
Abstract
A simple, sensitive, and reliable carboxylic group functionalized single-walled carbon nanotubes (f-SWCNTs)/poly(hydroxymethylated-3,4-ethylenedioxythiophene) (PEDOTM) modified glassy carbon electrode (GCE) was successfully developed for the electrochemical determination of catechin (CAT). In view of the merits of extraordinary conductivity of PEDOTM and excellent electrocatalytic property of f-SWCNTs, the f-SWCNTs/PEDOTM/GCE modified electrode exhibited a strong electrocatalytic activity for the oxidation of CAT. Under optimized conditions, the proposed modified electrode showed a wide linear response for CAT in the concentration range between 0.039 and 40.84 μM, with a low detection limit of 0.013 μM. Furthermore, the modified electrode also exhibited a good reproducibility and long-term stability, as well as high selectivity, which also was a good candidate for the electrochemical detection and analysis of CAT in commercial green tea.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Antioxidants have been attracting great interest on account of their important role in biological and industrial processes [1]. Catechins, or flavanols, belonging to naturally polyphenolic compounds, are well-known antioxidants because of their free radical scavenging abilities and are found extensively in tea, wine, fruits, and legumes [2, 3]. Their antioxidant activities are much higher than that of vitamin C and E. More importantly, a human diet containing a certain amount of catechins is potentially beneficial to human health: not only are they strong antioxidants but also are anti-carcinogens, anti-inflammatory agents, anti-mutagenic, anti-microbial, and inhibitor of platelet aggregation in in vivo or in vitro studies [2, 4]. Catechin (CAT), one of the most important catechins for human health researched thus far [5, 6], has been recognized as a radical scavenger and antioxidant [3]. Meanwhile, CAT has been reported to have peroxyl, hydroxyl, 1, 1-diphenyl-2-picrylhydrazyl, and superoxide radical scavenging activity, which is ten times higher than that of l-ascorbate and β-carotene [5, 7]. In addition, CAT is potentially beneficial to human health, for instance, autoxidation to form active oxygen species (hydrogen peroxide) and free radical intermediates (O2 •—) to induce fatty acid peroxidation, DNA damage, and diseases [3, 8–11].
In terms of human health, the quantitative determination of CAT in different products has attracted increasing interest. Different analytical technologies have been reported in the literature, and traditional analysis of CAT such as capillary electrophoresis [12–14], thin-layer chromatography [15], HPLC [16–18], and spectrophotometry [10] are mainly carried out through instrumental analysis. Generally, these methods are highly sensitive and efficient. However, such analysis methods are usually performed at centralized laboratories, requiring analytical resources and extensive labor, and are often costly and time consuming. Therefore, it is vital to establish a fast, simple, sensitive, and low-cost method for the detection of CAT. Electrochemical analysis has been proposed as efficient alternative for the determination of CAT on account of its advantages of simple operation, time-saving, low cost, sensitivity, selectivity, and real-time online detection. Furthermore, the sensitivity and selectivity of electrochemical analysis can be improved by chemically modified electrodes. So far, various kinds of electrochemical modified electrodes, including single-walled carbon nanotube-cetyramethylammonium bromide modified glassy carbon electrode (SWCNT-CATB/GCE) [3], beta-cyclodextrin modified carbon paste electrode (CD/CPE) [5], self-assembled monolayer of nickel (II) complex and thiol on gold electrode (Ni(II) complex–SAM-Au electrode) [9], ruthenium tris (2, 2′) bipyridyl modified boron-doped diamond electrode (Ru(bpy)3 3+/BDD) [19], hydroxypropyl-beta-cyclodextrin incorporated carbon paste modified electrode (HP-β-CD/CPE) [20], and laccase-modified electrode [21], have been proposed for the detection of CAT. However, using conducting polymers (CPs) as the electrode modified interface of efficient electrochemical sensors has not been reported for the voltammetric detection of CAT until now.
Compared to pure CPs, the composites of CPs with nanomaterials, such as carbon nanotubes (CNTs) or nanoparticles, typically have lower charge transfer resistance and higher surface area, which have been successfully employed as sensing elements [22–24]. Poly(3,4-ethylenedioxythiophene) (PEDOT), one of the successful commercially available CPs today, has already been widely used as promising electrode materials attributed to extraordinary conductivity, great environmental stability, and friendly biocompatibility [25–27]. However, the poor water solubility of 3,4-ethylenediox-ythiophene (EDOT) is a major drawback, limiting its further application in various fields. Recently, some studies proved that addition of an appropriate pendant side group onto the backbone of EDOT could improve its poor water-solubility [25, 28, 29]. Hydroxymethylated-3,4-ethylenedioxylthiophene (EDOTM), a polar derivative of EDOT, exhibited better water-solubility and a lower onset oxidation potential compared with EDOT monomer [28–32]. More importantly, PEDOTM, the polymer film of EDOTM, possesses similar excellent physicochemical properties of PEDOT and proves to display better biocompatibility, higher conductivity, and better electrochemical properties than PEDOT [28–32]. In addition, our previous work indicated that PEDOTM was a promising immobilization matrix for biologically active species and electrode materials for establishing efficient chemo-/biosensors [28, 32–34], which can be used alone or in combination with different materials (platinum nanoparticles, graphene oxide, or ascorbate oxidase) to obtain various chemo-/biosensors for electrochemical determination of anticancer herbal drug shikonin [28], vitamin C (VC) in commercial fruit juice[32], indole-3-acetic acid [33], and quercetin [34]. Until now, there has been no report about the electrochemical behaviors and determination of CAT based on PEDOTM modified electrode.
In this contribution, in view of the merits of conducting polymer PEDOTM and carbon nanotubes f-SWCNTs, a simple and sensitive f-SWCNTs/PEDOTM nanocomposite modified GCE was constructed for the electrochemical determination of CAT. The modified electrode exhibits good electrocatalytic activity and electrochemical performance for the voltammetric detection of CAT. Compared with PEDOT/GCE and carboxylic-functionalized poly(3,4-ethylenedioxythiophene) (PC4)/GCE electrodes, the PEDOTM/GCE exhibited a distinctly higher electrocatalytic activity for the oxidation of CAT. Moreover, f-SWCNTs/PEDOTM/GCE provided a large electrochemically active surface area for the adsorption of CAT, which effectively accelerated the electron transfer between electrode and solution, leading to a much more rapid and sensitive current response.
Experimental
Apparatus and regents
Cyclic voltammetric (CV) and chronocoulometry measurements were performed through a model CHI660B electrochemical workstation (Chenhua Instrument Company, Shanghai, China). A conventional three-electrode system was employed in the measurements, the working electrodes were bare GCE or composite modified GCE, a platinum wire was used as the auxiliary electrode, and the saturated calomel electrode (SCE) was used as the reference electrode. Scanning electron microscopy (SEM) measurements were taken with a VEGA\\TESCAN Digital Microscopy Imaging (Tescan, Czech) and the modified films were prepared on the indium tin oxide (ITO) transparent electrodes. All potentials were given versus SCE. pH values were measured with a portable pH meter PHB-5 (Qiwei Instrument, Hangzhou, China).
The CAT of 98 % purity was purchased from Shanghai Baoman Biotech. Co., Ltd. CAT stock solution (1 × 10−2 M) was prepared with deionized distilled water and stored at 4 °C. Carboxylic acid-functionalized single-walled carbon nanotubes (f-SWCNTs, 2.73 wt.%) suspensions were purchased from Chengdu Institute of Organic Chemistry, Chinese Academy of Sciences. EDOT (98 %) was purchased from Sigma–Aldrich (USA). EDOTM and 4-((2,3-dihydrothieno[3,4-b][1,4] dioxin-2-yl) methoxy)-4-oxobutanoic acid (C4-EDOT-COOH) were synthesized by our laboratory (as seen in Supporting material Fig. S1) [32, 35]. Sodium dihydrogen phosphate dihydrate (NaH2PO4·2H2O), disodium hydrogen phosphate dodecahydrate (Na2HPO4·12H2O), and lithium perchlorate trihydrate (LiClO4·3H2O) were obtained from Sinopharm Chemical Reagent Co., Ltd. 0.1 M phosphate-buffered solution (PBS) was prepared from 0.1 M NaH2PO4·2H2O and 0.1 M Na2HPO4·12H2O. The commercial green tea was purchased from a local supermarket with a trade mark of Jiangxi wuyuan. Two grams of green tea sample was washed with DDW and dried in an electro-thermostatic blast oven, powdered, placed in an Erlenmeyer flask containing 50 mL absolute ethanol, and constantly stirred for 12 h. Next, the extract was filtered and centrifuged, and then diluted to 100 mL with absolute ethanol and stored at 4 °C. All reagents were of analytical grade and used without further purification. All solutions were prepared by double-distilled deionized water (DDW).
Preparation of modified electrodes
Prior to modification, each bare GCE was carefully polished with 0.05 μm alumina slurry until a mirror-shine surface was obtained, followed by successive sonication in DDW and ethanol for 5 min, and then dried in air. The PEDOT/GCE, PEDOTM/GCE, and poly(4-((2,3-dihydrothieno[3,4-b][1,4] dioxin-2-yl) methoxy)-4-oxobutanoic acid)/GCE (PC4/GCE) electrodes were obtained individually by one-step electropolymerization of 10 mM monomers on GCE surface in 50 mM LiClO4 with a constant potential of 1.1 versus SCE, and the deposition time was 90 s. Then, the films were washed repeatedly using DDW in order to remove the electrolyte monomers and were then dried at room temperature. The f-SWCNTs/PEDOTM/GCE was prepared by directly drop-casting 5 μL f-SWCNTs (1.0 wt.%) on the surface of PEDOTM/GCE and then drying at room temperature.
Experimental measurements
A certain concentration of CAT solution was transferred into the sealed electrochemical cell containing 5 mL of 0.1 M PBS (pH 7.0) using micropipettes. Prior to each electrochemical measurement, solutions under test were deoxygenated by purging with nitrogen for 10 min, then the modified electrode was immersed into a stirring 0.1 M PBS (pH 7.0) solution containing the specific concentration of CAT for 150 s at 0.3 V. The fabrication process of the modified electrode and the mechanism for the determination of CAT are shown in Scheme 1.

The fabrication process of the f-SWCNTs/PEDOTM/GCE and the recognition mechanism for the detection of CAT
Results and discussion
The morphology of modified electrodes
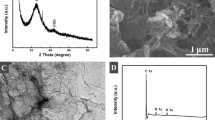
The surface morphology of PEDOTM film (A) and f-SWCNTs/PEDOTM composite (B) deposited on the indium tin oxide (ITO) transparent electrode was observed by SEM (Fig. 1). As can been seen from the high magnification (Fig. 1a), the image of PEDOTM film comprises numerous nanoparticles, which is beneficial to provide a large electroactive area and promote electron transfer. When drop-casting f-SWCNTs on the surface of PEDOTM/GCE (Fig. 1b), the f-SWCNTs/PEDOTM/GCE shows some nodular and highly porous morphology, which is good for improving the adsorption of analytes and increasing the sensitivity of the modified electrode.

SEM image of PEDOTM film (a) and f-SWCNTs/PEDOTM film (b) on ITO transparent electrode
Electrochemical behaviors of CAT
Influence of PEDOT derivates modified GCE
Figure 2 shows CV curves of CAT at PEDOT derivates modified GCE, PC4 (a), PEDOT (b), and PEDOTM (c). There is no redox peak observed in the absence of CAT based on these three modified electrodes, indicating that PEDOT derivates modified electrodes are non-electroactive in 0.1 M PBS (pH 7.0) (Fig. 2 inset). Only the oxidation peak, corresponding to the electrochemical oxidation of CAT, was observed on all the three modified electrodes within the potential window from −0.4 to 0.8 V, suggesting that the electrochemical reaction was a totally irreversible process. In comparison to PC4/GCE and PEDOT/GCE, the oxidation peak current of CAT at PEDOTM/GCE increased obviously, which can be attributed to the nodular and porous morphology of PEDOTM film that provides more active sites for the electrocatalysis of CAT.

CVs of 0.1 mM CAT at PC4/GCE (a), PEDOT/GCE (b), and PEDOTM/GCE (c) in 0.1 M PBS (pH 7.0). Scan rate: 50 mV s−1. Inset: CVs of PC4/GCE (a′), PEDOT/GCE (b′), and PEDOTM/GCE (c′) in the absence of CAT
Electrocatalytic behaviors of CAT
The electrochemical responses of CAT based on bare GCE (a), PEDOTM/GCE (b), and f-SWCNTs/PEDOTM/GCE (c) were studied using CVs. As shown in the inset of Fig. 3, the obvious redox peaks in Fig. 3c′ were produced by f-SWCNTs and no redox peaks of CAT are observed on three electrodes in the blank buffer solution, suggesting that these electrodes are non-electroactive in the selected potential region. The corresponding cathodic peaks do not appear on the listed electrodes, indicating that this electrochemical reaction is an irreversible process. The catalytic activity of different electrodes is also proved by CVs, CAT shows rather poor oxidation peak current at the bare GCE (curve a), while on the PEDOTM/GCE (curve b), the oxidation peak current is increased, suggesting the excellent catalytic activity and conductivity of PEDOTM layer. Compared with the PEDOTM/GCE, two obvious oxidation peaks are observed (i pa1 and i pa2), and the oxidation of the catechol moiety, 3′,4′-dihydroxyl groups at ring B, occurs first at very low positive potential corresponding to peak i pa1 [1]. The further scan to positive potential displayed an additional oxidation peak i pa2, known as the oxidation of other hydroxyl groups (Scheme 1). The oxidation peak current shows a remarkable increase on the f-SWCNTs/PEDOTM/GCE (curve c), which may be attributed to the synergistic effect of good conductivity of PEDOTM and excellent electrocatalytic property of f-SWCNTs. Therefore, f-SWCNTs/PEDOTM/GCE supplies a large specific surface area to increase the loading concentration of CAT, resulting in a remarkable enhancement of the oxidation peak current. Moreover, the large surface area of f-SWCNTs can accelerate the electron transfer on the electrode surface to amplify the electrochemical signal and improve the electrocatalytic performance of CAT on the as-prepared electrode. Thus, the composite modified electrode not only shows the extraordinary conductivity of PEDOTM but also exhibits an excellent electrocatalytic performance and large effective area of f-SWCNTs. Hence, the f-SWCNTs/PEDOTM composite modified electrode can provide an electron-transfer microenvironment to promote the electrochemical reaction of CAT.

CVs of 0.1 mM CAT based on bare GCE (a), PEDOTM/GCE (b), and f-SWCNTs/PEDOTM/GCE (c) in 0.1 M PBS (pH 7.0). Scan rate: 50 mV s−1. Inset: CVs of GCE (a′), PEDOTM/GCE (b′), and f-SWCNTs/PEDOTM/GCE (c′) in the absence of CAT
Optimization of experimental conditions
This work is aimed to establish a simple and highly sensitive electrochemical sensor for the detection of CAT. As mentioned above, the oxidation peak of CAT on the f-SWCNTs/PEDOTM/GCE is heightened, offering an enhanced sensitivity for the detection of CAT. However, it is still critically important to optimize experimental conditions for the detection of CAT. Thus, a systematic investigation of experimental parameters, such as accumulation potential, accumulation time, solution pH, and scanning rate, have been optimized.
Influence of accumulation conditions
Because the electrochemical reaction of CAT on f-SWCNTs/PEDOTM/GCE is an adsorption-controlled process, generally, the accumulation step is an effective way to increase the sensitivity of modified electrode. The oxidation peak current of 0.1 mM CAT in 0.1 M PBS (pH 7.0) was compared with different potentials. As shown in Fig. 4a, the oxidation current increases with the increasing accumulation potential at the range of 0.1–0.3 V, then decreases between 0.3 and 0.4 V. The highest oxidation peak current is achieved at 0.3 V. The influence of accumulation time on the oxidation peak current of 0.1 mM CAT in 0.1 M PBS (pH 7.0) was also investigated, and the solution was kept well stirred by a magnetically rotated sted rod in the enrichment process. Within the first 150 s, the oxidation current increases greatly and levels off subsequently (Fig. 4b), implying that the accumulation of CAT on the f-SWCNTs/PEDOTM/GCE reaches saturation. Therefore, 0.3 V of accumulation potential and 150 s of accumulation time are employed in this experiment.

a Influence of accumulation time on the oxidation peak current of 0.1 mM CAT in PBS (pH 7.0). Accumulation potential: 0.3 V. b Influence of accumulation potential on the oxidation peak current of 0.1 mM CAT in PBS (pH 7.0). Accumulation time: 150 s
Effect of pH and scan rates towards CAT
The effect of pH value on the current response of 0.1 mM CAT in 0.1 M PBS based on f-SWCNTs/PEDOTM/GCE was investigated by CVs, with the results shown in Fig. 5a. The redox peak currents as well as the oxidation peak potential (E pa) with the change of the buffer solution over the pH range from 6.0 to 8.0 are shown in Fig. 6. As shown in Fig. 6a, the peak current of CAT increases rapidly with the increase of pH value from 6.0 to 7.0, then decreases when pH value increases further, illustrating that protons were involved in the electrode reaction process [36]. When the solution was adjusted to the alkalinity, the residual carboxylic acid groups of f-SWCNTs deprotonated to anion, while CAT formed phenolic oxide anion, resulting in mutual repulsion between negatively charged CAT and acidic anions, which caused the decrease of current response [37]. Thus, pH 7.0 is chosen as the best pH condition in the following experiments. As can be seen from Fig. 6b, with the increase of pH from 6.0 to 8.0, the value of peak potential on the f-SWCNTs/PEDOTM/GCE shifts to a more negative potential, and two good linear relationships between E pa and pH are constructed with linear regression equation as E pa1 (V) = −0.0736 pH + 0.677 (R 2 = 0.9951) and E pa2 (V) = −0.0824 pH + 0.857 (R 2 = 0.9917). According to the formula: dE p/dpH = 2.303mRT/nF [38, 39], where m is the number of protons and n is the number of electrons. m/n is calculated to be 1.24 and 1.39 for two oxidation peak currents, indicating that the electron transfer of this electrode reaction is accompanied by an equal number of protons.

a CVs of 0.1 mM CAT at f-SWCNTs/PEDOTM/GCE with different pH values: 6.0, 6.5, 7.0, 7.5, 8.0. b CVs of 0.1 mM CAT at f-SWCNTs/PEDOTM/GCE with different scan rates (a to i): 10, 30, 50, 70, 90, 110, 130, 150, 170 mV s−1

Effect of pH value on the a anodic peak current and b anodic peak potential. c The plot of oxidation peak current versus scan rate. d The plot of anodic peak potential versus Napierian logarithm of scan rate (lnv)
Meanwhile, the effect of scan rate (v) on oxidation peak current of CAT was recorded over the range 10–170 mV s−1 (Fig. 5b). It can be seen that the oxidation peak current (i pa) increases gradually with the increase of v. Two linear relationships between i pa and v are constructed in the range of 10–170 mV s−1 with the linear regression equations as i pa1 (μA) = 22.8296 + 2.0116 v 1 (mV s−1) (R 2 = 0.9974) and i pa2 (μA) = 22.8296 + 2.0116 v 2 (mV s−1) (R 2 = 0.9961), respectively (shown in Fig. 6c), revealing that the oxidation of CAT is an adsorption-controlled electrode process. In addition, as shown in Fig. 6d, two linear relationships between E pa and Napierian logarithm of v (lnv) are also observed (Fig. 6d). Two equations are expressed as E pa1 (V) = 0.1150 + 0.0431 lnv 1 (mV s−1) (R 2 = 0.9988) and E pa2 (V) = 0.0447 + 0.0325 lnv 1 (mV s−1) (R 2 = 0.9985), respectively. According to Laviron’s equation [40]:
where E 0 is formal redox potential, R is the gas constant, T is the absolute temperature, α is transfer coefficient, n is electron transfer number involved in rate-determining step, F is the Faraday constant, k s is standard rate constant of the reaction, and v is scanning rate. According to the two linear correlations of E pa versus lnv mentioned previously, the slopes of two lines are equal to RT/αnF. Thus, corresponding to the slope of E pa-lnv, the values of αn are calculated to be 0.79 and 0.60, respectively (taking T = 298, R = 8.314, and F = 96485). Generally, α is assumed to be 0.5 in a totally irreversible electrode process; therefore, n is around 2 for the electrochemical oxidation of CAT. Considering the equal number of electrons and protons involved in the oxidation process of CAT, the electrooxidation of CAT on the f-SWCNTs/PEDOTM/GCE is a two-electron and two-proton process, described in Scheme 1.
Electrochemical effective area and k s
The electrochemically effective surface areas of bare GCE, PEDOTM/GCE, and f-SWCNTs/PEDOTM/GCE were determined by chronocoulometry using 0.1 mM K3[Fe(CN)6] as model complex. According to the Anson equation [41]:
where A is surface area of working electrode, c is concentration of substrate, D is diffusion coefficient, Q dl is double layer charge, and Q ads is Faradic charge. The D of 0.1 mM K3[Fe(CN)6] in 0.1 M KCl is 7.6 × 10−6 cm2 s−1 [36].
Based on the slopes of the linear relationship between Q and t 1/2 (Fig. 7a), A is calculated to be 0.0627, 1.1616, and 2.0429 cm2 for bare GCE, PEDOTM/GCE, and f-SWCNTs/PEDOTM/GCE, respectively. These results indicate that the electrodes’ effective surface areas can be increased obviously after electrodes modified by PEDOTM and f-SWCNTs, which can increase the electrochemical active site and enhance the total adsorption capacity of CAT, leading to the increase of current response of CAT and decreasing the limit of detection. Furthermore, the D and Q ads of CAT at different electrodes can also be determined based on the Anson equation. The chronocoulometry experiments were performed based on bare GCE (Fig. 7b), PEDOTM/GCE (Fig. 7c), and f-SWCNTs/PEDOTM/GCE (Fig. 7d) in 0.1 M PBS (pH 7.0) in the absence and presence of 0.1 mM CAT. These plots of Q versus t 1/2 show good linear relationships after background subtraction. These slopes are 9.795 × 10−6, 1.329 × 10−4, and 3.107 × 10−4 C s−1/2, and the intercepts (Q ads) are 3.609 × 10−7, 2.585 × 10−5, and 6.250 × 10−5 C for bare GCE PEDOTM/GCE and f-SWCNTs/PEDOTM/GCE, respectively. As can be seen from Table 1, according to the Anson equation and the equation Q ads = nFAГ s , the diffusion coefficient and adsorption capacity (Г s ) can be obtained. In comparison to bare GCE and PEDOTM/GCE, the f-SWCNTs/PEDOTM/GCE has larger adsorption capacity of CAT, which is beneficial to the improvement of the detection limit of CAT.

(A) The plot of Q–t curves of bare GCE (a), PEDOTM/GCE (b), and f-SWCNTs/PEDOTM/GCE (c) in 0.1 mM K3[Fe(CN)6] containing 0.1 M KCl. Insert: the plot of Q–t 1/2 curves on GCE (a′), PEDOTM/GCE (b′), and f-SWCNTs/PEDOTM/GCE (c′). The plot of Q–t curves of (B) bare GCE, (C) PEDOTM/GCE and (D) f-SWCNTs/PEDOTM/GCE in the absence (a) and presence (b) of 0.1 mM CAT. Insert: the plot of Q–t 1/2 curves of (B) bare GCE, (C) PEDOTM/GCE and (D) f-SWCNTs/PEDOTM/GCE in the absence (a′) and presence (b′) of 0.1 mM CAT
According to the totally irreversible oxidation, the standard heterogeneous rate constant (k s) at the f-SWCNTs/PEDOTM/GCE can be calculated based on the Velasco equation [42]:
where E p is the peak potential and E p/2 represents the potential when I = I p/2 in linear sweep voltammetry. In this experiment, E p − E p/2 = 172 mV, D = 4.880 × 10−5 cm2 s−1, F = 96485 C mol−1, v = 50 mV s−1, R = 8.314 J mol−1 K−1, and T = 298 K. The k s value was calculated to be 4.174 × 10−3 cm s−1, indicating a relatively fast electrode reaction process.
Electrochemical determination of CAT
Under the optimum conditions, various concentrations of CAT solutions were prepared to explore the relationship between peak currents and concentrations of CAT. The voltammetric response for different concentrations of CAT in 0.1 M PBS (pH 7.0) are illustrated in Fig. 8. As can be seen in the inset of Fig. 8, the oxidation peak current has a good linear relationship with the CAT concentration range from 0.039 to 40.84 μM. The regression equation is i pa (μA) = 64.0396 + 4.0492C (μM) (R 2 = 0.9997). The current sensitivity is calculated to be 4.05 μA μM−1. At a signal-to-noise ratio of 3 (S/N = 3), the detection limit is found to be 0.013 μM. The analytical performance of the f-SWCNTs/PEDOTM/GCE is compared with other modified electrodes reported in the literature. As shown in Table 2, the proposed electrode exhibited wider linear range and lower limit of detection. The satisfactory result can be attributed to the synergetic effect of f-SWCNTs and PEDOTM, in which f-SWCNTs provide a large surface area to increase the loading amount of CAT with excellent electrocatalytic performance for the determination of CAT. Meanwhile, the electron transfer on the electrode surface can be accelerated and the electrochemical signal is amplified due to the outstanding conductivity of PEDOTM layer. These results indicated that the proposed composite modified electrode is a good platform for the sensitive detection of CAT.

a CVs responses of f-SWCNTs/PEDOTM/GCE for the addition of 0.0398–40.843 μM CAT into continuously stirred 0.1 M PBS (pH 7.0). Insert: the linear calibration curve. Accumulation time: 150 s, accumulation potential: 0.3 V
Stability, selectivity, and applicability of f-SWCNTs/PEDOTM/GCE
In order to test the repeatability of the f-SWCNTs/PEDOTM/GCE, 0.1 mM CAT solution was successively measured ten times using the same f-SWCNTs/PEDOTM/GCE modified electrode. The relative standard deviation (RSD) of i pa1 and i pa2 are 1.19 and 1.37 %, respectively (Fig. S2), suggesting the good repeatability of the modified electrode. In addition, it also shows the excellent storage stability at room temperature, with a variation of response current less than 4 % for 7 days.
Moreover, interference studies were carried out by adding various foreign species to a fixed amount of 0.1 mM CAT in PBS (pH 7.0). The results showed that ions such as K+, Na+, Fe3+, Cu2+, SO4 2−, PO4 3−, and NO3 − in a 100-fold concentration; glucose, glycine, and citric acid in a 10-fold concentration; and ascorbic acid at a 5-fold concentration had no interference with the detection of CAT. The change of the peak currents of CAT was less than 5 %, suggesting that f-SWCNTs/PEDOTM/GCE has good selectivity.
In addition, under optimized conditions, the f-SWCNTs/PEDOTM/GCE was used to detect the content of CAT in commercial green tea three times. The content of CAT in commercial green tea is 158.2 μg mL−1 and the RSD is 0.25 %, indicating that the modified electrode can be sufficient for practical application without sample purification step. Satisfactory results revealed that the proposed sensor was stable, efficient, and selective for the detection of CAT in practical samples.
Conclusions
In this work, we constructed a simple, sensitive, and reliable CAT electrochemical sensor based on conducting polymer PEDOTM and carbon nanotubes f-SWCNTs. The f-SWCNTs/PEDOTM/GCE nanocomposite modified electrode exhibited a strong electrocatalytic activity for the oxidation of CAT due to the synergic effect of the electrocatalytic activity and strong adsorption ability of f-SWCNT with the advantages of PEDOTM (good water solubility and high conductivity). The sensing platform demonstrated very sensitive CAT detection capability, and less than 0.013 μM CAT could be detected. Furthermore, the modified electrode also exhibited good reproducibility and long-term stability, as well as high selectivity, which provided a promising sensing platform for the trace-level determination and analysis of CAT in the human diet.
References
Janeiro P, Bret AMO (2004) Catechin electrochemical oxidation mechanisms. Anal Chim Acta 518:109–115
Arts ICW, Hollman PCH (1998) Optimization of a quantitative method for the determination of catechins in fruits and legumes. J Agric Food Chem 46:5156–5162
Yang L-J, Cheng T, Xiong H-Y, Zhang X-H, Wan S-F (2009) Electrochemical properties of catechin at a single-walled carbon nanotubes–cetylramethylammonium bromide modified electrode. Bioelectrochemistry 75:158–162
Cook NC, Samman S (1996) Flavonoids: chemistry, metabolism, cardioprotective effects, and dietary sources. J Nutr Biochem 7:66–76
El-Hady D, El-Maali N (2008) Selective square wave voltammetric determination of (1)-catechin in commercial tea samples using beta-cyclodextrin modified carbon paste electrode. Microchim Acta 161:225–231
Higdona JV, Frei B (2003) Tea catechins and polyphenols: health effects, metabolism, and antioxidant functions. Crit Rev Food Sci Nutr 43(1):89–143
Fukumoto LR, Mazza G (2000) Assessing antioxidant and prooxidant activities of phenolic compounds. J Agric Food Chem 48(8):3597–3604
Wang R, Zhou W, Jiang X (2008) Mathematical modeling of the stability of green tea catechin epigallocatechin gallate (EGCG) during bread baking. J Food Eng 87:505–513
Moccelini SK, Fernandes SC, de Camargo TP, Ademir N, Iolanda Cruz V (2009) Self-assembled monolayer of nickel (II) complex and thiol on gold electrode for the determination of catechin. Talanta 78:1063–1068
Ozyurt D, Demirata B, Apak R (2007) Determination of total antioxidant capacity by a new spectrophotometric method based on Ce(IV) reducing capacity measurement. Talanta 71:1155–1165
Manabu M, Shin-ichi Y, Kenji K, Tokuji I (2002) Kinetic analysis and mechanistic aspects of autoxidation of catechins. Biochim Biophys Acta Gen Subj 1569:35–44
Chen Z, Zhang L, Chen G (2008) Microwave-assisted extraction followed by capillary electrophoresis-amperometric detection for the determination of antioxidant constituents in Folium Eriobotryae. J Chromatogr A 1193:178–181
El-Hady DA, El-Maali NA (2008) Determination of catechin isomers in human plasma subsequent to green tea ingestion using chiral capillary electrophoresis with a high-sensitivity cell. Talanta 76:138–145
Tsukagoshi K, Taniguchi T, Nakajima R (2007) Analysis of antioxidants using a capillary electrophoresis with chemiluminescence detection system. Anal Chim Acta 589:66–70
Dhalwal K, Shinde VM, Biradar YS, Mahadik KR (2008) Simultaneous quantification of bergenin, catechin, and gallic acid from Bergenia ciliata and Bergenia ligulata by using thin-layer chromatography. J Food Compos Anal 21:496–500
Khokhar S, Venema D, Hollman PCH, Dekker M, Jongen W (1997) A RP-HPLC method for the determination of tea catechins. Cancer Lett 114:171–172
Iacopini P, Baldi M, Storchi P, Sebastiani L (2008) Catechin, epicatechin, quercetin, rutin and resveratrol in red grape: content, in vitro antioxidant activity and interactions. J Food Compos Anal 21:589–598
Wang H, Provan GJ, Helliwell K (2003) Determination of rosmarinic acid and caffeic acid in aromatic herbs by HPLC. Food Chem 87:307–311
Wu J, Wang H, Liu F, Chen Z, Jiang JH, Shen G, Yu R (2005) Detection of catechin based on its electrochemical autoxidation. Talanta 65:511–517
El-Hady DA (2007) Selective and sensitive hydroxypropyl-beta-cyclodextrin based sensor for simple monitoring of (+)-catechin in some commercial drinks and biological fluids. Anal Chim Acta 593:178–187
Jarosz-Wilkołazka A, Ruzgas T, Gorton L (2004) Use of laccase-modified electrode for amperometric detection of plant flavonoids. Enzym Microb Technol 35:238–241
Nie G, Li C, Lin Z, Wang L (2014) Fabrication of a simple and sensitive QDs-based electrochemiluminescence immunosensor using a nanostructured composite material for the detection of tumor markers alpha-fetoprotein. J Mater Chem B 2:8321–8328
Nie G, Bai Z, Chen J, Yu W (2012) Simple label-free femtomolar DNA detection based on a nanostructure composite material: MWNT-doped poly(indole-6-carboxylic acid). ACS Macro Lett 1:1304–1307
Lin Z, Li C, Zhao D, Wu T, Nie G (2014) An electrochemical immunosensor for the tumor marker α-fetoprotein using a glassy carbon electrode modified with a poly(5-formylindole), single-wall carbon nanotubes, and coated with gold nanoparticles and antibody. Microchim Acta 181:1601–1608
Groenendaal LB, Jonas F, Freitag D, Pielartzik H, Reynolds JR (2000) Poly(3,4-ethylenedioxythiophene) and its derivatives: past, present, and future. Adv Mater 12:481–494
Elschner A, Kirchmeyer S, Lovenich W, Merker U, Reuter K (2011) PEDOT: principles and applications of an intrinsically conductive polymer. CRC Press, London
Jang J, Chang M, Yoon H (2005) Chemical sensors based on highly conductive poly(3,4-ethylenedioxythiophene) nanorods. Adv Mater 17:1616–1620
Wu L, Lu L, Zhang L, Xu J, Zhang K, Wen Y, Duan X, Yang F (2013) Electrochemical determination of the anticancer herbal drug shikonin at a nanostructured poly(hydroxymethylated-3,4-ethylenedioxythiophene) modified electrode. Electroanalysis 25:2244–2250
Stéphan O, Schottland P, Le Gall P-Y, Chevrot C, Mariet C, Carrier M (1998) Flectrochemical behaviour of 3,4-ethylenedioxythiophene functionalized by a sulphonate group. Application to the preparation of poly(3,4-ethylenedioxythiophene) having permanent cation-exchange properties. J Electroanal Chem 443:217–226
Lima A, Schottland P, Sadki S, Chevrot C (1998) Electropolymerization of 3,4-ethylenedioxythiophene and 3,4-ethylenedioxythiophene methanol in the presence of dodecylbenzenesulfonate. Synth Met 93:33–41
Xiao Y, Cui X, Hancock JM, Bouguettaya M, Reynolds JR, Martin DC (2004) Electrochemical polymerization of poly(hydroxymethylated-3,4-ethylenedioxythiophene) (PEDOT-MeOH) on multichannel neural probes. Sensors Actuators B 99:437–443
Wen Y, Dong L, Lu Y, He H, Xu J, Duan X, Liu M (2012) Poly(3,4-ethylenedioxythiophene methanol)/ascorbate oxidase/nafion-single-walled carbon nanotubes biosensor for voltammetric detection of vitamin C. Chin J Polym Sci 30:824–836
Feng Z, Yao Y, Xu J, Zhang L, Wang Z, Wen Y (2014) One-step co-electrodeposition of graphene oxide doped poly(hydroxymethylated-3,4-ethylenedioxythiophene) film and its electrochemical studies of indole-3-acetic acid. Chin Chem Lett 25:511–516
Yao Y, Zhang L, Wang Z, Xu J, Wen Y (2014) Electrochemical determination of quercetin by self-assembled platinum nanoparticles/poly(hydroxymethylated-3,4-ethylenedioxylthiophene) nanocomposite modified glassy carbon electrode. Chin Chem Lett 25:505–510
Zhang L, Wen Y, Yao Y, Xu J, Duan X, Zhang G (2014) Synthesis and characterization of PEDOT derivative with carboxyl group and its chemo/bio sensing application as nanocomposite, immobilized biological and enhanced optical materials. Electrochim Acta 116:343–354
Bard AJ, Faulkner LR (1980) Electrochemical methods: fundamentals and applications, 2nd edn. Wiley, New York
Yu YG, Cao DY, Luo X, Tang Y, Li H (2012) Sensitivity and selectivity determination of bisphenol A using SWCNT–CD conjugate modified glassy carbon electrode. J Hazard Mater 199–200:111–118
Zhou C, Liu Z, Dong Y, Li D (2009) Electrochemical behavior of o-nitrophenol at hexagonal mesoporous silica modified carbon paste electrodes. Electroanalysis 21:853–858
Yao Y, Wen Y, Zhang L, Wang Z, Zhang H, Xu J (2014) Electrochemical recognition and trace-level detection of bactericide carbendazim using carboxylic group functionalized poly(3,4-ethylenedioxythiophene) mimic electrode. Anal Chim Acta 831:38–49
Laviron E (1974) Adsorption, autoinhibition and autocatalysis in polarography and in linear potential sweep voltammetry. J Electroanal Chem Interfacial Electrochem 52:355–393
Anson FC (1964) Application of potentiostatic current integration to the study of the adsorption of cobalt (III)-(ethylenedinitrilo (tetraacetate) on mercury electrodes, Anal Chem 36(4):932–934
Velasco JG (1997) Determination of standard rate constants for electrochemical irreversible processes from linear sweep voltammograms. Electroanalysis 9:880–882
Acknowledgments
The authors would like to acknowledge the financial support of this work by the National Natural Science Foundation of China (No. 51463008, 51272096, and 51263010), GanPo Outstanding Talents 555 Projects, Jiangxi Provincial Department of Education (No. GJJ12595, GJJ13565), Youth Science and Technology Talent Training Plan of Chongqing Science and Technology Commission (cstc2014kjrc-qnrc10006).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Yuanyuan Yao and Long Zhang contributed equally to this work.
Rights and permissions
About this article

Cite this article
Yao, Y., Zhang, L., Wen, Y. et al. Voltammetric determination of catechin using single-walled carbon nanotubes/poly(hydroxymethylated-3,4-ethylenedioxythiophene) composite modified electrode. Ionics 21, 2927–2936 (2015). https://doi.org/10.1007/s11581-015-1494-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-015-1494-z