
Abstract
This study presents the synthesis and electrochemical characterization of a bimetallic PtCo electrocatalyst supported in a ZSM-5 zeolite and XC-72R Vulcan carbon 50 wt.% composite. Two synthesis methodologies were tested: reduction mediated by ethylene glycol and NaBH4. The catalyst electroactivity was assessed by measuring the electrochemical surface area, ESA, and the electrooxidation of methanol was evaluated by cyclic voltammetry and chronoamperometry in acid medium. The nanoelectrocatalysts were obtained with average particle size nearly 3.0 nm and ESA up to 44.7 m2 g−1, indicating the effectiveness of the synthesis methods and of the composite ZSM-5/C used as catalyst support. XPS studies showed that PtCo alloy was obtained by NaBH4 reduction. All samples presented good tolerance to carbonaceous species indicated by jf/jb ratio greater than 1 in methanol oxidation activity test. The 1-h chronoamperometry tests corroborated these results. Some samples obtained in the present work showed higher current density in steady state compared to the commercial sample used as reference and analyzed under the same experimental process. The results showed that the zeolite support was effective at improving catalytic activity independently of the alloy PtCo presence on the catalyst surface.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Over the last 30 years, the search for alternative energy-conversion techniques has been intensified with the aim of finding new ways to decrease greenhouse effects related to internal-combustion vehicles. Among many new green energy technologies, it is worthwhile to highlight fuel cells [1], which are very versatile and can be used in stationary, portable, and mobile applications. An example of fuel cell is the proton exchange membrane fuel cell (PEMFC), which is mainly employed in electric vehicles since it is lighter than solid-oxide fuel cells. The most commonly used electrocatalysts in PEMFC are carbon-supported Pt nanoparticles and carbon-supported PtM nanoparticles alloys (M, secondary metal). Carbon-supported electrocatalysts usually present problems concerning the conditions of cell operation, such as the separation of platinum contained in carbon support and its corrosion. These issues have motivated the search for new materials to be used as electrocatalyst supports [2].
The properties of the support employed are important to obtain catalysts with high dispersion and narrow distribution size of metal nanoparticles, which are the prerequisite for the good electroactive performance and durability of the catalyst. The employed support influences the shape, size, and dispersion of particles as well as the electronic interactions between catalysts and their support. An adequate support material must have the following properties: good electrical conductivity, large surface area, porous structure, good physicochemical stability, and stability to corrosion under oxidizing conditions [3]. According to the review work of Xingwen Yu and Siyu Ye [4], carbon is not an inert material when used as support in catalysts. It changes the galvanic potential of catalytic system, increases the electronic density of catalyst, and decreases the Fermi level. These factors accelerate electron transfer at electrode-electrolyte interface, thus accelerating the catalytic processes in electrode. The interaction between metal and support (Pt/C) occurs either through the transfer of Pt clusters electrons to oxygen atoms at support surface, by establishing chemical bonds, or through load transfer between the phases in contact. This Pt/C interaction is considered beneficial for catalyst because it improves the catalytic properties and stability. However, the use of carbon as catalyst support can decrease catalyst electroactivity due to the possible presence of organosulfur substances, impurities, and the existence of deep micropores, which could trap nanoparticles preventing their access to reagents and electrolyte. Carbon is also unstable under some operating conditions of fuel cells, resulting in its corrosion and detachment of catalytic nanoparticles [5].
Carbon-based nanomaterials like fullerene structures, carbon nanotubes, and graphene have been studied over the last 30 years to reduce the disadvantages related to carbon support. These materials can have different microstructures, morphologies, and excellent properties, such as high specific surface area, balanced pore distribution, and high conductivity [6]. In 2009, Jiang and coworkers used a microwave-polyol method to obtain Pt-Co alloyed nanoparticles supported onto the CNx nanotubes [7]. Recently, Zhang et al. reported a novel strategy for multi-walled carbon nanotube supported PtCo nanocatalysts, which exhibited much higher electrocatalytic activity, stability, and CO tolerance ability than the Pt/MWCNT catalyst for the methanol oxidation reaction. The catalyst was synthesized in deep eutectic solvents by a chemical reduction route [8]. As an example of PtCo nanocatalyst supported reduced graphene oxide matrix can be cited the work of Baronia and coworkers [9]. Additionally, using carbon support, recent work was published by Wang and coworkers. The authors developed nanocomposites based on Co@Pt core@shell nanoparticles encapsulated in nitrogen-doped porous carbons. It obtained a new type of high-performance electrocatalysts for oxygen reduction reaction [10]. However, the differential properties of these nanostructured systems depend on homogeneity and perfection of nanostructures, which may not be maintained when produced in large scale. The difficulty in large-scale production increases the cost of its application.
Zeolites also have well known structural advantages in their application as catalyst support for fuel cells. In 2014, Yeung and Han detailed various applications of zeolite and mesoporous materials in these devices [11]. The article cited a patent registered by Yasumoto et al., who claimed that using type-A zeolites as support for catalytic metals in fuel cell electrodes resulted in lower resistance and fewer ohmic power losses compared to electrodes containing catalysts that use only carbon as support [12]. These zeolitic materials also contain a channel matrix, which enables relatively high levels of gas permeability. Samant and Fernandes showed that Pt/HY and Pt-Ru/HY catalysts are efficient for methanol electrooxidation in fuel cells. These catalysts are significantly better than conventional Pt/C catalyst. The most probable cause of activity increase is due to the preferential CO cluster formation in zeolitic structure pores, facilitating CO2 oxidation during this interaction and, therefore, resulting a faster reaction. According to the authors, the increasing order of methanol electrooxidation activity is HY < Pt/C < Pt/HY < Pt-Ru/C < Pt-Ru/HY. It is important to point out that zeolites do not present electrical conductivity; therefore, they would not be the most adequate appropriate materials for electrocatalysis. To obtain a conductive composite, the porous electrode was prepared by mixing activated Vulcan-XC carbon with the catalyst previously synthesized [13].
In 2008, Pang et al. described Pt/ZSM-5/C catalyst activity in ethanol electrooxidation [14] preparing nanocomposite catalyst in one step. Due to its high stability in acid solutions, ZSM-5 zeolite, besides being a support, is also considered as a second catalyst for ethanol oxidation. The electrochemical performance of zeolitic catalyst (high electrocatalytic activity and good stability of long-term cycles) was better than conventional Pt/C catalyst.
It is noteworthy that the use of zeolites as support material for electrocatalysts employed in the electrode of fuel cells is promising and there is much to investigate in this area, such as the variation of the synthesis methodologies of zeolite-based electrocatalysts. The use of zeolite as catalyst’s support is not very common. Most of the authors present papers with only Pt, even in most recent articles, e. g., Ramires et al. [15] and Daas et al. [16]. A few authors present PtRu bimetallic catalysts like Samant and Fernandes [13] and Mojovic et al. [17]. The use of a secondary and non-noble metal, in a Pt alloy, has well known advantages. The CO is preferentially adsorbed by Pt atoms, and the other metal, which is less noble and hence more oxidizable, produces oxygenated species or hydrated oxides that act right in CO oxidation, then allowing Pt sites for fuel adsorption and oxidation [18].
Concerning the benefits of zeolitic support and the use of a secondary metal, the aim of this study is to synthesize the Pt-Co/ZSM-5/C catalyst through two different methodologies: (1) an adaptation of the method used by Pang et al. using NaBH4 as reduction agent and (2) an ethylene glycol reduction method. The PtCo alloy has not been studied until now, when supported on zeolite.
Materials and methods
Electrocatalyst preparation
Electrocatalysts were prepared using a composite consisting of ZSM-5 zeolite and Vulcan XC-72R carbon as support. It was used a ZSM-5 zeolite leased by CENPES-PETROBRAS with a surface area of 82 m2/g. In both tested methods, zeolite was previously washed with distilled water and dried in a hot-air oven at 80 °C. The catalysts were prepared with 19 wt.% of Pt in relation to support mass (ZSM-5 and C; 50 wt.%), with nominal atomic ratios Pt:Co 3:1 and 1:1, Pt3Co1/ZSM-5/C and Pt1Co1/ZSM-5/C.
In NaBH4 reduction method, zeolite mass was dispersed in distilled water and kept in ultrasonic treatment for 5 min. The previously prepared H2PtCl6.6H2O and Co(NO3)2.6H2O solutions were added to dispersion. Next, an NaBH4 excess solution was added drop by drop to mixture until the solution changed from yellow to black, what indicated the formation of Pt/ZSM-5. After 30-min ultrasonic treatment to complete H2PtCl6.6H2O reduction to Pt nanoparticles and NaBH4 decomposition, functionalized Vulcan carbon mass was added to suspension and submitted to ultrasonic treatment for 3 h. The product (Pt3Co1/ZSM-5/C-NaBH4 and Pt1Co1/ZSM-5/C-NaBH4) was washed with distilled water and dried at 100 °C. Furthermore, the synthesis of Pt3Co1/ZSM-5/C- NaBH4-pH and Pt1Co1/ZSM-5/C-NaBH4-pH catalysts was performed upon pH control. pH solution was adjusted to 9 by dropwise addition of NH4OH solution before NaBH4 reduction.
In the reduction method with ethylene glycol, it employed the same Pt concentration and metal atomic ratios previously used. Initially, a suspension was prepared with carbon and zeolite 50 wt.% in water and kept in ultrasonic treatment for 30 min. Salt solutions H2PtCl6.6H2O, Co(NO3)26H2O and 40 mL of ethylene glycol were added to suspension. The mixture was stirred at 80 °C for 80 min, vacuum filtered and dried at 100 °C resulting in Pt3Co1/ZSM-5/C-EG and Pt1Co1/ZSM-5/C-EG samples. Furthermore, Pt/ZSM-5/C-EG samples were prepared with 10 and 15 wt.% Pt in relation to support to verify the effect of Co introduction in catalyst nanoparticle size and ESA.
X-ray diffraction
X-ray diffraction analysis was conducted in a Rigaku Miniflex X-Ray Diffractometer (V = 15 kV, I = 30 mA) with copper kα radiation (λ = 15,418 Å). The scan rate was taken in steps of 0.05°.s−1, between 20 and 90 degrees in 2θ.
To estimate the crystallite size of samples it was used the Scherrer equation employing the XRD peak at 2θ~39.7° associated to (111) Pt plane of face cubic centered (fcc) Pt structure.
The peak attributed to (111) Pt plane was decomposed into Gaussian primitive functions, considering three different contributions for it in order to observe alloy formation effects in nanoparticles.
X-ray fluorescence
X-ray fluorescence experiments (XRF) were performed at room temperature (21.0 ± 1.5 °C) in a Bruker AXS device, model S4 Pioneer (wave dispersion), with a rhodium tube (E Kα1 = 20.2 KeV). These experiments were accomplished at Instituto Nacional de Metrologia e Qualidade (INMETRO). The fluorescense X-ray spectra were obtained in the energy bands consistent with investigated elements characteristic lines. Equipment calibration was done using reference samples provided by Bruker AXS manufacturer. The study of elements’ concentration calculation was performed through Eval program 1.7 version, using a standard-less method.
The samples were prepared in a tablet shape containing about 300 mg of catalyst, 13 mm diameter, and approximately 2 mm thickness.
X-ray photoelectron spectroscopy
XPS spectra were performed in a surface analysis station (Escaplus P System, Omicron Nanotecnology, Taunusstein, Germany) under ultra-high vacuum (P < 10−9 mbar) and using a Al Kα (1486.6 eV) monochromatic X-ray source, with 20 mA emission current and 15 kV tension.
Survey spectra, showing all elements present on surface, and individual high-resolution spectra of each element were performed. The surveys are used to find surface chemical composition and were measured using a 180 eV energy step in analyzer, while individuals spectra used a 20 eV step. All peaks were analyzed and adjusted after baseline subtraction using Shirley mathematical model, and applying a peak shape Gaussian-Lorentzian obtained from a package of CasaXPS (INMETRO).
The C 1s signal level, 284.6 eV, was chosen for calibration of all spectra. The surface elementary composition was obtained through the ratio of peaks areas and appropriate sensitivity factors. In order to improve the high-resolution peaks’ adjustment and deconvolution of Pt 4f levels, Al 2p contribution was accounted for, since these levels are overlapped.
Transmission electronic microscopy
For transmission electronic microscopy (TEM) analysis, a suspension of the sample in isopropanol was dropped onto Carbon Film 300 Mesh grid and imaged in a FEI Morgagni 268 transmission electron microscope (FEI Company, Netherlands) at accelerating voltage of 80 kV, Tecnai Spirit transmission electron microscope (FEI Company, Netherlands) at accelerating voltage of 120 kV and FEI Tecnai G2 FEG transmission electron microscope (FEI Company, Netherlands) at accelerating voltage of 200 kV for high-resolution image. Image J software was used to study TEM images determining particle sizes and their distribution.
Electrochemical characterization
The samples were characterized by cyclic voltammetry (CV) using a three-electrode cell linked to an Autolab PGSTAT-30 potenciostat/galvanostat. A spiral platinum wire was used as counter electrode, saturated calomel electrode (SCE) as reference electrode and glassy carbon containing a thin catalyst layer as working electrode. The potentials were correct against SHE.
The catalyst sample was dispersed in Nafion® solution in ultrasonic bath. Part of dispersion was carefully deposited onto a glassy carbon electrode surface. Solvent dispersion was evaporated to form a thin film.
The CV was performed in 0.5 mol L−1 H2SO4 aqueous solution to determine the ESA. In electrocatalytic test of methanol oxidation and chronoamperometry, measurements were performed in a 0.5 mol L−1 H2SO4 + 0.5 mol L−1 CH3OH aqueous solution previously saturated with argon. A rotating disk electrode at 1000 rpm was used to take the measurements.
From the hydrogen adsorption/desorption peaks, the ESA was calculated according to procedure described by Schmidt et al. [19]. The monocrystalline platinum reference value was 0.210 mC cm−2 [20, 21]. Calculations for ESA determination were performed considering the amount of Pt present in sample mass used to form the thin layer on electrode.
Results and discussions
XRD and XRF
The XRD of bimetallic catalysts Pt3Co1/ZSM-5/C-EG, Pt1Co1/ZSM-5/C-EG, Pt3Co1/ZSM-5/C-NaBH4 and Pt1Co1/ZSM-5/C- NaBH4 are shown in Fig. 1, where EG and NaBH4 indicate the reduction method employed. All diffractograms showed characteristic platinum peaks at 2θ = 39.7, 46.2, 67.7, and 81.9°, referring to planes (111), (200), (220), and (311), respectively. These indicate the face-centered cubic structure (fcc) of Pt. The peaks at 2θ = 34.9°, 35.9°, 38.4°, 45.4°, 55.2° and 62.4° are present in all samples and are attributed to ZSM-5 zeolite [22].

Diffractograms of Pt3Co1/ZSM-5/C-NaBH4, Pt1Co1/ZSM-5/C-NaBH4, Pt3Co1/ZSM-5/C-EG, Pt1Co1/ZSM-5/C-EG catalysts and ZSM-5 zeolite
The diffractograms showed an asymmetric peak at 2θ = 39.7° attributed to (111) Pt plane. Figure 2a illustrates this peak between 2θ = 36° and 43.5° for Pt3Co1/ZSM-5/C-EG and for ZSM-5. Figure 2b shows an application of decomposition treatment into Gaussian primitive functions considering three different contributions to the peak. The result is shown by dashed curves with maximum at 38.5°, 39.8°, and 40.5° representing the contribution of ZSM-5, (111) Pt plane and a displacement of (111) Pt peak, respectively, due to formation of alloys with Co in the sample. This fact is usually verified in catalyst systems that contain binary alloys [23].

a Diffractogram region related to (111) Pt plane of ZSM-5 and Pt3Co1/ZSM-5/C-EG. b Decomposition into Gaussian primitive curves of peak attributed to (111) Pt plane
Analysis results of samples’ real composition obtained by XRF showed that in NaBH4 reduction the real composition was identical to the nominal one, with proportions 1:1 for Pt1Co1/ZSM-5/C-NaBH4 and 3:1 for Pt3Co1/ZSM-5/C-NaBH4. For samples reduced by ethylene glycol, the real composition showed a significant different result, with 8:1 for Pt1Co1/ZSM-5/C-EG and 20:1 for Pt3Co1/ZSM-5/C-EG.
Ethylene glycol reduction is efficient for obtaining bimetallic catalyst supported in C [24, 25]. A similar method, using reduction by polyol with isopropyl alcohol, was employed by Ramirez et al. in obtaining Pt supported in FAU zeolite-C, a monometallic catalyst [15]. In this article, bimetallic catalysts were prepared in zeolitic support and the method was not effective to obtain the expected atomic ratios. Cobalt reduction was not favored by ethylene glycol reduction method even though the % Pt wt. load was the expected one. Samples are identified by their nominal ratios.
XPS
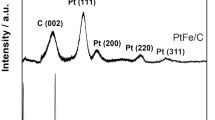
Catalysts’ surface composition of Pt3Co1/ZSM-5/C-NaBH4 and Pt3Co1/ZSM-5/C-EG was evaluated by XPS, as shown by survey spectra in Fig. 3. The spectra present signals of C 1s to 285 eV, O 1s to 533 eV, Pt 4f to 72 eV and Pt 4d to 315 eV [8].

XPS survey spectra of Pt3Co1/ZSM-5/C-NaBH4 and Pt3Co1/ZSM-5/C-EG
Figure 4 shows XPS high-resolution spectra of (a) Pt 4f and (b) Co 2p for Pt3Co1/ZSM-5/C-NaBH4 and (c) Pt 4f for Pt3Co1/ZSM-5/C-EG catalysts.

XPS high-resolution spectra of (a) Pt 4f and (b) Co 2p for Pt3Co1/ZSM-5/C-NaBH4. (c) Pt 4f XPS spectrum for Pt3Co1/ZSM-5/C-EG
The Pt 4f spectrum of catalyst Pt3Co1/ZSM-5/C-NaBH4 presents peaks of levels Pt 4f7/2 and Pt 4f5/2 at 71.7 and 75.1 eV, respectively. The doublet was adjusted to three components for each peak, subtracting the Alo2p level contribution which is located in the same binding energy interval. The three components indicate Pt existence related to different elements in catalysts surface, which enables different oxidation states. The two intense peaks with binding energy of 71.7 eV (Pt/Co 4f7/2) and 75.1 eV (Pt/Co 4f5/2) were stemmed by Pt bonded to Co in a metallic alloy form, Pt-Co. The peaks at 72.4 eV (Pt 4f7/2) and 75.8 eV (Pt 4f5/2) were attributed to Pt(II) species with Pt bonded to zeolitic structure Si, Pt-Si. The peaks corresponding to 73.8 eV (Pt 4f 7/2) and 77.3 eV (Pt 4f5/2) were related to species Pt(II), PtO.
The Co 2p spectrum from catalyst Pt3Co1/ZSM-5/C-NaBH4 shows that oxidized Co is the predominant species in sample surface. Co 2p3/2 peak (781.2 eV) and its satellite (786.3 eV) can be assigned to the Co(OH)2 and CoOOH species, respectively [8, 26]. Zhang et al. presented XPS of Co/MWCNT catalyst exhibiting such peaks Co2p3/2 (783.6 eV) and its satellite (788.4 eV). The displacement of these peaks to lower binding energy values verified in the XPS of the bimetallic catalyst Pt3Co1/ZSM-5/C-NaBH4 indicates the strong charge transfer interaction from Co to Pt atoms on the alloyed PtCo catalytic surface [8, 26].
In XPS Pt 4f spectrum of Pt3Co1/ZSM-5/C-EG catalyst, one notices a doublet with maxima at 72.1 and 75.4 eV attributed to Pt 4f7/2 and Pt 4f5/2, respectively. The doublets were adjusted with two components for each peak. These two components indicate the existence of two different Pt oxidation states in catalysts surface. The two intense signals at 72.1 eV (Pt 4f7/2) and 75.4 eV (Pt 4f5/2) are related to Pt(0). Peaks at 73.5 eV (Pt 4f7/2) and 76.9 eV (Pt 4f5/2) were assigned to Pt(II) species in PtO form. No signals indicating Co presence in surface were detected, as attributed to Co 3p (62.8 eV) and Co 2p between 775 e 810 eV present in NaBH4 reduced sample. This result agrees with XRF analysis which showed a low Co concentration in samples reduced by ethylene glycol.
TEM
TEM images of Pt3Co1/ZSM-5/C-EG, Pt1Co1/ZSM-5/C-EG, and Pt3Co1/ZSM-5/C-NaBH4-pH samples are depicted in Fig. 5.

TEM images and particle size distributions of (a) Pt3Co1/ZSM-5/C-EG, (b) Pt1Co1/ZSM-5/C-EG, (c) Pt3Co1/ZSM-5/C-NaBH4-pH, and (d) HRTEM of Pt3Co1/ZSM-5/C-NaBH4-pH
All samples show highly dispersed nanoparticles on catalytic support. The particle size distribution was calculated considering about 300 nanoparticles. For samples reduced by ethylene glycol, Pt3Co1/ZSM-5/C-EG, Pt1Co1/ZSM-5/C-EG, a large distribution of particle size ranging from 1.5 to 7 nm are illustrated in the insets. The sample reduced by NaBH4 under pH control, Pt3Co1/ZSM-5/C-NaBH4-pH, exhibits narrower distribution ranging from 1 to 4 nm. The average particle size values were 2.7, 2.8 and 2.3 nm for Pt3Co1/ZSM-5/C-EG (Fig. 5a), Pt1Co1/ZSM-5/C-EG (Fig. 5b), and Pt3Co1/ZSM-5/C-NaBH4-pH (Fig. 5c), respectively. Figure 5d shows HRTEM image of Pt3Co1/ZSM-5/C-NaBH4 showing smaller particles, as well as bigger agglomerates. Others authors as Xiao and coworkers obtained in their work average particle sizes from 1.3 to 2.4 nm, for a monometallic Pt/ZSM5 catalyst reduced by ethylene glycol, applied in toluene oxidation. [27].
Table 1 shows the crystallite size calculated through Scherrer equation for all samples including those previously studied by TEM.
The values of particle sizes determined by TEM and the Scherrer equation showed good agreement. There was no significant difference in nanoparticle size due changing Pt:Co ratios. However, it was observed that after introducing Co in Pt electrocatalysts, there was a decrease of particle mean size as verified by others authors previously [28].
Cyclical voltammetry
The cyclic voltammograms for Pt3Co1/ZSM-5/C-NaBH4, Pt1Co1/ZSM-5/C-NaBH4, Pt3Co1/ZSM-5/C-EG, and Pt1Co1/ZSM-5/C-EG are shown in Fig. 6. Table 1 illustrates calculated ESA values from cyclic voltammograms in acid medium and the crystallite size calculated through the Scherrer equation from X-ray diffractograms.

Cyclic voltammograms (20° cycle) in H2SO4(aq), 0.5 mol L−1, sweep speed 50 mV s−1 vs SHE for (a) Pt3Co1/ZSM-5/C-NaBH4 and Pt1Co1/ZSM-5/C-NaBH4, (b) Pt3Co1/ZSM-5/C-EG and Pt1Co1/ZSM-5/C-EG.
The cyclic voltammogram patterns showed the H2 adsorption and desorption occurring between 0 and 0.4 V for all analyzed samples. It is observed the anodic peak attributed to Pt oxide formation between 0.8 and 1.2 V. The reduction of formed oxide occurred at approximately 0.9 and 0.5 V, in direction of cathodic sweeping. This process can be avoided changing the potential swept range.
The catalysts’ ESA are shown in Table 1. Pt3Co1/ZSM5/C-EG sample presented greater ESA: 36.3 m2 g−1. This value is considered good, since Pt mass used in samples was 19 wt.%. Similar ESA results were obtained by Hsieh et al. [29] using Pt-Co nanoparticles supported on carbon nanotubes, which are considered prime support materials. The authors’ results for ESA were 35.4 m2 g−1 using 30 wt% Pt load and atomic ratio Pt:Co 3:1, with average particle size of 6.74 nm determined by MET. The present study emphasizes that zeolites are non-conductive materials; however, greater ESA was obtained with a smaller Pt load, even though particle size was 2.8 nm, as per the Scherrer equation.
The cyclic voltammograms of Pt/ZSM-5/C-EG samples containing 10 and 15 wt% of Pt, Pt/ZSM-5/C-EG-10% and Pt/ZSM-5/C-EG-15%, are shown in Fig. 7 and presented ESA 18.7 and 22.0 m2 g−1, respectively. The calculated ESA and Scherrer particle sizes are shown in Table 1. As observed in Table 1, a slightly higher mass of Pt and the Co introduction should result in smaller particles with higher ESA as observed for Pt3Co1/ZSM5/C-EG sample.

Cyclic voltammograms (20° cycle) in H2SO4(aq), 0.5 mol L−1, sweep speed 50 mV s−1 vs SHE for Pt/ZSM-5/C-EG-10% and Pt/ZSM-5/C-EG-15%
For NaBH4 reduction method, the greatest ESA obtained was 10.1 m2 g−1 for Pt1Co1/ZSM-5/C- NaBH4 sample, a low value when compared with 36.3 m2.g−1 for Pt3Co1/ZSM-5/C-EG. Although the Pt3Co1/ZSM-5/C-NaBH4 had a higher Pt load, it also had a smaller ESA. Considering only the Pt load used, one might expect that a higher load would correspond to a higher ESA as observed, for instance, in the study by Huang et al. [30]. They presented for PtCo/C obtained by H2 reduction, at 150–300 °C, using atomic ratios Pt:Co 5:1, 3:1 and 2:1 ESA values of 70.48, 63.99 and 54.82 m2 g−1, respectively. However, the electroactivity depends on various factors such as particles size, metal alloys formation, number of active Pt sites on catalyst surface, homogeneity of metal nanoparticle dispersion, interaction strength between metal nanoparticles and the support conductivity [31].
The non-conductivity of zeolite in zeolite-carbon nanocomposite electrocatalyst does not seem to be the predominant factor for such low ESA, since the sample reduced with ethylene glycol was more electroactive than the previously mentioned nanotube carbon system, which are usually more conductive than carbon.
Studies with carbon has pointed out that the conditions for synthesis and the nature of precursor metals are crucial in support impregnation process in both methods, the reduction method with chemical agents, such as NaBH4, and applying high-temperature hydrogen reductions [32, 33].
Using zeolites as support, adjusting solution pH promotes an increase in degree of ionic exchange with greater anchoring of metal ions in zeolite structure [34]. Figure 8 shows cyclic voltammograms for Pt3Co1/ZSM-5/C-NaBH4-pH and Pt3Co1/C-NaBH4-pH samples synthesized at pH 9 and Table 1 shows their ESA values and average particle size. It obtained ESA 44.7 m2 g−1 and particle size of 2.9 nm for Pt3Co1/ZSM-5/C-NaBH4- pH. The effect of zeolite 50 wt.% in support material can be observed by comparing the ESA of Pt3Co1/ZSM-5/C and Pt3Co1/C. The sample containing ZSM-5 presented ESA value 35% greater than samples containing only carbon.

Cyclic voltammograms (20o cycle) in H2SO4(aq), 0.5 mol L−1, sweep speed 50 mV s−1 vs SHE for Pt3Co1/ZSM-5/C- NaBH4-pH and Pt3Co1/C- NaBH4-pH
Pt3Co1/ZSM-5/C-EG, Pt3Co1/ZSM-5/C-NaBH4, Pt1Co1/ZSM-5/C-NaBH4, and commercial catalysts A (10%-Pt/C) and B (20%-Pt/C) were submitted to methanol electrooxidation tests by cyclical voltammetry, which are shown in Fig. 9.

Cyclic voltammograms using rotating disk electrode (1000 rpm) in 0.5 mol L−1 H2SO4(aq) + 0.5 mol.L−1 CH3OH(aq), sweep speed 10 mV s−1 vs SHE for (a) Pt3Co1/ZSM-5/C-EG; (b) Pt3Co1/ZSM5/C-NaBH4; (c) Pt1Co1/ZSM5/C-NaBH4; and (d) commercial A (10%-Pt/C) and commercial B 20%-Pt/C (inset)
The cyclic voltammograms’ patterns of electrocatalytic testing in methanol electrooxidation presented an anodic peak current density, jf, which is directly related to the reaction of methanol oxidation and a cathodic peak current density, jb, related to the removal of carbonaceous species which are not completely oxidized in the forward scan. Moreover, the ratio jf/jb provides another measure of catalytic performance: tolerance to carbonaceous species. Table 2 lists values of jf; jb, ratio jf/jb, anodic peak potential (Ef) and cathodic peak potential (Eb).
The cyclic voltammograms tests show the beginning of methanol oxidation occurs around 0.5 V, with an anodic peak potential between 0.88 and 0.92 V and cathodic peak potential between 0.68 and 0.73 V. Among the catalysts tested, Pt3Co1/ZSM-5/C-EG presented greater methanol oxidation activity with jf equal to 1.36 mA.cm−2.
Commercial Pt/C catalyst A and B analyzed under the same conditions showed methanol electrooxidation process initiating around 0 V and the oxidation peak potential (Ef) between 0.96 and 1.1 V. The ratio jf/jb of commercial catalyst A is 1.06 and for commercial catalyst B is 0.63.
The ratio jf/jb can be used to assess the catalysts tolerance to CO poisoning, second Manohara R. and Goodenough J. B. [35]. The authors attributed this anodic peak in the cathodic sweep (jb, reactivation) to removal of carbon species such as CO, for instance, which is partially oxidized during the anodic sweep (jf, activation). These carbon species are generally formed by linear Pt = C=O bonds. The increase in jf/jb ratio can be used to describe catalyst tolerance in terms of intermediate carbon species adsorbed on catalyst surface. A low jf/jb ratio indicates that there is little methanol oxidation for the formation of carbon dioxide during anodic sweep and an excessive accumulation of carbon residues on catalyst surface [36]. Thus, the higher the jf/jb ratio, the higher tolerance, and the lower the ratio, the greater the amount of intermediate carbon species remaining on electrode surface in the anodic sweep, resulting formation of an anodic peak in cathodic sweep with higher current density (jb).
All jf/jb values (Table 2) were greater than 1, indicating a good carbonaceous species tolerance, higher than commercials catalysts with lower jf/jb values, such as commercial catalyst B (0.63). The cyclic voltammograms of commercials catalysts shown in Fig. 9d indicate that jb current was significantly higher than jf. This was not observed in cyclic voltammograms for the samples prepared in this work. This indicates that samples prepared with zeolitic composite might benefit itself from the strong adsorption of intermediates produced during methanol electrooxidation process, enabling a more efficient methanol oxidation. The bimetallic samples would have the role of decreasing the concentration of adsorbed intermediates on Pt surface. Among the catalysts tested, Pt3Co1/ZSM-5/C-NaBH4 had greater jf/jb, 1.79, thus being considered the catalyst with the highest tolerance.
Chronoamperometry
Figure 10 illustrates chronoamperograms displaying electrocatalytic performances for methanol oxidation in 1.0 mol L−1 of CH3OH in 0.5 mol L−1 H2SO4 at anodic potential of 0.5 V versus SHE for Pt3Co1/ZSM-5/C-EG, Pt1Co1/ZSM-5/C-EG, Pt3Co1/ZSM-5/C-NaBH4, Pt1Co1/ZSM-5/C-NaBH4, Pt3Co1/ZSM-5/C-NaBH4-pH, Pt3Co1/C-NaBH4-pH, and commercial B catalyst—20% Pt/C.

Chronoamperograms at 0.5 V using rotating disk electrode (1000 rpm) in 0.5 mol L−1 H2SO4(aq) + 0.5 mol L−1 CH3OH(aq), for (a) Pt3Co1/ZSM-5/C-EG and Pt1Co1/ZSM-5/C-EG; (b) Pt3Co1/ZSM-5/C-NaBH4 and Pt1Co1/ZSM-5/C-NaBH4; (c) Pt3Co1/ZSM-5/C-NaBH4-pH and Pt3Co1/C-NaBH4-pH. Details show, for all chronoamperogram, the initial j values until 900 s
The insets of Fig. 10 show the current density (j) decreasing suddenly after the initial potential jump for all samples. This initial j decrease is due to double layer charging and others fast processes at electrode surface [37]. For commercial sample B (20% Pt/C), this sudden j decrease is followed by a slow change towards a steady state after 25-min polarization. For bimetallic zeolitic catalysts, a quasi steady state is reached after only 5 min except for the Pt3Co1/C-NaBH4, which shows a slow j decrease over all time range measured. All j were between 8 and 12 mA cm−2, values higher than those of commercial sample (j = 4 mA.cm−2). Similar chronoamperograms’ patterns were obtained in recent work of Daas and Ghosh [16], where Pt/ZSM-5/C showed similar stability behavior, although with a lower j value (2 and 4 mA cm−2) for electrodeposited Pt nanoparticles. The higher j, higher the number of electrochemical reactions occurring per unit of time in the system, indicating good electrocatalytic properties of sample. Lower currents could occur due to increase of poisoning species on catalysts surface.
The current values obtained for Pt1Co1/ZSM-5/C-EG and Pt1Co1/ZSM-5/C-NaBH4 eletrocatalysts were higher than those obtained for Pt3Co1/ZSM-5/C-EG and Pt3Co1/ZSM-5/C-NaBH4. The samples reduced under pH control did not present good electrochemical stability results as seen in Fig. 10c. Although Pt3Co1/ZSM-5/C-NaBH4-pH and Pt3Co1/C-NaBH4-pH present the best methanol electrooxidation properties in previous study by cyclic voltammetry, the chronoamperometric tests showed that pH adjustment in zeolitic supports was not a good synthesis strategy to obtain stable catalysts.
The chronoamperometric results corroborate with jf/jb ratios’ results obtained by cyclic voltammetric, indicating the good carbonaceous tolerance for majority of the electrocatalysts tested comparing to commercial sample B.
Conclusion
Zeolite application proved to be efficient in enabling a homogeneous dispersion of nanoparticles in the zeolitic support according to TEM images. Both reduction methods employed allowed to obtain nanoparticles with average size of up to 3.0 nm with narrow sizes distribution. XPS analyzes show the presence of PtCo alloy on the surface of samples reduced by NaBH4 and the absence on the surface of samples reduced by the ethylene glycol method.
All electrocatalysts showed electroactivity, which confirms the electrical conduction capacity of C/ZSM-5 composite support. The samples with higher ESA were Pt3Co1/ZSM-5/C-NaBH4-pH and Pt3Co1/ZSM-5/C-EG. It was noted that the increase in Pt:Co ratio tends to also raises ESA values.
The methanol electrooxidation tests indicated that zeolitic catalysts had a good carbonaceous species tolerance. The 1-h chronoamperometric tests confirmed these results. Some samples showed higher current density in quasi steady state than B commercial sample used as reference and analyzed under the same experimental conditions.
References
Serrano E, Rus G, García-Martínez J (2009) Nanotechnology for sustainable energy. Renew Sust Energ Rev 13:2373–2384
Vignarooban K, Lin J, Arvay A, Kolli S, Kruusenberg I, Tammeveski K, Munukutla L, Kannan AM (2015) Nano-electrocatalyst materials for low temperature fuel cells: a review. Chin J Catal 36:458–472
Zhang S, Shao Y, Yin G, Lin Y (2013) Recent progress in nanostructured electrocatalysts for PEM fuel cells. J Mater Chem A 1:4631–4641
Yu X, Ye S (2007) Recent advances in activity and durability enhancement of Pt/C catalytic cathode in PEMFC part I. Physico-chemical and electronic interaction between Pt and carbon support, and activity enhancement of Pt/C catalyst. J Power Sources 172:133–144
Trogadas P, Fuller TF, Strasser P (2014) Carbon as catalyst and support for electrochemical energy conversion. Carbon 75:5–42
Wu Q, Yang L, Wang X, Hu H (2017) From carbon-based nanotubes to nanocages for advanced energy conversion and storage. Acc Chem Res 50:435–444
Jiang S, Ma Y, Jian G, Tao H, Wang X, Fan Y, Lu Y, Hu Z, Chen Y (2009) Facile construction of Pt-co/CNx nanotube electrocatalysts and their application to the oxygen reduction reaction. Adv Mater 21:4953–4956
Zhang JM, Sun SN, Li Y, Zhang XJ, Zhang PY, Fan YJ (2017) A strategy in deep eutectic solvents for carbon nanotube-supported PtCo nanocatalysts with enhanced performance toward methanol electrooxidation. Int J Hydrog Energy 42:26744–26751
Baronia R, Goel J, Tiwari S, Singh P, Singh D, Singh SP, Singhal SK (2017) Efficient electro-oxidation of methanol using PtCo nanocatalysts supported reduced graphene oxide matrix as anode for DMFC. Int J Hydrog Energy 42:10238–10247
Wang L, Tang Z, Yan W, Wang Q, Yang H, Chena S (2017) Co@Pt Core@Shell nanoparticles encapsulated in porous carbon derived from zeolitic imidazolate framework 67 for oxygen lectroreduction in alkaline media. J Power Sources 343:458–466
Yeung KL, Han W (2014) Zeolites and mesoporous materials in fuel cell applications. Catal Today 236:182–205
Yasumoto E, Hatoh K, Gamou T (1997) U.S. Patent No. 5,702,838
Samant PV, Fernandes JB (2004) Enhanced activity of Pt(HY) and Pt–Ru(HY) zeolite catalysts for electrooxidation of methanol in fuel cells. J Power Sources 125:172–177
Pang H, Chen J, Yang L, Liu B, Zhong X, Wei X (2008) Ethanol electrooxidation on Pt/ZSM-5 zeolite-C catalyst. J Solid State Electrochem 12:237–243
Ramírez AM, Aguilera MV, López-Badillo CM, Ruiz-Camacho B (2017) Synthesis of FAU zeolite-C composite as catalyst support for methanol electro-oxidation. Int J Hydrog Energy 42:30291–30300
Daas BM, Ghosh S (2017) Electro-oxidation of methanol and ethanol catalyzed by Pt/ZSM-5/C. Electroanalysis 29:2516–2525
Mojovic Z, Bancóvic P, Jovic-Jovicic N, Rabi-Stankovic AA, Milutinovic-Nikolic A, Jovanovic D (2012) Carbon monoxide electrooxidation on Pt and PtRu modified zeolite X. J Porous Mater 19:695–703
Camara GA, Giz MJ, Paganin VA, Ticianelli EA (2002) Correlation of electrochemical and physical properties of PtRu alloy electrocatalysts for PEM fuel cells. J Electroanal Chem 537:21–29
Schimidt J, Gasteiger HA, Stab GD, Urban PM, Koib DM, Behm RJ (1998) Characterization of high-surface-area electrocatalysts using a rotating disk electrode configuration. J Electrochem Soc 145:2354–2358
Bal L, Gao L, Conway BE (1993) Problem of in situ real area determination in evaluation of performance of rough or porous, gas-evolving electrocatalysts. Part 1—basis for distinction between capacitance of the double layer and pseudocapacitance due to absorbed H in the H2 evolution reaction at Pt. J Chem Soc 89:235–242
Bal L, Gao L, Conway BE (1993) Problem of in situ real-area determination in evaluation of performance of rough or porous, gas-evolving electrocatalysts. Part 2—unfolding of the electrochemically accessible surface of rough or porous electrodes: a case-study with an electrodeposited porous Pt electrode. J Chem Soc 89:243–249
Yun H, Kim SD, Park DW, Lee JY, Yi SC, Kim WJ (2010) Cell performance of MEA fabricated with Pt-ZSM-5-carbon electrode for PEMFC. Microporous Mesoporous Mater 131:122–127
Salgado JRC, Antolini E, Gonzalez ER (2004) Preparation of Pt-Co/C electrocatalysts by reduction with borohydride in acid and alkaline media: the effect on the performance of the catalyst. J Power Sources Short Commun 138:56–60
Neto O, Dias RR, Tusi MM, Linardi M, Spinacé E (2007) Electro-oxidation of methanol and ethanol using PtRu/C, PtSn/C and PtSnRu/C electrocatalysts prepared by an alcohol-reduction process. J Power Sources 166:87–91
Ribeiro VA, Correa OV, Neto AO, Linardi M, Spinacé EV (2010) Preparation of PtRuNi/C electrocatalysts by an alcohol-reduction process for electro-oxidation of methanol. Appl Catal A Gen 372:162–166
Lee E, Kim S, Jang JH, Park HU, Matin MA, Kim YT, Kwon YU (2015) Effects of particle proximity and composition of Pt-M (M= Mn, Fe, Co) nanoparticles on electrocatalysis in methanol oxidation reaction. J Power Sources 294:75–81
Chen C, Chen F, Zhang L, Pan S, Bian C, Zheng X, Meng X, Xiao F (2015) Importance of platinum particle size for complete oxidation of toluene over Pt/ZSM-5 catalysts. Chem Commun 51:5936–5938
Fetohi AE, Amin RS, Hameed RMA, El-Khatib KM (2017) Effect of nickel loading in Ni@Pt/C electrocatalysts on their activity for ethanol oxidation in alkaline medium. Electrochim Acta 242:187–201
Hsieh CT, Lin JY, Wei JL (2009) Deposition and electrochemical activity of Pt-based bimetallic nanocatalysts on carbon nanotube electrodes. Int J Hydrog Energy 34:685–693
Huang Q, Yang H, Tang Y, Lu T, Akins DL (2006) Carbon-supported Pt-co alloy nanoparticles for oxygen reduction reaction. Electrochem Commun 8:1220–1224
Lai J, Luque R, Xu G (2015) Recent advances in the synthesis and electrocatalytic applications of platinum-based bimetallic alloy nanostructures. ChemCatChem 7:3206–3228
Fugiwara N, Yasuda K, Ioroi T, Siroma Z, Miyazaki Y (2002) Preparation of platinum-ruthenium onto solid polymer electrolyte membrane and the applucation to a DMFC anode. Electrochim Acta 47:4079–4084
Dickinson AJ, Carrette LPL, Collins JA, Friedrich KA, Stimming U (2000) Preparation of a Pt-Ru/C catalyst from carbonil complexes for fuel cell applications. Electrochim Acta 47:3733–3739
Foletto EL, Kuhnen NC, Jose HJ (2000) Synthesis of the ZSM-5 zeolite and its structural properties after copper ion-exchange. Cerâmica 46:210–213
Manohara R, Goodenough JB (2004) Methanol oxidation in acid on ordered NiTi. J Mater Chem 2:875–887
Liu Z, Ling XY, Su X, Lee JY (2004) Carbon-supported Pt and PtRu nanoparticles as catalysts for a direct methanol fuel cell. J Phys Chem B 108:8234–8240
Bergamaski K, Gonzalez ER, Nart FC (2008) Ethanol oxidation on carbon supported platinum-rhodium bimetallic catalysts. Electrochim Acta 53:4396–4406
Acknowledgements
The authors would like to thank Oleksii Kuznetsov for XRF measurements and CENABIO for TEM images.
Funding
This work was supported by the Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (Nos. E-26/110.665/2013 and E-26/010.001095/2015).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Rocco, A.M., Melo, K.V., de Araújo Mota, C.J. et al. Synthesis of PtCo/ZSM-5/C electrocatalyst and electrochemical activity. Ionics 25, 253–264 (2019). https://doi.org/10.1007/s11581-018-2572-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-018-2572-9