
Abstract
Herein, we report the structural, bonding, dielectric, and electrical transport properties of Ca3(MgTa2)O9 (CMT) and Ca3(ZnTa2)O9 (CZT) with the chemical formula Ca(Mg1/3Ta2/3)O3 and Ca(Zn1/3Ta2/3)O3, respectively, synthesized by the solid-state reaction technique. The Rietveld refinement of powder X-ray diffraction (PXRD) patterns has confirmed that the CMT and CZT are triple perovskites belonging to monoclinic P2 1 /c space group with 1:2 B-site ordering. The unit cell contains four formula units of CMT and CZT. The results of the structural study are corroborated by Fourier transform infrared (FTIR) and Raman spectroscopic studies. Comparing the crystal structures of 1:2 ordered perovskites with the chemical formula Ba(B′1/3B″2/3)O3 and Ca(B′1/3B″2/3)O3, we have predicted that for this group of perovskites, only calcium-based systems exhibit a monoclinic crystal structure of P2 1 /c space group due to the smaller ionic radii of Ca2+ compared to those of Ba2+. The grain size ranges between 0.38 and 2.66 μm for CMT and 0.11 and 1.60 μm for CZT, respectively. The analysis of the dielectric permittivity in the framework of the modified Cole-Cole model has revealed that the dielectric relaxation in CMT and CZT is strongly temperature dependent and polydispersive in nature. The activation energies associated with dielectric and electrical transport properties are ≈0.35 and 0.33 eV for CMT and CZT, respectively. The polaron hopping governs the electrical and dielectric response of the samples. It has been found that CMT and CZT exhibit enhancement in dielectric properties compared to their niobate counterparts and 1:1 ordered tantalum-based perovskite oxides.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Studies on the dielectric, structural, and microstructural properties of complex perovskite oxides have attracted paramount attention in the recent past owing to their widespread technological applications in memory devices (dynamic RAM), wireless communication systems, cellular phones, global positioning systems, etc. in the form of capacitors, filters, and resonators [1–17]. The development of materials with high dielectric constant and low loss tangent has immense importance for miniaturization of electronic devices. The heterovalent perovskites with the general formula A(B′ x B″1 − x )O3 exhibit various types of cation ordering at B site of the perovskite unit cell [9–17]. Moreover, these materials have high dielectric constant and they have applications in multilayer capacitors and piezoelectric and pyroelectric transducers [9, 10]. For exploring widespread applications, the proper understanding of the structural property and ac electrical behavior of these materials is very crucial. In recent years, the alternating current impedance spectroscopy (ACIS), which probes the electrical phenomena taking place in a system by applying an ac signal, has emerged as an authoritative tool to study the electrical properties of complex perovskite oxides [9–17]. This method also enables us to separate out the intrinsic (bulk) and extrinsic (grain boundary, surface layer, and electrode) contributions of polycrystalline materials.
In case of A(B′ x B″1 − x )O3-type perovskite oxides, the dielectric property significantly depends upon the B-site ion ordering, atomic radius, and valence of B-site ions as well as on the presence of defect and oxygen vacancies. A large body of work has been carried out on the dielectric property and relaxation mechanism of A(B′1/3B″2/3)O3 perovskite oxides where A = Pb, Ba, Sr, Ca; B′ = Mg, Zn, Ni, Sr; and B″ = Nb, Ta [11–18] in the frequency range of 100 Hz to 1 MHz over a wide range of temperature. It may be noted that the dielectric property of perovskite oxides can be indirectly probed by using infrared spectroscopy. The real and imaginary parts of dielectric permittivity, loss tangent, and quality factor of these materials can be estimated using phonon parameters obtained from infrared spectroscopic study. If the experimental error is low, the values of dielectric parameter so obtained match well with the experimentally measured values [19–22]. To the best of our knowledge, there is no such report on the analysis of variation of real and imaginary parts of the dielectric permittivity with frequency of the perovskite systems having the chemical composition Ca(B′1/3Ta″2/3)O3 (where B′ = Mg and Zn). In this background, an analysis of the real and imaginary parts of the dielectric permittivity spectra of 1:2 ordered Ca3(MgTa2)O9 (CMT) and Ca3(ZnTa2)O9 (CZT) with the chemical formula Ca(Mg1/3Ta2/3)O3 and Ca(Zn1/3Ta2/3)O3 considering the distribution of relaxation time and effect of dc conductivity appears to be very interesting. Further, comparison of the results with other perovskite oxides of the same kind may provide fruitful information regarding the dielectric properties of Ta-based perovskite systems.
The complex perovskite oxides exhibit a wide variety of crystal structure depending on the difference in the radii and valence of B-site cations [23–29]. The A(B′1/3B″2/3)O3-type perovskite oxides show hexagonal crystal structure of \( Pm\overline{3}1 \), P6 3 /mmc and monoclinic structure of P2 1 /c space groups [30–37], and they exhibit both the 1:1 (e.g., Pb(Mg1/3Nb2/3)O3 [23, 24] and 1:2 (e.g., Ba(Mg1/3Ta1/3)O3 [29, 38]) B-site ordering patterns. The study on the crystal structure, microstructure, and cation ordering of Sr(Mg1/3Nb2/3)O3 and Ca(Mg1/3Nb2/3)O3 using X-ray diffractometry, selected area electron diffractometry, and transmission electron microscopy revealed that both samples had 1:2 ordered monoclinic unit cell [28, 29]. However, both Park et al. and Lee et al. did not analyze the powder X-ray diffraction (PXRD) patterns of the samples by the Rietveld refinement method [32, 33]. Relying upon the results of these works, Fu et al. indexed the PXRD pattern of Ca(Mg1/3Ta2/3)O3 in monoclinic P2 1 /c space group and confirmed the validity of space group assignment by Raman spectroscopic study [34, 35]. Again, Fu et al. did not analyze the PXRD pattern of Ca(Mg1/3Ta2/3)O3 by the Rietveld refinement method, although they had shown that the PXRD pattern of the sample matched well with that of Ca[(Mg1/3Ta2/3)0.9Ti0.1]O3. The monoclinic P2 1 /c symmetry of Ca(Mg1/3Ta2/3)O3 was proven by the Rietveld refinement of PXRD pattern and Raman spectroscopic study [34–36]. However, to the best of our knowledge, there is no report on the crystal structure and bonding property of CMT and CZT. In this context, the aim of the present work is to thoroughly investigate the crystal structure, bonding property, and B-site cation ordering of CMT and CZT along with their dielectric behavior in the frequency range of 50 Hz to 1 MHz.
In this paper, we have reported the crystal structure, microstructure, and dielectric relaxation property of CMT and CZT along with the results of dc conductivity measurements. We have employed powder X-ray diffractometry, scanning electron microscopy (SEM), Fourier transform infrared (FTIR) spectroscopy, Raman spectroscopy, and ACIS technique to fulfill the goal. The present study has revealed that the dielectric relaxations in the samples are polydispersive in nature and the polaron hopping mechanism controls the electrical response of the samples. Results also indicate that the dielectric property of perovskite oxides can be tuned by the modification of crystallite size and judicious choice of B-site metal ions.
Experimental
The polycrystalline CMT was synthesized through conventional ceramic method by mixing powders of CaCO3 (reagent grade), Ta2O5 (reagent grade), and MgO (reagent grade) in stoichiometric ratio, while for CZT, ZnO (reagent grade) was used instead of MgO. The blended powders were calcined in a Pt crucible at 1623 K in air for 16 h and thereafter cooled down to room temperature at the rate of 100 K h−1. The phase formation was confirmed by recording the powder PXRD patterns of the calcined samples. Finally, the calcined samples were pelletized into a disc using polyvinyl alcohol as binder, and the discs were sintered at 1673 K for 10 h and cooled down to room temperature at the rate of 1 K/min. The thickness and the diameter of these discs were 1.63 and 9.56 mm, respectively, for CMT and 2.87 and 9.72 mm, respectively, for CZT. The structural, morphological, and electrical characterizations were carried out using these discs. For electrical measurements, both the flat surfaces of these discs were polished thoroughly and were coated with silver paste and then heated at 473 K for 2 h. After electrical measurement, the silver paste was thoroughly removed by rubbing these discs by fine emery paper and then cleaned by acetone and distilled water. Afterward, to check whether the porosity will contribute to the dielectric property or not, we had measured the density of these discs using the Archimedes principle. The Archimedes density of CMT is 5.967 g cm−3 and CZT is 6.342 g cm−3, while the crystalline densities of CMT and CZT are 5.971 and 6.348 g cm−3, respectively. This indicates that the amount of porosities in the sample discs is negligible.
The PXRD patterns of CMT and CZT were recorded at room temperature by Bruker D8 Advanced Diffractometer using Cu Kα (λ = 1.54184 Å) radiation over a range of Bragg angles 10° ≤ 2θ ≤ 70° with the step size of 0.02° and scan time of 6 s/step. The generator was set at 40 kV and 40 mA. We have determined the space group of the samples by using FINDSPACE of EXPO2009 package [39] through statistical analysis of the PXRD data. The GSAS program with EXPGUI interface was used for Rietveld structural refinement of the PXRD patterns [40]. The background was described by the shifted Chebyshev function of first kind with 36 points regularly distributed over the entire 2θ range. The field emission scanning electron microscope (FESEM) (FEI, INSPECT F50) was used for the morphological and microstructural characterizations. The chemical compositions of the samples were determined by the energy dispersive X-ray spectrometer (EDS, BRUKER) attached with the FESEM equipment.
The FTIR spectra of the samples were recorded in transmission mode within the wavenumber range 400–1300 cm−1 at room temperature using Perkin Elmer Spectrum Two FTIR spectrometer. The Raman spectra of CMT and CZT were recorded by WITEC alpha, 300R Raman spectrometer equipped with diode laser source of λ = 532 nm. To have a good profile, a thin film sample over a glass slide was used instead of a powder sample. For preparing the films, the powders of the sample were dispersed in acetone. Then, the films of the sample were developed on a clean glass slide by the drop cast technique. Afterward, the solvent was removed by drying the films in vacuum at room temperature.
The capacitance (C), conductance (G), impedance (Z), and phase angle (φ) were recorded by an LCR meter (HIOKI 3532) at different temperatures between 393 and 573 K as a function of frequency (50 Hz to 1 MHz) and at the oscillation voltage of 1.0 V. During data collection, the sample was heated at a rate of 0.5 K/min and the measurement temperatures were kept constant with an accuracy of ±0.5 K using a programmable vacuum oven. The dc conductivity of the samples was measured by the four probe method.
Results and discussion
Structural and morphological properties
In case of perovskite oxides, subtle structural variation leads to drastic changes in their physical properties. Thus, determination of the crystal structure of perovskite oxides has immense importance. For many ABO3-type perovskites, the oxygen octahedra gets tilted due to the misfit of the ionic radii of the A and B cations in the cubic lattice, and in consequence, distortions from ideal cubic structure occur in such systems [41, 42]. The degree of such distortion can be estimated by calculating the Goldschmidt tolerance factor (T f) using the formula \( {T}_{\mathrm{f}}=\frac{r_{\mathrm{A}}+{r}_0}{\sqrt{2}\left(\left\langle {r}_{\mathrm{B}}\right\rangle +{r}_0\right)} \), where r A, r B, and r 0 are the ionic radii of A site, B site, and O ions, respectively [43]. The cubic structure is obtained if the value of T f is close to unity, whereas for monoclinic and orthorhombic perovskite, T f is less than 1 [16–18]. The values of the tolerance factor (T f) for CMT and CZT are 0.942 and 0.934, respectively. T f has been calculated by using Shannon effective ionic radii [44] and taking the \( {r}_{\mathrm{B}}=\frac{2{r}_1+{r}_2}{3} \), where r 1 is the radius of Ta5+ ion and r 2 is the radius of Mg2+ ion for CMT and Zn2+ ion for CZT. According to Reaney et al., perovskites with 0.985 < T f < 1.06 are expected to have an untilted structure, while perovskites with 0.964 < T f < 0.985 exhibit antiphase tilting and those with T f < 0.964 show both inphase and antiphase tilting distortions [45, 46]. Thus, CMT and CZT may possess a monoclinic structure with both inphase and antiphase tilting.
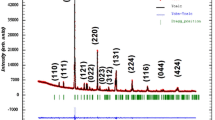
Here, we have determined the crystal structure of CMT and CZT through the Rietveld refinement of PXRD data. It is noteworthy that the PXRD patterns of CMT and CZT (Fig. 1) match very well with that of Ca(Zn1/2Nb2/3)O3 [33]. According to the literature, Ca(Zn1/3Nb2/3)O3 possesses a monoclinic structure of P2 1 /c symmetry, which results from an inphase and antiphase octahedral tilting along the [111] direction and in the basal plane of perovskite pseudocubic cell [33]. It may be noted that the ionic radius of Nb (0.641) ≈ Ta (0.640) and that of Zn(0.74) ≈ Mg(0.72). As the crystal structure of ABO3 type of perovskite oxide mainly depends upon the ionic radii of A and B cations, it may therefore be inferred that the crystal structure of CMT and CZT may be analogous to that of Ca(Zn1/2Nb2/3)O3. Further, the analysis of PXRD data by FINDSPACE indicates that both CMT and CZT possess a monoclinic structure of P2 1 /c space group. Given this background, the crystal structures of both CMT and CZT have been refined by the Rietveld-based method by assigning the atomic coordinates using the crystal structure of Ca(Zn1/2Nb2/3)O3 [37] as the trial structural model.

Rietveld refinement plots of a CMT and b CZT. The experimental data are represented by green solid circles and the continuous red line represents the simulated XRD pattern. The difference between the experimental and simulated PXRD patterns is shown by the continuous pink line. The magnified view of the highest peaks of CMT and CZT is shown in the insets of the corresponding figures
To determine the lattice parameters, bond lengths, and bond angles, we have fitted the PXRD patterns of CMT and CZT by the Rietveld refinement package GSAS [40]. The refinement was performed by placing the atoms (Ca, Mg, Zn, Ta, and O) at Wyckoff positions as depicted in Tables 1 and 2. In the refinement process, positions of Mg ions for CMT and Zn ions for CZT are set fixed and the positions of all the other atoms have been refined. After a few cycles, the refinement successfully converges with an excellent agreement between the experimental and calculated patterns (Fig. 1). No trace of any impurity phase has been detected. The refined values of atomic coordinate and isotropic thermal parameter are presented in Tables 1 and 2. The values of lattice, structural, and refinement parameters are listed in Table 3. These values are nearly equal to those reported for Ca(Zn1/3Nb2/3)O3. The bond lengths for CMT and CZT are presented in Tables 4 and 5, respectively. The asymmetric units of only CMT along with the coordination environment of different metal ions are shown in Fig. 2 as CMT and CZT are isostructural. According to the results of the Rietveld refinement, the crystal structure of both samples is monoclinic, belonging to P2 1 /c space group with 60 atoms in the unit cell. The unit cell of CMT and CZT contains four formula units of Ca3(Mg1Nb2)O9 and Ca3(Zn1Nb2)O9, respectively. In the unit cell of CMT, there are two Mg ions at 2a site and two Mg ions at 2d site, while for CZT, there are two Zn ions at 2a site and two Zn ions at 2d site in addition to 12 Ca, 8 Ta, and 36 O ions occupying the 4e Wyckoff positions in the unit cell of both crystal structures.

Unit cell of CMT along with the coordination environment of Mg2+ and Ta5+ ions
The A-site Ca2+ ions are coordinated with eight nearest neighboring oxygen ions. The A-site cations of perovskite oxides usually exhibit 12 coordination mode, but eight coordination of A cations is unique for perovskites belonging to monoclinic P2 1 /c space group and has been observed earlier for Ca(Zn1/3Nb2/3)O3 and Ca(Ca1/3Nb2/3)O3 [37, 47]. In monoclinic P2 1 /c structure, the highly distorted/deformed B-site octahedrons force the A cations at body center position with respect to the nearest neighbor oxygen ions, and in turn, eight oxygen ions will be available in the first coordination sphere of A cations. Thus, for perovskite oxides with monoclinic P2 1 /c structure, eight oxygen ions are closer to A cations than the next nearest neighbor B cations and therefore the A cations exhibit eight coordination mode. The average bond valence sum (BVS) of Ca2+ ion at A site is calculated to be equal to 1.86 for CMT and 1.91 for CZT, both of which are less than the ideal BVS value for Ca2+ ion. This indicates that the A-site Ca2+ ions in both CMT and CZT are underbounded and the stretching of Ca-O bonds has taken place.
Considering the structural similarity of CMT and CZT, the schematic projections of TaO6 and ZnO6 octahedra on ac plane are illustrated only for CZT (Fig. 3). The TaO6 and ZnO6 octahedrons exhibit both inphase and antiphase rotation. Within the crystal structure, six coordinated Ta5+ ions and Mg2+ ions of CMT/Zn2+ ions of CZT are linked with six nearest neighboring oxygen ions in octahedral fashion, and the cavities between the B′O6 (B′ = Mg for CMT and Zn for CZT) and TaO6 octahedrons are occupied by A-site Ca2+ ions located at 4(e) Wyckoff position. Each TaO6 octahedron is surrounded by two nearest neighbor TaO6 and two B′O6 octahedra, while each B′O6 octahedron is surrounded by four TaO6 octahedra. Thus, the octahedra are arranged in sequence {Ta-Ta-B′} and repeat. This suggests that both CZT and CMT exhibit 1:2 cation ordering at B site. In both samples, the B-site metal oxygen bonding network is asymmetric (Tables 4 and 5). Moreover, the BVS calculations suggest that the Ta-O bonds are compressed, while the Zn-O bonds of CZT and Mg-O bonds of CMT are in extension. These indicate that the B′O6 and TaO6 octahedra are distorted in nature.

The schematic projections of TaO6 and ZnO6 octahedra of CZT on the ac plane
The FESEM micrographs of CMT and CZT, which are shown in Fig. 4a, b, respectively, show well-defined particles with irregular shape and assorted size. The grain size for CMT and CZT is between the range of 0.38 to 2.66 μm and 0.11 to 1.60 μm, respectively. Only the characteristic peaks for constituent elements are observed in the EDS spectra (Fig. 5) of the samples. This implies that there is no impurity in the samples.

The FESEM micrographs of a CMT and b CZT

EDAX spectra of a CMT and b CZT
It is noteworthy that 1:2 ordered perovskites having the general chemical formula Ba(B′1/3B″2/3)O3 possess hexagonal crystal structure of either \( Pm\overline{3}1 \) or P6 3 /mmc space group and the value of the tolerance factor for this type of perovskites is ≈1 [29–31, 38]. On the hand, Ca(Zn1/3Nb2/3)O3, Ca(Ca1/3Nb2/3)O3, Ca[(Mg1/3Ta2/3)0.9Ti0.1]O3, and the solid solution of 0.9Ca[(Mg1/3Ta2/3)]O3/0.1CaTiO3 have monoclinic crystal structure of P2 1 /c space symmetry [34–37, 47]. To the best of our knowledge, no more example of calcium-based 1:2 ordered perovskite is available in the literature. It may further be noted that the crystal structures of both CMT and CZT belong to monoclinic P2 1 /c symmetry. The value of the tolerance factor of perovskites with the general formula Ca(B′1/3B″2/3)O3 is <0.965. It is well known that the tolerance factor (which depends on the radius of A- and B-site metal cations and oxygen anion) has a strong influence on the crystal structure and physical properties of perovskite oxides. According to previous reports, the crystal symmetry of perovskite oxide reduces as the tolerance factor decreases [45, 46]. The value of the tolerance factor of the members of Ca(B′1/3B″2/3)O3 group of perovskites is less than those of perovskites with the general formula Ba(B′1/3B″2/3)O3 mainly due to the smaller radius of Ca2+ (1.34 Å) ions compared to that of Ba2+ (1.61 Å) ions. It may therefore be inferred that 1:2 ordered Ca(B′1/3B″2/3)O3 systems crystallize in monoclinic P2 1 /c space group (lower symmetry), while the crystal structure of the members of Ba(B′1/3B″2/3)O3 family of perovskites with 1:2 ordering at B site possesses a hexagonal crystal structure of either \( Pm\overline{3}1 \) or P6 3 /mmc space group (higher symmetry) due to the difference in ionic radius of A-site cations.
FTIR and Raman spectroscopic investigation
Several reports on the assignment of the IR and the Raman active modes for 1:2 ordered perovskite oxide with space group Fm3m (O s h ), \( Pm\overline{3}1 \) (D 33d ), Pm3m (O 1h ), Pbnm (D 162h ), P21/n (C s2h ), and P2 1/C (C s2h ) are available in the literature [31, 48–53]. The details of the site symmetry and corresponding modes of vibration for 1:2 ordered perovskites belonging to P2 1/C (C s2h ) space group were reported by Fu et al. [35]. According to them, perovskites belonging to this category present 180 normal modes of vibration out of which 3 (Au + 2Bu) are acoustic, 93 (47 Au + 46Bu) are IR active, and 84 (42 Ag + 42Bg) are Raman active.
The FTIR spectra of CMT and CZT in the wavenumber range of 1300 to 400 cm−1 are shown in Fig. 6. Three strong and broad bands are observed in the spectrum of both samples. It may be noted that the IR bands due to symmetric stretching vibration of Mg-O and Zn-O bonds appear at ∼436 and 490 cm−1, respectively [54, 55]. Further, asymmetric stretching vibration of Ta-O bonds in 1:2 ordered perovskite oxides having Ta5+ ions at B site produces a IR band at ∼630 cm−1 [18]. The strong and maximum energy band around 636 cm−1 for CMT and 627 cm−1 CZT can be assigned to the asymmetric stretching mode of Ta-O bonds of TaO6 octahedra due to higher charge of Ta5+ ions [18]. The lowest energy bands around 472 cm−1 for CMT and 469 cm−1 for CZT are related to the symmetric stretching vibration of Mg-O and Zn-O bonds of the MgO6 and ZnO6 octahedra of CMT and CZT, respectively. The broad nature of the IR bands at ∼636 and 472 cm−1 for CMT and 627 and 469 cm−1 for CZT can be attributed to the deformity in the Ta-O, Mg-O, and Zn-O bonding networks due to unequal length of Ta-O, Mg-O, and Zn-O bonds forming TaO6 and MgO6 (for CMT) and ZnO6 (for CZT) octahedra, respectively, as revealed by the structural study (see Tables 4 and 5). The broad medium energy bands at around 554 cm−1 for CMT and 555 cm−1 for CZT may be allocated to the symmetric stretching vibration of Ta-O and some deformed modes of MgO6 octahedra for CMT and ZnO6 octahedra for CZT. The broad nature of these peaks is due to the occurrence of absorption peaks due to symmetric stretching of Ta-O and deformed modes of MgO6 (for CMT) and ZnO6 (for CZT) octahedra at nearly the same wavenumbers.

FTIR spectra of a CMT and b CZT
The Raman spectroscopy is regarded as an authentic tool to probe the crystal symmetry and B-site cation ordering of perovskite oxides, even when this ordering is confined to the nanoscale level [35, 47, 56, 57]. The Raman spectra of CMT and CZT are displayed in Fig. 7. Eleven distinct Raman modes centered about 143.6, 233.9, 276.3, 318.5, 379.2, 411.2, 480.9, 563.3, 631.3, 725.6, and 832.2 cm−1 for CMT and 148.4, 230.5, 276.3, 323.2, 382.5, 410.6, 471.7, 558.7, 629.9, 716.6, and 827.7 cm−1 for CZT are observed. In addition, five weak peaks are observed in the Raman spectra of both CMT and CZT. According to the factor group analysis for 1:2 ordered A(B′1/3B″2/3)O3 perovskite oxides with monoclinic P2 1 /c symmetry, 84 (42 Ag + 42 Bg) Raman active modes are expected out of which only 29 modes are detected experimentally till date[47]. Mode assignment for perovskite oxides with monoclinic P2 1 /c symmetry is very difficult due to the presence of a large number of modes in close proximity and serious overlapping of the modes, because the Raman spectrum of such perovskites is analyzed by comparing them with those of other analogous crystal structures [35]. Usually nine to ten Raman active modes are observed for 1:2 ordered perovskite oxides with monoclinic P2 1 /c symmetry in the range of 100–1000 cm−1 [35, 47]. It is noteworthy that for the perovskite oxides having monoclinic P2 1 /c crystal structure with 1:2 ordering at B site and Ca2+ ions at A site, a characteristic Raman mode appears at about 408 cm−1 [35]. Thus, the Raman mode at about 411.2 cm−1 for CMT and 410.6 cm−1 for CZT gives evidence in favor of long range 1:2 ordering at B site of the samples. Moreover, the appearance of a large number of (16) Raman modes indicates that both CMT and CZT have a monoclinic crystal structure of P2 1 /c space group [35, 47]. The Raman shifts of CZT and CMT match well with that of Ca[(Mg1/3Ta2/3)0.9Ti0.1]O3, which also possesses a monoclinic crystal structure of P2 1 /c space group [35]. A Raman mode is observed at about 800 cm−1 for both 1:1 and 1:2 ordered perovskite oxides, and this nonspecific mode corresponds to short range cation ordering [35]. The strong peak at about 830 cm−1 for CMT and 825 cm−1 for CZT corresponds to Ag mode and confirms the formation of a complex perovskite phase and the existence of a short range cation ordering in the samples. Thus, the Raman study has confirmed that both samples have crystallized in P2 1 /c space group and exhibit 1:2 ordering.

Raman spectra of a CMT and b CZT
Investigation on dielectric permittivity
The variation of the real (ε′) and imaginary (ε″) parts of complex dielectric permittivity (ε*) with the logarithmic angular frequency ω (=2πυ) for CMT and CZT at different temperatures is shown in Figs. 8 and 9, respectively. The peak in the ε″ versus log ω curves shifts toward higher frequencies on increasing the temperature (Figs. 8b and 9b), i.e., at higher temperatures, relaxation occurs at higher frequencies. This implies that the relaxation mechanism governing the dielectric properties of the samples is temperature dependent and the rate of polarization formed increases with the increase of temperature.

The a ε′ versus log ω and b ε″ versus log ω curves for CMT at various temperatures, where the solid lines are the fit of the experimental data (shown by symbols) by the modified Cole-Cole equations

The a ε′ versus log ω and b ε″ versus log ω curves for CZT at various temperatures, where the solid lines are the fit of the experimental data (shown by symbols) by the modified Cole-Cole equations
A close look at Figs. 8 and 9 reveals the presence of a strong dielectric relaxation in CMT and CZT. According to the literature, in perovskite crystals, various relaxation processes with different relaxation times exist simultaneously due to the presence of various types of defects arising in the course of the crystal growth process [18]. The large width of the loss peaks in Figs. 8b and 9b points toward the possibility of participation of a large number of relaxation processes with their own discrete relaxation times. It seems that the dielectric property of CMT and CZT cannot be accounted in terms of the Debye-type monodispersive relaxation process as the dielectric relaxation in the samples is polydispersive in nature. The polydispersive character (distribution of relaxation time) of the dielectric relaxation can be examined by the Cole-Cole model. According to this model, the complex dielectric constant can be expressed as [58, 59]
where ε s and ε ∞ are low- and high-frequency values of ε′, respectively, and α (a constant) is a measure of the distribution of relaxation times. For an ideal Debye-type (monodispersive) relaxation process, α = 0, while for polydispersive relaxation, α > 0. It may be noted that although the polydispersive nature of dielectric relaxation can be successfully accounted by the Cole-Cole model, it cannot follow the low-frequency dielectric behavior of the sample when ε″ increases very rapidly with the decrease of frequency due to the influence of dc conductivity [18]. Under such circumstances, a term accounting for the contribution of the dc electrical conductivity is added in the Cole-Cole equation and the complex dielectric permittivity is described by the empirical equation [60, 61]:
where n lies in the range between 0 and 1 and σ* (σ* = σ 1 + iσ 2) is the complex conductivity. Here, σ 1 is the dc conductivity and σ 2 is the conductivity due to localized charges. From Eq. (2), on equating the real and imaginary parts of ε*, ε′ and ε″ can be written as
We have fitted the dielectric spectra (ε′ versus log ω and ε″ versus log ω curves) obtained at 393, 433, 463, and 480 K using Eqs. (3) and (4). The calculated curves are shown by solid lines in Figs. 8 and 9. The experimentally obtained and calculated ε′ versus log ω and ε″ versus log ω curves match very well with each other (see Figs. 8 and 9). The values of the fitting parameters at different temperatures are listed in Table 6. The nonzero value of α discards the possibility of the existence of a Debye-type monodispersive relaxation process in the samples and indicates that the relaxation process is polydispersive in nature. As temperature increases, the value of the Cole-Cole constant α for both CMT and CZT decreases slightly. Thus, for both CMT and CZT, the width of the relaxation time distribution profile decreases with the increase of temperature.
It may be noted that the dielectric permittivity of CZT is higher than that of CMT, whereas the loss component of CMT is less than that of CZT. The value of dielectric permittivity of CZT is higher, while CMT is comparable to that of the 1:2 ordered niobium-based perovskites (A(B′1/3Nb2/3)O3, where A = Ba, Ca, and Sr and B′ = Ni, Mg, Sr, Fe, and Zn) perovskites and 1:1 ordered tantalum-based systems reported in the literature [12–18]. The value of loss tangent of CMT is less than that obtained for niobium- and tantalum-based perovskites. Taking into account our recent works on tantalum-based A(B′1/3B″2/3)O3-type perovskite oxides [16, 17], it may be concluded that for Ca(B′1/3Ta2/3)O3 (where B′ = Ni, Mg, and Zn) series of perovskite oxides, the highest value of dielectric permittivity is obtained for Ca(Zn1/3Ta2/3)O3 (CZT), while CMT exhibits the lowest dielectric loss. It is well known that the dielectric property of ABO3-type perovskite oxides strongly depends on the radii of A- and B-site metal ions and the crystal structure. Thus, the lower dielectric permittivity of CMT compared to CZT can be attributed to the lattice contribution to permittivity.
Investigation on impedance
Figure 10 depicts the complex plane impedance plots of CMT and CZT, where the imaginary part (Z″) of complex impedance is plotted against the real part (Z′) of complex impedance at different temperatures. In the complex plane impedance plots of the samples, only one semicircular arc has been observed and there is no signature of any low-frequency arc. It may therefore be inferred that there is no grain boundary (interfacial boundary layer) contribution in the impedance spectrum of the samples [62–64]. The high-frequency semicircular arcs in the complex plane impedance plots are therefore ascribed to the bulk effect and can be modeled by parallel combination of bulk resistance (r g) and bulk capacitance (c g) of the material as shown in the insets of Fig. 10a, b. The impedance Z* for the equivalent circuit is

The complex impedance plane plots of a CMT and b CZT. The solid lines represent the fitted data using the RC equivalent circuit shown in the inset
where,
and
We have fitted our experimental data using Eqs. (6) and (7) and the fitted parameters are given in Table 7. A good agreement between the experimental and fitted curves reveals that only the bulk effect contributes to the polarization process in these materials.
Activation energy
In order to estimate the activation energy and to determine the electrical character of CMT and CZT, we have measured the dc conductivity (σ dc) of the samples at different temperatures by the four probe method. The reciprocal temperature dependence of σ dc for CMT and CZT is presented in Fig. 11a. The σ dc versus 103/T plots of CMT and CZT obey the Arrhenius law, σ dc = σ 0 exp(−E σ /k B T), where σ 0 is the pre-exponential factor and E σ is the activation energy. According to the numerical fitting, E σ = 0.35 and 0.33 eV for CMT and CZT, respectively. Moreover, the value of σ dc for both samples increases with increasing temperature.

a Temperature dependence of dc conductivity for CMT and CZT. The symbols represent the experimental data and solid lines are least-square straight-line fit. b Temperature dependence of the most probable relaxation frequency obtained from the logarithmic frequency dependent plots of ε″ for CMT and CZT. The symbols represent the experimental data and the solid lines are the least-square straight-line fit
The activation energy can also be estimated from the temperature dependence of characteristic relaxation frequency, ω m (the frequency corresponds to loss peak). The log ω m versus 103/T plots of CMT and CZT are presented in Fig. 11b. It has been found that log ω m of both samples follows the Arrhenius law: \( {\omega}_{\mathrm{m}}={\omega}_0 \exp \left(-{E}_{\varepsilon^{{\prime\prime} }}/{k}_BT\right) \) (where ω 0 is the pre-exponential factor) with activation energy (\( {E}_{\varepsilon^{{\prime\prime} }} \)) 0.34 eV for CMT and 0.33 eV for CZT, which are almost equal to the activation energy obtained from temperature dependence of σ dc. For the metallic oxides which are p-type polaronic conductors, the activation energy lies in the range from 0.2 to 0.9 eV [61, 65, 66]. The numerical values of activation energy of CMT and CZT indicate that the electrical conduction process in both samples is governed by the polaron hopping. Further, it may be noted that the values of dc conductivity obtained by fitting the dielectric spectra of CZT and CMT at different temperatures and those obtained experimentally agree well. Good matching of the activation energies obtained from log ω m versus 103/T plots and temperature dependence of σ dc along with good agreement between the values of σ dc determined by fitting the dielectric spectra and obtained experimentally suggests that relaxation and conductivity have same microscopic origin (hopping) [67].
Conclusion
The structural, morphological, dielectric, and electrical transport properties of CMT and CZT with grain size ranging between 0.38 and 2.66 μm and between 0.11 and 1.60 μm, respectively, have been investigated by PXRD, FESEM, and impedance spectroscopic techniques. The compounds have crystallized in monoclinic structure of P2 1 /c symmetry. The 1:2 ordering and formation of monoclinic perovskite oxide phase with P2 1 /c symmetry are confirmed by FTIR and Raman spectroscopic studies. Comparing the crystal structures of 1:2 ordered Ba(B′1/3B″2/3)O3- and Ca(B′1/3B″2/3)O3-type perovskite systems, we have shown that for these two classes of perovskites, the calcium-based systems crystallize in monoclinic P2 1 /c space group while their barium-based counterparts possess a hexagonal crystal structure of either \( Pm\overline{3}1 \) or P6 3 /mmc due to the smaller ionic radius of Ca2+ compared to that of Ba2+.
The variation of ε″ with frequency at different temperatures indicates the presence of dielectric relaxation in the samples, and the characteristic relaxation frequency is strongly temperature dependent. The analysis of the loss component of the dielectric constant in the framework of the modified Cole-Cole model points toward the polydispersive character of the relaxation process. The dielectric and electrical transport properties of the samples are governed by the same mechanism (polaron hopping). It has been shown that the p-type polaron hopping is responsible for the dielectric behavior and electrical response of the samples. Finally, for their high dielectric permittivity and low loss component, CMT and CZT appear to be suitable for various technological applications in capacitors, resonators, filters, and integrated circuits.
References
Fu MS, Liu XQ, Chen XM, Zeng YW (2010) Effects of Mg substitution on microstructures and microwave dielectric properties of Ba(Zn1/3Nb2/3)O3 perovskite ceramics. J Am Ceram Soc 93:787–795
Shah MR, AktherHossain AKM (2013) Structural, dielectric and complex impedance spectroscopy studies of lead free Ca0.5+xNd0.5-x(Ti0.5Fe0.5)O3. J Mater Sci Technol 29:323–329
Chen CT, Huang CY, Lin YM, Lee CT (2011) Structure and microwave dielectric property relations in barium cobalt magnesium niobate ceramics. Jpn J Appl Phys 50:091503
Belous A, Ovchar O, Kramarenko O, Mischuk D, Jancar B, Spreitzer M, Suvorov D, Annino G, Grebennikov D, Mascher P (2009) Low-loss perovskite niobates Ba(M1/32+Nb2/3)O3: composition, structure, and microwave dielectric properties. Ferroelectrics 387:36–45
Ovchar O, Belous A, Kramarenko O, Mischuk D, Jancar B, Spreitzer M, Suvorov D, Annino G, Grebennikov D, Mascher P (2009) The effect of impurity phases on the structure and properties of microwave dielectrics based on complex perovskites Ba(Co1/32+Nb2/3)O3. Ferroelectrics 387:189–196
Yang Z, Zhang Y, You G, Zhang K, Xiong R, Shi J (2012) Dielectric and electrical transport properties of the Fe3+− doped CaCu3Ti4O12. J Mater Sci Technol 28:1145–1150
Hungria T, Alguero M, Castro A (2007) Grain growth control in NaNbO3–SrTiO3 ceramics by mechanosynthesis and spark plasma sintering. J Am Ceram Soc 90:2122–2127
Lu H, Zhu L, Kim JP, Son SH, Park JH (2012) Structural, sintering and electrical properties of Cr-doped La06S.r0.4Crx Fe1−xO3−δ(x=0.10, 0.20) oxides. J Mater Sci Technol 28:654–660
Agrawal L, Dutta A, Shannigrahi S, Singh BP, Sinha TP (2011) Impedance spectroscopy study and ground state electronic properties of In(Mg1/2Ti1/2)O3. Physica B 406:1081–1087
Prakash V, Dutta A, Choudhary SN, Sinha TP (2007) Dielectric relaxation in perovskite Ba(Zn1/2W1/2)O3. Mater Sci Eng B 142:98–105
Bajpai PK, Singh KN (2011) Dielectric relaxation and ac conductivity study of Ba(Sr1/3Nb2/3)O3. Physica B 406:1226–1232
Bajpai PK, Pastor M, Singh KN (2011) Relaxor behavior and dielectric relaxation in Pb(Ba1/3Nb2/3)O3: a phase pure new relaxor material. J Appl Phys 109:014114
Dutta A, Bharti C, Sinha TP (2008) Dielectric relaxation in Sr(Mg1/3Nb2/3)O3. Physica B 403:3389–3393
Dutta A, Sinha TP (2010) Impedance spectroscopy study of BaMg1/3Nb2/3O3: frequency and time domain analyses. Physica B 405:1475–1479
Dutta A, Bharti C, Sinha TP (2008) AC conductivity and dielectric relaxation in CaMg1/3Nb2/3O3. Mater Res Bull 43:1246–1254
Hoque MM, Dutta A, Kumar S, Sinha TP (2012) The impedance spectroscopic study and dielectric relaxation in A(Ni1/3Ta2/3)O3 [A=Ba, Ca and Sr]. Physica B 407:3740–3748
Hoque MM, Dutta A, Kumar S, Sinha TP (2014) Dielectric relaxation and conductivity of Ba(Mg1/3Ta2/3)O3 and Ba(Zn Ta)O. J Mater Sci Technol 30:311–320
Dutta A, Sinha TP (2011) Structural and dielectric properties of A(Fe1/2Ta1/2)O3[A = Ba, Sr, Ca]. Mater Res Bull 46:518–524
Dias A, Paschoal CWA, Moreira RL (2003) Infrared spectroscopic investigations in ordered barium magnesium niobate ceramics. J Am Ceram Soc 86(11):1985–1987
Silva RX, Moreira RL, Almeida RM, Paniago R, Paschoal CWA (2015) Intrinsic dielectric properties of magnetodielectric La CoMnO. J Appl Phys 117:214105
Sagala DA, Koyasu S (1993) Infrared reflection of Ba(Mg1/3Ta2/3)O3 ceramics. J Am Ceram Soc 76(10):2433–2436
Dias A, Khalam LA, Sebastian MT, Paschoal CWA, Moreira RL (2006) Chemical substitution in Ba (RE1/2Nb1/2) O3(RE= La, Nd, Sm, Gd, Tb, and Y) microwave ceramics and its influence on the crystal structure and phonon modes. Chem Mater 18:214–220
Bhalla AS, Guo R, Roy R (2000) The perovskite structure – a review of its role in ceramic science and technology. Mat Res Innov 4:3–26
Galasso F (1990) Perovskites and high-Tc superconductors. Gordon and Breach Science, New York, pp 3–58
Galasso F, Pyle J (1963) Ordering in compounds of the A(B´0.33Ta0.67)O3 type. Inorg Chem 2:482–484
Galasso F, Pyle J (1963) Preparation and study of ordering in A(B´0.33Nb0.67)O3perovskite-type compounds. J Phys Chem 67:1561–1562
Takahashi J, Fujii T, Shimada S, Kodaira K (1999) Changes in ordered structure and dielectric properties with the A-site and B-site cation ratios of complex perovskites (Sr1-xBax)(Sr0.33Ta0.67)O3. J Eur Ceram Soc 19:1089–1093
Lee C, Chou C, Tsai D (1997) Effect of La/K A-site substitutions on the ordering of Ba(Zn1/3Ta2/3)O3. J Am Ceram Soc 80:2885–2890
Akbas MA, Davies PK (1998) Cation ordering transformations in the Ba(Zn1/3Nb1/3)O3–La(Zn2/3Nb2/3)O3 system. J Am Ceram Soc 81:1061–1064
Lufaso MW (2004) Crystal structures, modeling, and dielectric property relationships of 2:1 ordered Ba3MM′2O9(M=Mg, Ni, Zn; M′ =Nb, Ta) perovskites. Chem Mater 16:2148–2156
Mani R, Selvamani P, Joy JE, Gopalakrishnan J (2007) Study of Ba3MIIMIVWO9(MII= Ca, Zn; MIV= Ti, Zr) perovskite oxides: competition between 3C and 6H perovskite structures. Inorg Chem 46:6661–6667
Park CS, Paik JH, Nahm S, Lee HJ, Park HM, Kim KY (1999) Crystal structure of A2+(Mg1/3Nb2/3)O3, (A2+=Sr2+and Ca2+) ceramics. J Mater Sci Lett 18:691–694
Lee HJ, Park HM, Cho YK, Song YW, Nahm S, Byun JD (2001) Microstructure characterizations in calcium magnesium niobate. J Am Ceram Soc 84:1632–1636
Fu MS, Liu XQ, Chen XM, Zeng YW (2008) Microstructure and microwave dielectric properties of (1-x)Ca(Mg1/3Ta2/3)O3/xCaTiO3 ceramics. J Am Ceram Soc 91:1163–1168
Fu MS, Liu XQ, Chen XM (2008) Raman spectra analysis for Ca(B′1/3B′′2/3)O3-based complex perovskite ceramics. J Appl Phys 104:104108
Fu MS, Liu XQ, Chen XM, Zeng YW (2008) Cation ordering and domain boundaries in Ca[(Mg1/3Ta2/3)1-xTi]O3 microwave dielectric ceramics. J Am Ceram Soc 91:2581–2587
Chen XM, Liu D, Hou RZ, Hu X, Liu XQ (2004) Microstructures and microwave dielectric characteristics of Ca(Zn1/3Nb2/3)O3complex perovskite ceramics. J Am Ceram Soc 87:2208–2212
Akbas MA, Davies PK (1998) Ordering-induced microstructures and microwave dielectric properties of the Ba(Mg1/3Nb2/3)O3–BaZrO3 system. J Am Ceram Soc 81:670–676
Altomare A, Caliandro R, Camalli M, Cuocci C, Giacovazzo C, Moliterni AGG, Rizzi R (2004) J Appl Crystallogr 37:1025–1028
Larson AC, Von Dreele RB (2000) General structure analysis system (GSAS). Los Alamos National Laboratory, Report LAUR 86-784
Glazer AM (1975) Simple ways of determining perovskite structures. Acta Crystallogr A31:756–762
Zhou JS, Goodenough JB (2005) Universal octahedral-site distortion in orthorhombic perovskite oxides. Phys Rev Lett 94:065501
Goldschmidt VM (1926) Naturwissenschaften 14:477–485
Shannon RD (1976) Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr A 32:751–767
Reaney IM, Colla EL, Setter N (1994) Dielectric and structural characteristics of Ba- and Sr-based complex perovskites as a function of tolerance factor. Jpn J Appl Phys 33:3984–3990
Woodward DI, Reaney IM (2005) Electron diffraction of tilted perovskites. Acta Crystallogr B61:387–399
Levin I, Chan JY, Geyer RG, Maslar JE, Vanderah TA (2001) Cation ordering types and dielectric properties in the complex perovskiteCa(Ca1/3Nb2/3)O3. J Solid State Chem 156:122–134
Siny IG, Tao R, Katiyar RS, Bhalla AS, Guo R (1998) Raman spectroscopy of Mg-Ta order–disorder in BaMg1/3Ta2/3O3. J Phys Chem Solids 59(2):181–195
Dias A, William C, Paschoal A, Moreira RL (2003) Infrared spectroscopic investigations in ordered barium magnesium niobate ceramics. J Am Ceram Soc 86(11):1985–1987
Rodrigues JEFS, Moreira E, Bezerra DM, Maciel AP, Paschoal CWA (2013) Ordering and phonons in Ba3CaNb2O9 complex perovskite. Mater Res Bull 48:3298–3303
Moreira RL, Matinaga FM, Dias A (2001) Raman-spectroscopic evaluation of the long-range order in Ba(B′1/3B″2/3)O3 ceramics. Appl Phys Lett 78:428
Siny IG, Katiyar RS, Bhalla AS (1998) Cation arrangement in the complex perovskites and vibrational spectra. J Raman Spectrosc 29:385–390
Rodrigues JEFS, Bezerra DM, Maciel AP, Paschoal CWA (2014) Synthesis and structural ordering of nano-sized Ba3B′Nb2O9(B′ = Ca and Zn) powders. Ceram Int 40:5921–5930
Li LX, Xu D, Li XQ, Liuc WC, Jia Y (2014) Excellent fluoride removal properties of porous hollow MgO microspheres. New J Chem 38:5445–5452
Maensiria S, Laokula P, Promarak V (2006) Synthesis and optical properties of nanocrystalline ZnO powders by a simple method using zinc acetate dihydrate and poly (vinyl pyrrolidone). J Cryst Growth 289:102–106
Zheng H, Reaney IM, Csete de Györgyfalva GDC, Ubic R, Seabra MP, Ferreira VM, Yarwood J (2004) Raman spectroscopy of CaTiO3-based perovskite solid solutions. J Mater Res 19:488–495
Zheng H, Bagshaw H, Csete de Györgyfalva GDC, Reaney IM, Ubic R, Yarwood J (2003) Raman spectroscopy and microwave properties of CaTiO3-based ceramics. J Appl Phys 94:2948–2956
Cole KS, Cole RH (1941) Dispersion and absorption in dielectrics I. Alternating current characteristics. J Chem Phys 9:341–351
Cole KS, Cole RH (1942) Dispersion and absorption in dielectrics II. Direct current characteristics. J Chem Phys 10:98–105
Coelho R (1978) Physics of dielectrics. Elsevier, New York
Maity SK, Dutta A, Kumar S, Sinha TP (2013) Electrical properties of Ba2YbNbO6: an impedance spectroscopy study. Phys Scr 88:065702
Gerhardt R (1994) Impedance and dielectric spectroscopy revisited: distinguishing localized relaxation from long-range conductivity. J Phys Chem Solids 55:1491–1506
Mckubre MCH, Macdonald JR (1987) Impedance spectroscopy emphasizing solid materials and systems. In: JR Macdonald (ed.) Wiley, New York, pp 191
Bharti C, Sinha TP (2010) Dielectric properties of rare earth double perovskite oxide Sr2CeSbO6. Solid State Sci 12:498–502
Idrees M, Nadeem M, Hassan MM (2010) Investigation of conduction and relaxation phenomena in LaFe0.9Ni0.1O3 by impedance spectroscopy. J Phys D Appl Phys 43:155401
Jung WH (2001) Dielectric loss anomaly and polaron hopping conduction of Gd1/3Sr2/3FeO3. J Appl Phys 90:2455–2458
Schönhals A, Kremer F (2003) Broadband dielectric spectroscopy. Springer, Berlin, pp 59–98
Acknowledgments
We gratefully acknowledge the financial support of CSIR, New Delhi, Government of India, through grant number 60(0106)/13-EMR-II. The authors gratefully acknowledge the financial support of the Department of Science & Technology, Govt. of India through FIST and PURSE program of the Department of Physics, Jadavpur University. The financial assistance granted by UGC, Govt. of India through SAP and UPE program is also acknowledged. We gratefully acknowledge Prof. R. N. Joardar for helpful discussion.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Hoque, M.M., Barua, A., Dutta, A. et al. Study on the structural, spectroscopic, and dielectric properties of 1:2 ordered Ca3(B′Ta2)O9 (B′ = Mg and Zn). Ionics 23, 471–483 (2017). https://doi.org/10.1007/s11581-016-1916-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-016-1916-6