery test system (Neware CT-4008 W, China).
Results and discussion
The MnO nanoparticle/RGO nanocomposite was prepared by a low-temperature annealing method with the synthesis process illustrated in Fig. 1. In a typical procedure, the precursor was first fabricated by precipitation of Mn ions in the GO solution. The Mn(NO3)2 50 wt.% aqueous solution was used as ion source of Mn2+, and the ammonium hydroxide was used as alkali source for Mn2+ precipitation. The abundant oxygen functional groups on GO surface enhance the interaction of Mn2+ with the GO, leading to the uniform nucleation of MnO precursor on GO surface and thereby the formation of strongly anchored MnO precursor on GO. For the next step, the precursor was transformed to MnO/RGO nanocomposite by annealing in the reducing atmosphere of H2/Ar mixed gas. Both of MnO precursor and GO were decomposed and reduced in the reducing gas under the high temperature condition, giving rise to the formation of MnO/RGO nanocomposite. The dispersion of MnO on high-surface area RGO prevents the MnO from aggregation; otherwise, the bare MnO will grow into huge and uniform particles, as illustrated in Fig. 1.

Schematic illustration for the preparation of MnO/RGO composite and bare MnO
The morphology and microstructure of MnO/RGO sample were investigated by FESEM and TEM technologies. Figure 2 a shows the low-resolution FESEM image to characterize the overall morphology of MnO/RGO composite. Both of MnO nanoparticles and RGO sheets can be observed in the FESEM image. It is found that the MnO nanoparticles are all uniformly and strongly anchored on graphene nanosheets surface, and this composite structure prevents both the aggregation of MnO particles and the restacking of graphene nanosheets at the same time. As shown in Fig. 2b, MnO particles prevent the mutual contact of the graphene sheets. From the TEM images in Fig. 2c, d, it is confirmed that the MnO nanoparticles are all anchored on the graphene surface. The specific role of graphene that prevents the aggregation of MnO can be verified by the FESEM images of bare MnO sample (Fig. 2e). The bare MnO product shows a morphology of uneven particles, and some of the particles show a size even higher than 500 nm. The high surface area of RGO serves as a substrate dispersing the MnO nanoparticles, which effectively avoids the aggregation of MnO. The high-magnified TEM image shown in Fig. 2f demonstrates the crystal fringes from the (111) and (200) crystal planes of MnO phase, indicating the synthesis of MnO sample. The composition of the MnO/RGO sample was also measured by the EDS elemental mapping test that equipped on FESEM. As shown in Fig. 2g, the elemental mapping images suggest the existence of C, Mn, and O elements in the MnO/RGO composite. Figure 2 h shows the XRD patterns of the MnO/RGO and the bare MnO products. The MnO/RGO and bare MnO samples show similar diffraction patterns. The diffraction peaks at 35.28°, 40.92°, 59.02°, 70.58°, and 74.12° can be readily indexed to the diffraction from 111, 200, 220, 311, and 222 crystal plane systems of a cubic MnO (JCPDS no. 07–0230), indicating synthesis of the MnO sample. In addition, the weak diffraction peak at 36.42° can be ascribed to the residual Mn3O4 phase from the precursor or the surface oxidation of the sample. The Raman spectroscopy was used to characterize the MnO/RGO sample (Fig. S1). The Raman peak located at 653 cm−1 can be ascribed to the Mn–O vibrational band of MnO. By contrast, the MnO/RGO and its precursor both display two typical D and G bands at 1346 and 1603 cm−1, which can be attributed to sp3-type disordered carbon and sp2-type ordered graphitic carbon, respectively. The areal ratios between D and G bands (AD/AG) of the MnO/RGO and its precursor are 2.65 and 2.46, respectively. The increased AD/AG of MnO/RGO suggests enhanced defects and disordered structure than its precursor, indicating the reduction of GO under the annealing condition.

a, b FESEM and c, d TEM images of the MnO/RGO composite. e FESEM image of the bare MnO. f High-resolution TEM image of a MnO particle in MnO/RGO composite. g The EDS elemental mapping images of the MnO/RGO composite. h XRD patterns of MnO/RGO composite and bare MnO
Figure 3 a shows the wide-scan survey XPS spectrum of the MnO/RGO sample, where the signals from Mn, C, and O elements can be positively observed. The high-resolution XPS spectra was measured to analyze the chemical valence of each composition. As shown in Fig. 3b, in addition to XPS peak at 291.5 eV from the plasmon loss feature, the C 1s spectrum can be best fitted into three peaks. The fitted peaks at 284.8 eV can be ascribed to graphitic carbon in graphene, and the fitted peaks at 286.0 and 288.8 eV can be ascribed to the C–O–C and C–C=O components of oxygenated carbon atoms [22]. From the C 1 s XPS peak, it is found that the XPS peaks from oxygenated carbon atoms are much lower than that from graphitic carbon atoms, which verifies that the GO used for the synthesis procedure has been reduced to RGO under the annealing condition. The high-resolution Mn 2p spectrum shows the XPS peaks at 653.4 eV and 641.8 eV from the Mn 2p1/2 and 2p3/2 electrons of MnO, respectively [24]. In addition, the Mn 3s XPS spectrum was measured to investigate the chemical valences of Mn elements. The Mn 3s spectrum shows two XPS peaks because the coupling of non-ionized 3s electron with 3d valence-band electrons of Mn element, [25, 26] and the component at low binding energy region can be best fitted into two peaks, which exhibit peak splitting of 6.0 and 4.9 eV relative to the XPS peak at high binding energy. The peak splitting of 6.0 eV can be attributed to the Mn2+ in MnO, while the peak splitting of 4.9 eV indicates the existence of high valence Mn ions owing to the surface oxidation of MnO [27, 28]. The high-resolution O 1s XPS spectra shows the peak at 530.0 eV is from the O-containing species in MnO, and the peaks at 531.5 and 533.0 eV can be ascribed to the C=O and C–O species in RGO [22, 23]. The TGA curves for both MnO/RGO nanocomposite and bare MnO were measured in air to determine the mass percent of RGO. As shown in Fig. 3a, the first step of weight loss starts from 30 to 170 °C, which can be attributed to the loss of absorbed water. The TGA curves show increased mass retention when further elevating the temperature owing to the oxidation of MnO. The MnO/RGO sample shows decreased weight owing to the decomposition of RGO. From the TGA curves, the weight ration of RGO in MnO/RGO can be calculated to be 18%. The porous structure of MnO/RGO was investigated by nitrogen adsorption/desorption analysis (Fig. S2). The pore size distribution curve obtained using the Barrett-Joyner-Halenda (BJH) method reveals an average pore size of 1.97 nm, and the Brunauer–Emmett–Teller (BET) surface area of MnO/RGO is 21.5 m2 g−1.

a Wide-scan survey XPS spectrum and the high-resolution b C 1s, c Mn 2p, d Mn 3s, and e O 1s XPS spectra of MnO/RGO. f TGA curves of the MnO/RGO and bare MnO measured in air
The electrochemical lithium-storage properties of MnO/RGO composite were investigated as anode material for lithium-ion battery. Figure 4 a shows the representative cyclic voltammetry (CV) profiles of MnO/RGO electrode for the initial ten cycles at a scan rate of 0.3 mV s−1 within a potential region of 0.01–3 V vs. Li/Li+. In the first cathodic scan, the sharp caudate peak at the potentials lower than 0.25 V can be ascribed to the initial reduction of MnO to metallic Mn (MnO + 2Li+ + 2e− → Mn + Li2O) [24]. An irreversible cathodic peak at around 0.55 V can be attributed to the irreversible reduction of electrolyte and the formation of a solid electrolyte interphase (SEI) layer [12]. In addition, two weak anodic peaks can be also observed at potentials of about 1.44 and 1.84 V, which can be assigned to the reduction of higher oxidation state manganese (e.g., Mn3+, Mn4+) originating from the residual MnOx (1 < x ≤ 2) impurities in MnO. For the first anodic scan, the cathodic peak located at about 1.33 V can be attributed to the reversible oxidization of Mn to MnO (Mn + Li2O → MnO + 2Li+ + 2e−) [29]. Additionally, an oxidation peak at about 2.12 V was observed, indicating the Mn2+ ions could be re-oxidized to a higher oxidation state. In the subsequent CV cycles, the dominant cathodic peak shifts to higher potentials of about 0.26 V and becomes stable at above potential after the third cycle, indicating improved kinetics of lithiation process. At the same time, the dominant anodic peak only slightly shifts to higher potential after the second cycle with a stable anodic peak located at around 1.38 V. The constant CV profiles of MnO/RGO suggest the stabilization of SEI layer after the third lithiation process. The initial ten CV cycles of bare MnO were also measured for comparison (Fig. S3); it is found that the bare MnO sample shows rapidly increased currents in the first four cycles for both cathode and anodic peaks; however, the redox peaks are gradually decreased in the subsequent six cycles. The remarkable change in CV curves of bare MnO suggests the irreversible redox reaction during the lithiation/delithiation process. The stable CV curves of MnO/RGO electrode verifies the significant contribution from RGO.

a CV curves for the initial ten cycles and b CV curves at different scan rates of the MnO/RGO electrode. c Specific capacity as a function of scan rates of the MnO/RGO and bare MnO electrodes. EIS spectra of d MnO/RGO and e, f bare MnO electrodes at different CV cycles with a testing scan rate of 0.5 mV s−1
Figure 4 b shows the CV curves of MnO/RGO at scan rates ranging from 0.2 to 1.0 mV s−1. The Mn/RGO electrode was first cycled at a scan rate of 0.3 mV s−1 for 10 CV cycles to activate the electrode. The CV curves at different scan rates all exhibit strong reduction and oxidation peaks during the cathodic and anodic sweeps, respectively. The reduction peaks shift from 0.30 to 0.15 V with a polarizing voltage of 0.15 V when the specific current increases five times, which shift from 1.34 to 1.46 V with a polarizing voltage of 0.12 V for the oxidation peaks. The small polarization of redox peaks indicates the fast kinetics for the redox reaction, indicating high-rate performance of the MnO/RGO electrode. The CV curves at different scan rates of bare MnO were also measured for comparison (Fig. S4). The bare MnO exhibits a higher polarization than the MnO/RGO, and the reduction peaks of bare MnO rapidly shifted to low potentials, leaving a caudate reduction peak behind for each CV curve when the scan rate is higher than 0.4 mV s−1. The lithium-ions storage capacities of both MnO/RGO and bare MnO were calculated from CV curves according to the equation of C = ∫IdV/(7.2vm) = S/(7.2vm), where S (in W) is the integral area of a CV curve, v (in V s−1) is the scan rate and m (in g) is the mass of electroactive material [30, 31]. The MnO/RGO delivers a specific capacity of 534 mA h g−1 at 0.2 mV s−1, which is much higher than the bare MnO with a specific capacity of 348 mA h g−1. The MnO/RGO electrode exhibits 68% of capacity retention after the specific current increases five times, which is also higher than the bare MnO electrode with only 40% of the capacity retention. Therefore, the RGO can enhance the specific capacity and the rate capability of MnO at the same time.
The transport kinetics of MnO/RGO and bare MnO electrodes was also investigated using the electrochemical impedance spectra (EIS). Figures 4 d–f show Nyquist plots of the EIS spectra of the MnO/RGO and bare MnO electrodes at different CV cycles. The EIS spectra of fresh MnO/RGO and bare MnO electrodes both exhibit an intercept of the high-frequency semi-circular pattern on the real axis attributed to the bulk electrolyte resistance (Rs), a high-frequency semi-circular related to the charge transfer resistance (Rct) and the double-layer capacitance, and a slope line in the low frequency region attributed to the Warburg impedance (Rw) of the diffusion process of lithium-ions. But after the first cycle, another semi-circle was observed at high-frequency range, which is related to the migration resistance (Rf) of lithium-ions through the solid electrolyte interface. The EIS spectra are fitted using the equivalent circuit model in Fig. S5, and the fitting results are compared in Table 1. It is found that the MnO/RGO electrode exhibits remarkably reduced Rct, indicating the RGO should be more effective to construct the electrically conductive networks for charge transfer. In addition, the MnO/RGO electrode also demonstrates greatly reduced Rf compared to the bare MnO electrode, indicating the formation of more stable lithium transfer channels owing to the use of RGO. The reduced Rf of MnO/RGO can be attributed to better contact between MnO and electrolyte, leading to larger reaction area and faster reaction kinetics.
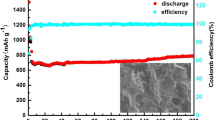
Figure 5 a compares the first, second, and fifth cycles of galvanostatic discharge/charge curves of MnO/RGO electrode. In the discharge curves, a sustained voltage plateau at about 0.5 V is presented, which can be assigned to the reduction from high valence state of manganese ions to metallic Mn during the Li insertion process. The subsequent charge curves show no distinct plateaus but a slope from 1.0 to 1.3 V due to the oxidation of Mn to high valence state. The MnO/RGO electrode exhibits the first cycle discharge/charge capacity of 919/552 mA h g−1 at 0.3 A g−1, showing an initial coulombic efficiency of 60%. The low coulombic efficiency of electrode is mainly attributed to the electrolyte decomposition and inevitable formation of the SEI layer. The lithium storage capability of MnO/RGO electrode tends to be stable in the subsequent cycles. As shown in Fig. 5a, the MnO/RGO electrode shows a charge/discharge capacity of 544/583 mA h g−1 with a coulombic efficiency of 93% in the second cycle, while the electrode exhibits a charge/discharge capacity of 574/580 mA h g−1 with a coulombic efficiency as high as 99% in the fifth cycle. The bare MnO electrode shows rapidly reduced specific capacity in the GCD curves (Fig. S6), so the enhanced cycling stability of MnO/RGO can be ascribed to the specific role of RGO. The high cycling stability of MnO/RGO electrode can be further verified by measuring the specific capacity values at different charge/discharge cycles, as shown in Fig. 5b. The MnO/RGO electrode demonstrates improved cycling stability than the bare MnO electrode. After the second cycle, the MnO/RGO electrode experiences slowly capacity arising in the subsequent 30 cycles and then slowly declining in the subsequent cycles while maintaining coulombic efficiency values higher than 98%, and the electrode showing a specific capacity of 544 mA h g−1 after 60 cycles. By contrast, the bare MnO electrode shows a rapidly declined specific capacity at different charge/discharge cycles, and the bare MnO electrode only retains 249 mA h g−1 after 60 cycles.

a The first, second, and fifth cycles of galvanostatic discharge/charge curves of MnO/RGO electrode. b Cycling performance of MnO/RGO and bare MnO electrodes at a specific current of 0.3 A g−1 and the coulombic efficiency of MnO/RGO electrode at different cycles. c Galvanostatic discharge/charge profiles of MnO/RGO electrode at different specific currents. d Rate performance of MnO/RGO and bare MnO electrodes at specific currents ranging from 0.2 to 0.5 A g−1 and the coulombic efficiency of MnO/RGO electrode at different cycles
The rate performance of MnO/RGO electrode was also evaluated. Figure 5 c shows the GCD curves at different specific currents. The MnO/RGO electrode shows similar galvanostatic discharge/charge curves at different specific currents. Compared to the MnO/RGO electrode, the bare MnO electrode shows rapidly decreased specific capacities at high specific currents (Fig. S7). Figure 5 d shows the rate performance of MnO/RGO and bare MnO electrodes at stepped specific currents between 0.2 and 0.5 A g−1. The MnO/RGO electrode delivers reversible capacities of 656, 621, 580, and 532 mA h g−1 at specific currents of 0.2, 0.3, 0.4, and 0.5 A g−1, respectively. The MnO/RGO shows higher specific capacities than the previously reported MnO-based active materials, such as MnO/graphene aerogel (363 mAh g−1 at 0.2 A g−1) [32], MnO/Graphite (305 mAh g−1 at 0.05 A g−1) [33], carbon-coated MnO microparticulate porous nanocomposites (479 mAh g−1 at 0.2 A g−1) [34], and MnO/carbon nanorods (223 mAh g−1 at 0.05 A g−1) [35]. After 45 cycles, the MnO/RGO electrode displays a specific capacity of 691 mA h g−1 when the specific current returned to the initial specific current of 0.2 A g−1, which is higher than that in the initial 15 cycles. By contrast, the bare MnO electrode shows rapid declined specific capacities at these specific currents, and the electrode only shows a specific capacity of 145 mA h g−1 when the specific current returned to the initial specific current of 0.2 A g−1. Therefore, the MnO/RGO electrode displays superior rate capability against cycling at different specific currents than the bare MnO electrode because of the improved performance from RGO.
Based on the above analysis, it can be found that the MnO/RGO electrode achieves improved performance of higher specific capacity, superior cycling stability, and enhanced rate capability than the bare MnO electrode. The superior performance of MnO/RGO electrode can be ascribed by the following reasons: first, the RGO serves as the substrate for the MnO growth, which effectively prevents the aggregation and increases the contact area of MnO with the electrolyte; second, the RGO provides high electrically conductive networks for fast charge transfer, which increase the charge transportation kinetics; third, the MnO nanoparticles strongly anchored on the RGO surface, and the RGO buffers the volume change of MnO during the lithium intercalation. As a result, superior performance of MnO/RGO has been achieved compared to bare MnO.
Conclusions
In conclusion, uniform MnO nanoparticles strongly anchored on RGO surface have been synthesized by precipitating MnO precursor on graphene oxide followed by annealing process. The RGO prevents the aggregation of MnO, increases the contact area of MnO with the electrolyte, provides high electrically conductive networks for fast charge transfer, and buffers the volume change of MnO during the lithium intercalation. As a result, the MnO/RGO electrode achieves improved performance of higher specific capacity, superior cycling stability, and enhanced rate capability than the bare MnO electrode. Our work provides an effective method for the synthesis of composite electrodes with high LIB performance, and relative investigation gives insights for the electrochemical behaviour of composite electrodes and the fabrication of high-performance anode materials for LIBs.
References
Liu N, Lu Z, Zhao J, McDowell MT, Lee HW, Zhao W, Cui Y (2014) A pomegranate-inspired nanoscale design for large-volume-change lithium battery anodes. Nat Nanotechnol 9:187–192
Stephenson T, Li Z, Olsen B, Mitlin D (2014) Lithium ion battery applications of molybdenum disulfide (MoS2) nanocomposites. Energ Environ Sci 7:209–231
Nitta N, Wu F, Lee JT, Yushin G (2015) Li-ion battery materials: present and future. Mater Today 18(5):252–264
Tan QK, Kong Z, Guan XG, Zhang LY, Jiao ZB, Chen HC, Wu GL, Xu BH (2019) Hierarchical zinc oxide/reduced graphene oxide composite: preparation route, mechanism study and lithium ion storage. J Colloid Interface Sci 548:233–243
Liu Z, Yu Q, Zhao Y, He R, Xu M, Feng S, Li S, Zhou L, Mai L (2019) Silicon oxides: a promising family of anode materials for lithium-ion batteries. Chem Soc Rev 48(1):285–309
Ni S, Liu J, Chao D, Mai L (2019) Vanadate-based materials for Li-ion batteries: the search for anodes for practical applications. Adv Energy Mater 9(14):1803324
Xiao YC, Xu CY, Wang PP, Fang HT, Sun XY, Ma FX, Pei Y, Zhen L (2018) Encapsulating MnO nanoparticles within foam-like carbon nanosheet matrix for fast and durable lithium storage. Nano Energy 50:675–684
Li Z, Wang C, Chen X, Wang X, Li X, Yamauchi Y, Xu X, Wang J, Lin C, Luo D, Wang X, Zhao XS (2020) MoOx nanoparticles anchored on N-doped porous carbon as Li-ion battery electrode. Chem Eng J 381:122588
Wan HR, Hu XF (2020) Sulfur-doped honeycomb-like carbon with outstanding electrochemical performance as an anode material for lithium and sodium ion batteries. J Colloid Interface Sci 558:242–250
Zhang Y, Chen P, Gao X, Wang B, Liu H, Wu H, Liu H, Dou S (2016) Nitrogen-doped graphene ribbon assembled core-sheath MnO@graphene scrolls as hierarchically ordered 3D porous electrodes for fast and durable lithium storage. Adv Funct Mater 26:7754–7765
Zhang S, Zhu L, Song H, Chen X, Zhou J (2014) Enhanced electrochemical performance of MnO nanowire/graphene composite during cycling as the anode material for lithium-ion batteries. Nano Energy 10:172–180
Dubal DP, Jayaramulu K, Sunil J, Kment S, Gomez-Romero P, Narayana C, Zboril R, Fischer RA (2019) Metal–organic framework (MOF) derived electrodes with robust and fast lithium storage for Li-ion hybrid capacitors. Adv Funct Mater 29:1900532
Liu MC, Xu Y, Hu YX, Yang QQ, Kong LB, Liu WW, Niu WJ, Chueh YL (2018) Electrostatically charged MoS2/graphene oxide hybrid composites for excellent electrochemical energy storage devices. ACS Appl Mater Interfaces 10:35571–35579
Li Y, Ye DX, Liu W, Shi B, Guo R, Pei H, Xie JY (2017) A three-dimensional core-shell nanostructured composite of polypyrrole wrapped MnO2/reduced graphene oxide/carbon nanotube for high performance lithium ion batteries. J Colloid Interface Sci 493:241–248
Huang CH, Song XX, Qin YL, Xu BH, Chen HC (2018) Cation exchange reaction derived amorphous bimetal hydroxides as advanced battery materials for hybrid supercapacitors. J Mater Chem A 6:21047–21055
Song XX, Huang CH, Qin YL, Li HL, Chen HC (2018) Hierarchical hollow, sea-urchin-like and porous Ni0.5Co0.5Se2 as advanced battery material for hybrid supercapacitors. J Mater Chem A 6:16205–16212
Bai T, Zhou H, Zhou X, Liao Q, Chen S, Yang J (2017) N-doped carbon-encapsulated MnO@graphene nanosheet as high-performance anode material for lithium-ion batteries. J Mater Sci 52:11608–11619
Wang H, Xu Z, Li Z, Cui K, Ding J, Kohandehghan A, Tan X, Zahiri B, Olsen BC, Holt CM, Mitlin D (2014) Hybrid device employing three-dimensional arrays of MnO in carbon nanosheets bridges battery-supercapacitor divide. Nano Lett 14:1987–1994
Wu Q, Jiang M, Zhang X, Cai J, Lin S (2017) A novel octahedral MnO/RGO composite prepared by thermal decomposition as a noble-metal free electrocatalyst for ORR. J Mater Sci 52:6656–6669
Zhang J, Zhang W, He T, Amiinu IS, Kou Z, Li J, Mu S (2017) Smart reconstruction of dual-carbon decorated MnO for anode with high-capacity and ultralong-life lithium storage properties. Carbon 115:95–104
Zeng F, Zhang X, Pan Y, Xu M, Yang L, Qu Y, Guo M, Yuan C (2018) Graphene-templated growth of vertical MnO nanosheets with open macroporous architectures as anode materials for fast lithium storage. J Alloys Compd 769:10–17
Sun Y, Hu X, Luo W, Xia F, Huang Y (2013) Reconstruction of conformal nanoscale MnO on graphene as a high-capacity and long-life anode material for Lithium ion batteries. Adv Funct Mater 23:2436–2444
Liu DH, Lu HY, Wu XL, Hou BH, Wan F, Bao SD, Yan Q, Xie HM, Wang RS (2015) Constructing the optimal conductive network in MnO-based nanohybrids as high-rate and long-life anode materials for lithium-ion batteries. J Mater Chem A 3:19738–19746
Zhu G, Wang L, Lin H, Ma L, Zhao P, Hu Y, Chen T, Chen R, Wang Y, Tie Z, Liu J, Jin Z (2018) Walnut-like multicore-shell MnO encapsulated nitrogen-rich carbon nanocapsules as anode material for long-cycling and soft-packed lithium-ion batteries. Adv Funct Mater 28:1800003
Zhu YY, Huang CH, Li C, Fan MQ, Shu KY, Chen HC (2019) Strong synergetic electrochemistry between transition metals of α phase Ni-Co-Mn hydroxide contributed superior performance for hybrid supercapacitors. J Power Sources 412:559–567
Chen HC, Qin YL, Cao HJ, Song XX, Huang CH, Feng HB, Zhao XS (2019) Synthesis of amorphous nickel–cobalt–manganese hydroxides for supercapacitor-battery hybrid energy storage system. Energy Storage Mater 17:194–203
Bag S, Roy K, Gopinath CS, Raj CR (2014) Facile single-step synthesis of nitrogen-doped reduced graphene oxide-Mn3O4 hybrid functional material for the electrocatalytic reduction of oxygen. ACS Appl Mater Interfaces 6:2692–2699
Lee JW, Hall AS, Kim JD, Mallouk TE (2012) A facile and template-free hydrothermal synthesis of Mn3O4 nanorods on graphene sheets for supercapacitor electrodes with long cycle stability. Chem Mater 24:1158–1164
Zheng F, Yin Z, Xia H, Bai G, Zhang Y (2017) Porous MnO@C nanocomposite derived from metal-organic frameworks as anode materials for long-life lithium-ion batteries. Chem Eng J 327:474–480
Chen HC, Jiang S, Xu B, Huang C, Hu Y, Qin Y, He M, Cao H (2019) Sea-urchin-like nickel–cobalt phosphide/ phosphate composites as advanced battery materials for hybrid supercapacitors. J Mater Chem A 7:6241–6249
Hu YZ, Huang CH, Jiang SP, Qin YL, Chen HC (2020) Hierarchical nickel-cobalt selenide nanoparticles/nanosheets as advanced electroactive battery materials for hybrid supercapacitors. J Colloid Interface Sci 558:291–300
Yang HN, Zhang CK, Meng QH, Cao B, Tian GY (2019) Pre-lithiated manganous oxide/graphene aerogel composites as anode materials for high energy density lithium ion capacitors. J Power Sources 431:114–124
Tian XD, Du L, Yan YR, Wu SP (2019) An investigation into the charge-storage mechanism of MnO@graphite as anode for lithium-ion batteries at low temperature. ChemElectroChem 6:2248–2253
Guo SM, Lu GX, Qiu S, Liu JR, Wang XZ, He CZ, Wei HG, Yan XR, Guo ZH (2014) Carbon-coated MnO microparticulate porous nanocomposites serving as anode materials with enhanced electrochemical performances. Nano Energy 9:41–49
Zhang XJ, Zhu G, Yan D, Lu T, Pan LK (2017) MnO@C nanorods derived from metal-organic frameworks as anode for superiorly stable and long-life sodium-ion batteries. J Alloys Compd 710:575–580
Acknowledgments
The authors also acknowledge the support of the 1000-talents plan, the World-class discipline program, and the Taishan scholars advantageous and distinctive discipline program of Shandong province for supporting the research team of energy storage materials.
Funding
This work is financially supported by the National Natural Science Foundation of China (no. 21905148), China Postdoctoral Science Foundation (no. 2019T120567 and 2017M612184) and Open Project from Jiangsu Laboratory of Lake Environment Remote Sensing Technologies, Huaiyin Institute of Technology (no. JSLERS-2018-002).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOC 3886 kb)
Rights and permissions
About this article

Cite this article
Qin, Y., Wang, B., Jiang, S. et al. Strongly anchored MnO nanoparticles on graphene as high-performance anode materials for lithium-ion batteries. Ionics 26, 3315–3323 (2020). https://doi.org/10.1007/s11581-020-03515-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-020-03515-7